

Abstract
CTCF is a highly conserved zinc finger protein implicated in diverse genomic regulatory functions, including transcriptional activation/repression, insulation, imprinting, and X-chromosome inactivation. Here we re-evaluate data supporting these roles in the context of mechanistic insights provided by recent genome-wide studies and highlight evidence for CTCF-mediated intra- and inter-chromosomal contacts at several developmentally regulated genomic loci. These analyses support a primary role for CTCF in the global organization of chromatin architecture and suggest that CTCF may be a heritable component of an epigenetic system regulating the interplay between DNA methylation, higher-order chromatin structure, and lineage-specific gene expression.
Introduction
Genomes of higher eukaryotes are intricately packaged into several hierarchical levels of organization. Within each chromosome, DNA is wrapped around histones to form the 10 nm nucleosomal fiber, which is subsequently folded and looped into sophisticated higher-order structures. Although the precise geometrical configurations have not yet been definitively elucidated, emerging evidence suggests that chromatin structure has a marked effect on how the DNA sequence is interpreted during a vast array of cellular processes (Misteli, 2007). This complex structure-function relationship is best understood at the level of the 10 nm fiber, where chromatin accessibility to regulatory factors is modulated by the interplay between DNA sequence and a secondary layer of potentially heritable epigenetic marks (such as histone modifications and DNA methylation) that are dynamically accumulated throughout the lifetime of an organism in response to developmental and/or environmental cues (Bernstein et al., 2007).
In addition to chromatin structure, emerging evidence suggests that spatial positioning of genomic segments within the three-dimensional nuclear space also has an important influence on genome function (Fraser and Bickmore, 2007; Lanctot et al., 2007). Globally, chromosomes occupy distinct territories with respect to each other in interphase nuclei. Chromatin is not static within these territories, but is dynamically condensed and decondensed in a manner that generally correlates with transcriptional activity. More recently, advanced imaging technologies in combination with new molecular approaches have uncovered an extensive, and previously underestimated, network of local and long-range intra-chromosomal loops and inter-chromosomal contacts. Many of these interactions, both in cis and in trans, are likely stochastic and a consequence of the need to share common resources within nuclear sub-compartments. However, in specific instances long-range chromatin contacts have been linked to important biological processes such as olfactory receptor choice (Fuss et al., 2007; Lomvardas et al., 2006), monoallelic gene expression (Apostolou and Thanos, 2008; Ling et al., 2006), X-chromosome inactivation (Bacher et al., 2006; Xu et al., 2006), and developmentally regulated transcription (Spilianakis et al., 2005). As a consequence of these discoveries, the field is shifting from the study of transcription at a specific gene locus in a linear manner to three-dimensional models of gene regulation.
The complexity of genomic interactions within these mammalian ‘chromatin networks’ raises the possibility that factors exist with a sole and/or primary purpose of mediating intra- and inter-chromosomal contacts. Here, we discuss evidence that CCCTC-binding factor (CTCF) is a leading candidate for this role. Mechanistic insights and unique distribution patterns revealed by recent genome-wide analyses across multiple cell types suggest a global role for CTCF that departs significantly from canonical regulatory functions. We review these data in combination with evidence for CTCF-mediated loops at several developmentally regulated loci in order to support a principal role for CTCF in genome-wide organization of chromatin architecture. We conclude by highlighting compelling recent data suggesting that CTCF may be a heritable component of an epigenetic system regulating the complex interplay between DNA methylation, higher-order chromatin structure, and developmentally regulated gene expression.
Multivalent Factor, Widespread Regulatory Functions
CTCF is highly conserved in higher eukaryotes. The full-length protein contains an eleven zinc finger central DNA binding domain displaying close to 100% homology between mouse, chicken, and human embedded within slightly more divergent N- and C-termini (Ohlsson et al., 2001). On the basis of its ability to bind to a wide range of variant sequences as well as specific co-regulatory proteins through combinatorial use of different zinc fingers, CTCF was originally described as a ‘multivalent factor’ (Filippova et al., 1996). This unique structural feature provided the first clue suggesting a versatile role in genome regulation distinct from most zinc finger proteins.
Several lines of evidence highlight the critical importance of CTCF during diverse cellular processes. First, CTCF homozygous knockout mice exhibit early embryonic lethality prior to implantation (Heath et al., 2008; Splinter et al., 2006). Maternal depletion of CTCF in oocytes prior to fertilization markedly disrupts normal progression to the blastocyst stage (Fedoriw et al., 2004). In adult organisms, the protein is ubiquitously expressed in a manner similar to a housekeeping gene across most metazoan tissues. Expression levels and nuclear distribution patterns vary in a cell type-specific manner, indicating an important role in maintenance of phenotypic diversity and gene expression patterns in adult tissues. CTCF levels impact cellular function, as ectopic overexpression or RNA interference (RNAi)-based depletion in mammalian cell culture results in lineage-specific effects on growth, proliferation, differentiation, and apoptosis (Torrano et al., 2005). Tissue-specific CTCF depletion results in misregulated transcription of hundreds of genes in oocytes (Wan et al., 2008) and dramatically deregulates cell-cycle progression during T lymphocyte lineage commitment within the thymus (Heath et al., 2008).
Recent studies mapping genome-wide occupancy and distribution patterns in multiple divergent cell types have further reinforced the concept that downstream effects on cellular function are a consequence of the essential role for CTCF in genome regulation. Ren and colleagues reported 13,804 CTCF binding sites in IMR90 human fibroblasts by employing chromatin immunoprecipitation (ChIP) in combination with a series of tiling arrays representing non-repeat sequences of the human genome (ChIP-chip). In these cells, the global distribution patterns were reported as 46% intergenic, 22% intronic, 12% exonic, and 20% within 2.5 kb of promoters (Kim et al., 2007). In an independent study (Barski et al., 2007), Zhao and colleagues identified 20,262 CTCF target sites in resting human CD4+ T cells using ChIP in combination with high-throughput sequencing (ChIP-Seq). This particular dataset was subsequently re-analyzed (Jothi et al., 2008) with a new algorithm enabling detection of binding events with enhanced sensitivity and specificity, resulting in a refined number of binding sites to 26,814 and a genome-wide distribution of 45% intergenic, 7% 5′UTR, 3% exonic, 29% intronic, 2% 3′UTR, and 13% within 5kb of the transcription start site (TSS). Most recently, ChIP-Seq analyses reveal 39,609 CTCF binding sites in mouse embryonic stem (ES) cells (Chen et al., 2008), as well as 19,308, and 19,572 in HeLa and Jurkat cells, respectively (Cuddapah et al., 2009). It is not clear whether these cell-type specific differences in occupancy are functionally significant or merely due to differences in computational and experimental procedures employed by independent investigators.
Genome-wide data sets have enabled the identification of a ~11–15 bp core consensus sequence that is remarkably consistent in all cell types assessed by independent studies using different motif discovery algorithms. Given the ability of CTCF to bind numerous variant sequences, it is surprising at first glance that such a high percentage of genome-wide binding sites (~75% in fibroblasts and >90% in resting CD4+ T cells) can be represented by a core consensus. Recently, however, Renda et al. demonstrate high affinity binding (KD~10−10) to a specific 12bp variation of the core consensus with only 4–5 central zinc fingers (Renda et al., 2007). By contrast, early studies in which fingers are deleted in a stepwise manner from either end have reported the association of many more fingers with an extended 50–60bp sequence (Ohlsson et al., 2001), suggesting that binding may be partially stabilized by interactions between peripheral fingers and nucleotides surrounding the core consensus. Systematic characterization of zinc finger binding as a function of each consensus variant and surrounding divergent 50bp sequence, as well as the elucidation of the crystal structure in each scenario will be important to reconcile these discrepancies.
CTCF’s capacity to confer vastly different functions has been attributed to the interplay between zinc finger engagement and the underlying sequence. Soon after its initial discovery, it was proposed that “during formation of a CTCF-DNA complex, both DNA and CTCF polypeptide allosterically ‘customize’ their conformation to engage different zinc fingers, either for making base contacts or to make a target-specific surface that determines interactions with other nuclear proteins” (Ohlsson et al., 2001). Based on this property, the versatile functions of CTCF are generally described according to a model in which CTCF conformation is a function of differential zinc finger binding to divergent consensus sequences, resulting in different binding partners, different post-translational modifications, and, ultimately, multiple functional roles.
Activator, Repressor, or Insulator?
CTCF is implicated in diverse roles in gene regulation ranging from context-dependent promoter activation/repression, enhancer blocking and/or barrier insulation, hormone-responsive silencing, genomic imprinting, and, most recently, long-range chromatin interactions. These functions can now be critically assessed in the context of recent genome-wide analyses.
Early studies: evidence for transcription factor role
CTCF was first isolated and cloned by Lobanenkov and colleagues on the basis of its ability to bind to highly divergent 50–60 bp sequences within the promoter-proximal regulatory region of the chicken c-myc gene (Klenova et al., 1993; Lobanenkov et al., 1990) and immediately downstream of two conserved alternative transcriptional start sites (TSSs) in the human/mouse c-myc gene (Filippova et al., 1996). In both studies, heterologous reporter gene plasmids driven by a small portion of the c-myc promoter are used to support the conclusion that CTCF is a transcriptional repressor. In parallel with this work, Negative protein 1 (NeP1) was identified by Renkawitz and colleagues as a result of its ability to bind to a modular silencing element -2.4kb upstream of the chicken lysozyme gene (Baniahmad et al., 1990). Subsequent cloning and characterization of this protein revealed it was identical to CTCF (Burcin et al., 1997). Here CTCF was also reported as a transcriptional repressor based on the observation that the full composite silencer, containing a CTCF binding site adjacent to a thyroid hormone response element, synergistically decreased reporter gene expression (Kohne et al., 1993). However, it should be noted that the CTCF binding site alone had minimal effect on reporter gene expression and the full composite element displayed highly variable results dependent on the cell line used as a model system. In a subsequent study, CTCF was also purified from HeLa cell nuclear extracts for its ability to bind immediately upstream from the Amyloid β-Protein Precursor TSS. In this case, in vitro experiments and transgene assays were used to support the conclusion that CTCF can also serve as a transcriptional activator (Vostrov and Quitschke, 1997).
These seminal studies continue to be widely referenced as evidence supporting the direct role for CTCF as a classic transcription factor. Although useful in suggesting that CTCF can have an effect on transcription, it is important to point out that they rely on heterologous transgene assays that do not represent the native in vivo context of the endogenous genomic locus. Therefore, we ask if this putative role as a transcriptional activator/repressor is consistent with the global view for CTCF provided by genome-wide studies.
Genome-wide distribution compared to transcription factors
Global distribution patterns reveal key differences between CTCF and factors thought to function via canonical transcriptional mechanisms. CTCF binding sites strongly correlate with gene density with a correlation coefficient essentially equivalent to that of the general transcription factor TAF1. However, closer inspection of the distribution patterns revealed a notable difference; the majority (~85%) of TAF1 binding sites are localized within 2.5kb of the TSS, whereas the average CTCF distance from promoters is 48 kb, with only ~20% displaying promoter-proximal localization (Kim et al., 2007). Not all data are consistent with this theme, however, as CTCF displays markedly similar distribution patterns to the transcriptional repressor NRSF and activator STAT1 (Jothi et al., 2008). Further work will be necessary to reconcile these differences and to solidify the functional insights that are appropriately speculated from distribution patterns.
A powerful insight into the unique nature of CTCF is provided by the concurrent ChIP-Seq analyses of genome-wide binding sites for 15 transcription factors and/or co-regulatory proteins in mouse ES cells (Chen et al., 2008). Ng and colleagues identify over 3500 “Multiple-transcription factor binding loci” (MTL) which are associated with four or more transcription factors. MTLs are classified according to two general groups, the first containing Nanog, Sox2, Oct4, Smad1, and STAT3 and the second containing n-myc, c-myc, E2f1, and Zfx. By contrast to these transcriptional regulators, only a very small percentage of CTCF binding sites coincide with MTLs. Although correlations are established between transcription factor occupancy and gene expression levels for many proteins, the binding pattern of CTCF does not appear to predict ES cell-specific gene expression. Furthermore, whereas most factors associate near a particular class of genes, it appears that CTCF does not correlate with any particular gene type. Taken together, these differences suggest a role for CTCF distinct from traditional regulatory proteins.
Evidence for an insulator role
Insulators are classically defined by two experimental properties, namely the ability to block communication between adjacent regulatory elements in a position-dependent manner (enhancer blocking (EB)) and the capacity to buffer transgenes from position effects caused by the spread of repressive heterochromatin from adjacent sequences (barrier). As recently pointed out (Gaszner and Felsenfeld, 2006), most of our knowledge about insulators comes from experiments that rely on transgene constructs. For example, the “enhancer blocking assay” involves placement of the putative insulator sequence between an enhancer and a promoter driving a reporter gene. This transgene is transiently transfected or stably integrated into a defined cellular phenotype and the position-dependent ability to impede the enhancer, as determined by the level of reporter gene expression, is reported as the degree of insulation. The major limitations of this assay are that it typically involves heterologous enhancer/promoter sequences and an ectopically expressed plasmid outside of its native genomic context.
The first link between EB insulation and CTCF was proposed by Felsenfeld and colleagues based on their discovery that CTCF binds to the 5′HS4 insulator sequence upstream of the chicken β-globin locus (Bell et al., 1999). Another CTCF-dependent insulator sequence was subsequently identified by the same group at the 3′ end of this domain (Saitoh et al., 2000) in parallel with the discovery of four CTCF binding sites within the Imprinted Control Region (ICR) of the mammalian H19/Igf2 locus (Bell and Felsenfeld, 2000; Hark et al., 2000; Kanduri et al., 2000; Szabo et al., 2000). EB transgene assays provided the first evidence that CTCF is necessary for functional insulation by these sequences. After these initial discoveries, and prior to genome-wide queries, many independent investigators have reported a number of CTCF-bound DNA sequences that behave according to the experimental definition of EB insulation in transgene systems. Beyond these experimental systems, however, the actual endogenous enhancers that are functionally blocked by each individual CTCF insulator within their endogenous genomic location have not yet been identified. One possible exception may be the unique case at the imprinted H19/Igf2 ICR. This raises the question: Is CTCF-based insulation specific to a small number of genes, or is it a global regulatory mechanism?
A global role for insulation in vivo
Evidence supporting a widespread role for CTCF-based insulation is provided by an elegant computational analysis conducted by Lander and colleagues (Xie et al., 2007). In this approach, a database of conserved non-coding elements demonstrating strong conservation across 12 mammalian species is used to discover >200 new regulatory motifs (12–22 nt). Among this list, three similar sequences together represent an unusually large number of conserved instances (~15,000) in the human genome. Subsequent affinity capture experiments prove CTCF binding to these motifs, which displayed remarkable similarity to the core CTCF consensus identified in parallel with ChIP-chip and ChIP-Seq studies. A powerful component of this analysis is the use of a computational method to assess functional insulator activity of these sequences in their native genomic context without using a heterologous transgene system. Specifically, a data set of divergent gene pairs <20kb apart is divided into two groups: those separated by a CTCF binding site (n=80) and those that are not (n=883). Using this approach, they demonstrate that co-regulated expression of divergent gene pairs is decoupled to background levels if CTCF binds between genes. The high conservation of the ~15,000 motifs identified with these computational algorithms without searching for CTCF a priori increases the likelihood that they represent functional insulator elements.
CTCF is generally considered to function solely via EB mechanisms with no direct role in barrier insulation. This conclusion is based on early transgene experiments demonstrating that CTCF binding could be abrogated without affecting functional barrier activity of the extended 1.2kb chicken β-globin insulator (Recillas-Targa et al., 2002). More recently, a genome-wide mapping study uncovers a small but statistically significant proportion of CTCF binding sites localized to boundaries between active and repressive chromatin domains marked by histoneH2A lysine 5 acetylation (H2AK5Ac) and H3 lysine 27 trimethylation (H3K27me3), respectively (Cuddapah et al., 2009). Interestingly, the genomic regions demarcated by CTCF sites show minimal overlap between HeLa and CD4+ T cells, supporting a compelling new mechanistic explanation for CTCF-mediated lineage-specific regulation. Although it now appears likely that CTCF is directly acting as a boundary/barrier element in some subset of genome-wide binding sites, we cannot rule out the possibility that in many or all instances CTCF is merely adjacent to an independent sequence conferring barrier function in the same manner as the chicken β-globin locus. For the purpose of this review, we use a broader definition for CTCF-based insulators as a subclass of DNA sequences that interfere with inappropriate communication between neighboring regulatory elements and/or independent chromatin domains.
Overall, the percentage of genome-wide CTCF binding sites that represent truly functional EB and/or barrier elements remains to be determined. Mechanistic insights provided by genome-wide studies are all consistent with a global role for CTCF as an insulator, but do not rule out the possibility that a smaller, more obscure number of sites may be related to instances of non-canonical transcriptional regulation via protection against DNA methylation (Boumil et al., 2006; Engel et al., 2006; Filippova et al., 2005). Is this model sufficient to explain the widespread and multifunctional roles for CTCF across all tissue types at thousands of different regulatory regions and genomic contexts? Could there be a more unifying mechanism?
Insulation via Chromatin Loops
Traditional linear models of transcriptional regulation provide only a partial picture and have recently been broadened to account for the three-dimensional structure of chromatin within the nuclear space. Accumulating data suggest that CTCF mediates long-range chromatin interactions between insulator elements. First, homodimers and multimers formed by Flag-tagged CTCF have been detected in HeLa cells with mass spectroscopy and these physical interactions have been confirmed in vitro by yeast 2-hybrid (Yusufzai et al., 2004). This result was first interpreted by Felsenfeld and colleagues as evidence that vertebrate CTCF can mediate chromatin loop formation in a manner similar to insulators in D. melanogaster (Gerasimova et al., 2000). In parallel, evidence has been presented that CTCF-bound DNA probes will dimerize into complexes in vitro, but only if the probes encode divergent sequences (Pant et al., 2004). These data, coupled with evidence that the C-terminus of CTCF co-associates with the zinc finger domain in GST-pull down assays, suggest that CTCF molecules binding to different sequences have conformations permissive for direct and/or indirect interactions and, as a consequence, looping out of the intervening DNA. Genome-wide data are also consistent with the possibility of CTCF-mediated loops. Because the ChIP assay generally cannot distinguish between direct and indirect binding, we cannot rule out the possibility that the marked differences in CTCF binding sites identified by computational methods (~15,000) vs. Chip-based approaches (e.g. ~ 26000 in T cells; ~40000 in ES cells) are in part due to indirect interactions caused by long-range chromatin contacts.
These data provide indirect evidence that CTCF can mediate chromatin contacts, but what is the evidence that insulation in vertebrates requires loop formation? It was first proposed that CTCF confers EB insulation by tethering chromatin to subnuclear structures on the basis of observations that nucleophosmin/B23 binds to the chicken β-globin 5′HS4 and that localization of this insulator sequence to the nucleolar periphery is dependent on CTCF binding (Yusufzai et al., 2004). More recently, the first direct evidence that loop formation can occur via contact between two CTCF-bound insulators in vivo has been provided by Dean and colleagues (Hou et al., 2008). Specifically, this study demonstrates that an ectopically inserted human insulator in transgenic mice forms an aberrant loop that disrupts transcription in vivo. Beyond this transgenic model, however, direct evidence that functional enhancer blocking requires loop formation between two endogenous CTCF-bound insulators in their native genomic context has not been definitively demonstrated to date. A notable exception may be the imprinted H19/Igf2 locus.
Taken together, these results suggest that CTCF-bound insulators have the capacity to form genome-wide loops. Are all CTCF binding sites insulators that confer their EB and/or barrier function by the formation of loops? Or is it possible that the primary role for CTCF is as a chromatin looper, with all downstream effects on transcription as secondary consequences of these physical interactions? Because this mechanism is likely dependent on the genomic context, we examine experimental evidence for CTCF-mediated loop formation at endogenous loci.
Beyond Insulation: Nothing But a Looper?
Direct evidence for CTCF-mediated intra-chromosomal interactions between distal regulatory elements has been reported at specific developmentally regulated genes. These studies predominantly rely on fluorescence in situ hybridization (FISH) and “chromosome conformation capture” (3C) for the detection of long-range interactions. Principles behind these techniques and their limitations are discussed in detail elsewhere (Dekker, 2006; Fraser and Bickmore, 2007).
Mouse H19/Igf2: Loops essential for allele-specific silencing at an imprinted locus
The molecular mechanism by which mammalian CTCF confers EB function and the potential link between insulation and long-range chromatin interactions is best understood at the imprinted Igf2/H19 locus. At this genomic region the role of CTCF in the establishment and maintenance of imprinting and parent-of-origin gene expression patterns during development. The imprinting control region (ICR) immediately upstream of the H19 gene is essential for regulation of the entire locus and contains four CTCF binding sites. DNA methylation abrogates CTCF binding to these sequences, and all four sites demonstrate methylation-sensitive insulator activity in EB assays (Bell and Felsenfeld, 2000; Hark et al., 2000; Kanduri et al., 2000; Szabo et al., 2000). CTCF binding to the maternal ICR is also essential for imprint maintenance in somatic cells, as well protection against aberrant, de novo methylation at multiple differentially methylated regions (DMRs) throughout the extended locus (Fedoriw et al., 2004; Kurukuti et al., 2006; Rand et al., 2004; Schoenherr et al., 2003; Szabo et al., 2004). Here we present a simplified model of this locus in order to more clearly illustrate the role for CTCF in chromatin organization (Figure 1a,b).
Figure 1. Allele-specific chromatin contacts at an imprinted locus.
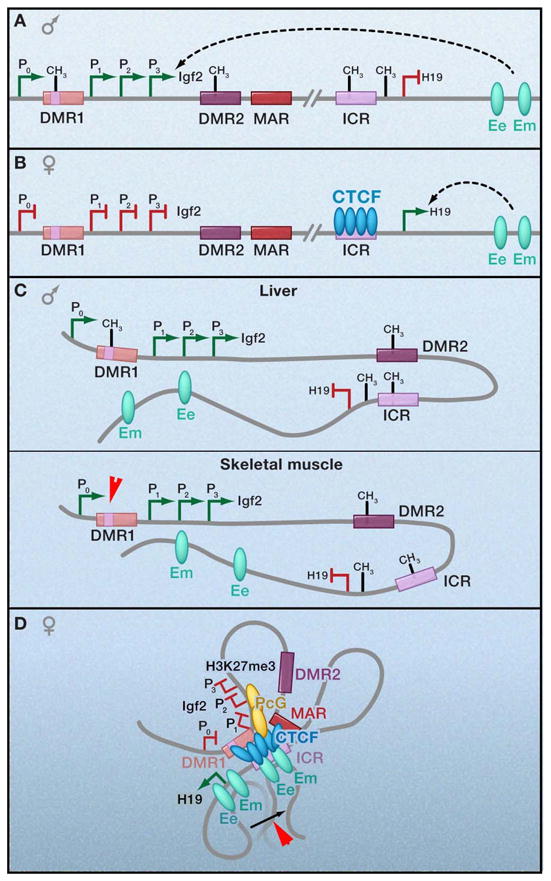
(a,b) Linear depiction of the mouse H19/Igf2 locus. The maternally expressed non-coding H19 gene is located approximately 90kb downstream from the gene encoding Insulin-like growth-factor 2 (Igf2) that is expressed exclusively from the paternal allele. The imprinting control region (ICR) ~2kb upstream of H19 contains four CTCF binding sites and is essential for regulation of the entire locus. Differentially methylated regions (DMRs), such as DMR1 upstream of Igf2 promoters (P1, 2, 3) and DMR2 within Igf2 exon 6, act in concert to regulate reciprocal, allele-specific expression patterns from a shared set of downstream enhancers at 8kb (Ee: endodermal tissue enhancer) and 25kb (Em: mesodermal tissue enhancer) downstream of the H19 gene. –CH3, DNA methylation. Green ovals, enhancers. (c,d) Schematic 3-D models illustrating allele-specific patterns of CTCF binding, DNA methylation, and chromatin looping. Although loops are illustrated here via CTCF multimerization, it is not yet clear if these long-range interactions can be attributed to CTCF binding to all sites and subsequent dimerization or if CTCF detection via chromatin immunoprecipitation is due to indirect interactions via looping.
Prior to the view of transcriptional regulation in three-dimensions, the mechanism by which methylation-sensitive CTCF binding serves as a functional EB insulator was poorly understood. On the maternal allele the ICR is unmethylated, CTCF is bound, and the Igf2 promoter is prevented from accessing the enhancers downstream of H19 (Figure 1b). On the paternal allele, the ICR is methylated, which abrogates CTCF binding and ICR-mediated insulation, resulting in functional communication between the promoters and the enhancers in order to activate Igf2 expression (Figure 1a). H19 expression is repressed on the paternal allele due to promoter methylation, suggested to be linked to loss of CTCF binding at the methylated ICR (Pant et al., 2003). Importantly, we note that this offers an explanation for any potential direct role for CTCF as a transcription factor, in that the promoter-proximal placement may merely protect genes from methylation-dependent silencing.
Experimental evidence that CTCF confers these allele-specific effects on transcription via long-range interactions has accumulated since the invention of 3C (Dekker et al., 2002). Reik, Ohlsson, and colleagues first reported CTCF-loops at this locus using an elegant transgenic model that enables detection of parent-of-origin chromatin interactions in cells derived from mouse fetal liver (Kurukuti et al., 2006; Murrell et al., 2004). On the maternal allele (Figure 1d), where Igf2 expression is silent, 3C data are generally consistent with a model in which the CTCF-bound ICR contacts both the upstream DMR1 and a downstream matrix attachment region (MAR3). Genetic studies confirm that CTCF binding to the ICR is required for both the formation of ICR-DMR1-MAR3 contacts and prevention of maternal-specific enhancer-promoter interactions. In a subsequent study, Yoon et al. provide evidence that downstream enhancers and proximal Igf2 promoters can also be detected in close spatial proximity to the maternal ICR (Yoon et al., 2007). ChIP experiments confirm CTCF enrichment specifically on the maternal allele at the ICR and DMR1, as well as within the unmethylated P2/P3 Igf2 promoters and the downstream enhancers (Li et al., 2008; Yoon et al., 2007). Notably, CTCF binding to both DMR1 and the proximal Igf2 promoter is abrogated upon genetic deletion or mutation of the maternal ICR, suggesting that CTCF binding to these distal regulatory elements in vivo occurs through a mechanism dependent on ICR-mediated loop structures. Taken together, these results strongly indicate that multiple CTCF-mediated contacts form a tightly coiled loop around the maternal Igf2 gene, making the proximal promoter inaccessible to downstream enhancers.
By contrast, on the paternal allele (Figure 1c), in which Igf2 expression is active, all DMR sequences are methylated, CTCF is not bound, and the majority of this region appears to be more fluidly accessible for contact with the enhancer (Kurukuti et al., 2006). Tissue-specific enhancer-promoter interactions have been detected, as the endodermal enhancer is markedly enriched at the active paternal Igf2 gene promoter in liver cells, while mesodermal enhancer–promoter contacts are enriched in muscle cells (Yoon et al., 2007). It is necessary to note that some conflicting data exist between studies regarding the ability of the maternal Igf2 promoter to contact the ICR, as well as the fluidity of the downstream enhancers to contact the entire length of the paternal Igf2 gene. These relatively minor discrepancies likely reflect the dynamic nature of chromatin contacts within a heterogeneous population of cells and may also be due in part to differences in 3C procedures or cell lines used by independent investigators. For the purposes of this review, we assume that interactions detected in these seminal reports are not mutually exclusive and can be integrated into one unified working model.
A recent study by Hoffman and colleagues (Li et al., 2008) has shed light into a mechanism by which CTCF-mediated loops confer silencing around the imprinted maternal Igf2 gene. They report that contact between the CTCF-bound ICR and P2/P3 promoters facilitates the recruitment of Suz12, a member of Polycomb Repressor Complex 2, and the subsequent acquisition of the silencing chromatin modification H3K27 trimethylation (H3K27me3) on the maternal allele. Genetic experiments indicate that CTCF binding at the ICR is essential, and potentially sufficient, to coordinate both CTCF and Suz12 binding at the Igf2 promoters and to also regulate the finely-tuned balance between additional activating and repressive chromatin modifications throughout the entire domain in an allele-specific manner (Han et al., 2008). Notably, RNAi-mediated Suz12 knockdown results in de-repression of the maternal Igf2 gene, suggesting that loop formation alone is not sufficient for silencing and may require Suz12. It will be important to determine if this polycomb-based silencing mechanism is more broadly required beyond this specific imprinted locus.
Overall, CTCF has multiple roles at the H19/Igf2 ICR, including: (1) allele-specific insulation of maternal Igf2 promoter from downstream enhancers, (2) initiation of H19 transcription, (3) maintenance of allele-specific DNA methylation imprints, and (4) organization of locus-wide chromatin modifications. These data are consistent with the idea that the CTCF-bound ICR confers multiple functions via its primary role as a chromatin looper. Understanding the kinetics of CTCF binding, Suz12 recruitment, loop formation, and chromatin modification will be important to critically assess if CTCF functions as a canonical EB insulator at this locus.
Mouse β-globin: Active chromatin hub for developmentally regulated activation
The murine β-globin locus has been extensively characterized (Figure 2a) and is an excellent model system for the role of CTCF-based chromatin contacts during developmentally regulated expression of a lineage-specific gene cluster. By contrast to the maternal H19/Igf2 allele, these CTCF-mediated contacts are associated with transcriptional activation. Two highly conserved CTCF consensus sequence variants, 5′HS5 within the locus control region (LCR) and 3′HS1 20kb downstream, were first reported by Felsenfeld and colleagues (Farrell et al., 2002). Subsequent ChIP experiments confirmed direct CTCF binding to three sites upstream (5′HS85, 5′HS62/60, and 5′HS5) and one downstream (3′HS1) of the mouse β-globin locus (Bulger et al., 2003; Splinter et al., 2006). Hypersensitivity and CTCF occupancy patterns at these sites vary in a cell type-specific manner (Figure 2b–d).
Figure 2. Cell type-specific interchromosomal interactions at a developmentally regulated locus.
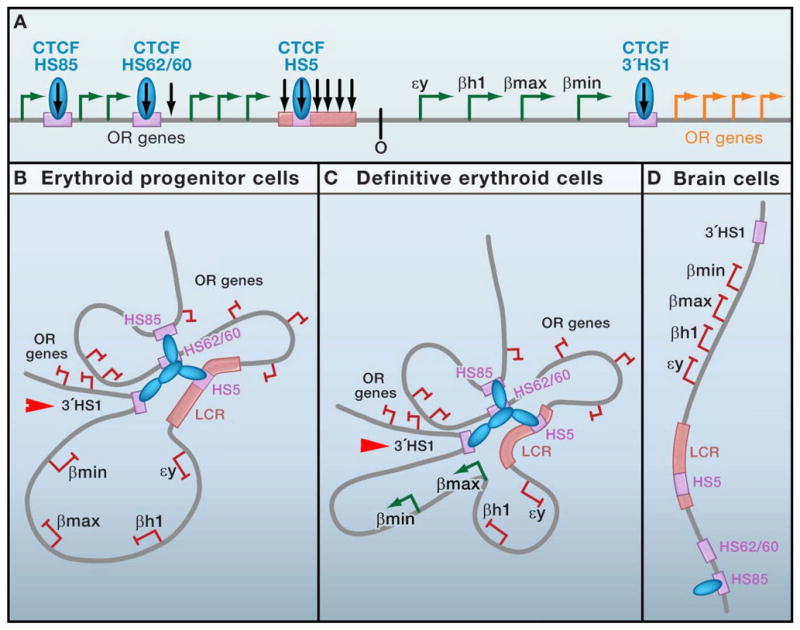
(a) Schematic representation of the mouse β-globin locus. Four globin genes (solid green arrows) are embedded within a larger olfactory receptor genes cluster (open arrows). Developmentally regulated globin expression (εy and βh1 in primitive erythroid cells; β-major and β-minor in definitive erythroid cells) is regulated in part by a series of cis-acting regulatory elements surrounding the locus. An upstream locus control region (LCR) containing 6 DNase I-hypersensitive sites (HSs) is required for high-level transcription. Three CTCF binding sites have been identified upstream (5′HS85, 5′62/60, and 5′HS5) and one 20kb downstream (3′ HS1) of the gene. Black arrows, HSs. (b–d) Diagrams illustrating lineage-specific CTCF binding patterns, 3C-based intra-chromosomal interactions, and globin gene expression profiles in erythroid progenitors (b), definitive erythroid cells (c), and non-erythroid brain cells (d). Although loops are illustrated here via CTCF multimerization, it is not yet clear if CTCF binds directly to DNA at each site or if detection by chromatin immunoprecipitation is due to indirect interactions via looping.
De Laat and colleagues have used 3C to demonstrate that CTCF-bound regulatory sequences throughout the β-globin locus come into spatial proximity to form an ‘active chromatin hub’ (ACH) during tissue-specific activation of specific globin genes. In mouse erythroid progenitors that do not yet express β-globin (Figure 2d), physical contact between distal upstream elements (HS85, HS62/60), the 5′ portion of the LCR (including HS5), and the downstream 3′HS1 site are detected prior to gene activation (Palstra et al., 2003; Splinter et al., 2006). These pre-established contacts are maintained in definitive erythroid cells (Figure 2c), where active β-major and β-minor genes also preferentially interact with the LCR, resulting in looping out of transcriptionally silent embryonic isoforms (βh1 and εy). By contrast, in non-globin expressing cells derived from embryonic brain tissue (Figure 2b), long-range interactions between CTCF binding sites in a ~200kb region surrounding the locus are not detected (Tolhuis et al., 2002). ChIP analysis confirm CTCF binding only to HS85 in these cells, suggesting that developmentally-regulated occupancy can in part modulate the formation of appropriate chromatin contacts (Splinter et al., 2006).
Most recently, the putative functional link between CTCF binding, loop formation, and globin gene expression has been explored (Splinter et al., 2006). Conditional knockdown as well as genetic experiments mutating the 3′HS1 CTCF binding site results in destabilization of CTCF contacts in erythroid progenitors to levels equivalent to the linear topology found in non-erythroid brain cells. Surprisingly, disruption of this hub via 3′HS1 mutation has no effect on kinetics or levels of globin gene expression during erythroid differentiation. An explanation for this surprising result is provided by a recent genome-wide analysis detecting >60 inter- and intra-chromosomal contacts with the LCR 5′HS2 in cells derived from tissues where the globin genes are both transcriptionally active (liver) and inactive (brain) (Simonis et al., 2006). This observation coupled with the high number of CTCF binding sites throughout the larger context of this locus suggests that there may be some redundancy in chromatin contacts that may not be readily abrogated by deletion of one regulatory element. Notably, CTCF-mediated contacts around this locus occur prior to gene activation and are not disrupted by inhibition of RNA polymerase II (Palstra et al., 2008), indicating that loop formation is not simply a consequence of transcription.
The functional significance of CTCF-based insulation at this locus is equally unclear. CTCF-bound 3′HS1 displays strong EB activity in transgene assays (Farrell et al., 2002) and, in principle, could prevent inappropriate LCR activation of downstream olfactory receptor (OR) genes. However, genetic disruption of CTCF binding to 3′HS1 has no effect on OR transcription in erythroid cells (Splinter et al., 2006). Furthermore, in transgenic mice, both 3′HS1 and 5′HS5 are dispensable for normal globin expression patterns, suggesting that EB transgene assays may not be sufficient to predict functional insulation in vivo (Bender et al., 2006; Bender et al., 1998). It will be interesting to see if future work identifies specific distal enhancers blocked by this putative insulator element in the endogenous locus or provides evidence for a marked transition in chromatin structure demarcated by CTCF (Bulger et al., 2003).
Taken together, these data support a critical role for CTCF in gathering together regulatory elements into an active chromatin hub, but suggest that the exact loops formed by a specific CTCF binding site may be dispensable to create chromatin conformations favorable for transcription. This is a very different picture than loops at the imprinted H19/Igf2 locus and suggests that, at least in some loci, CTCF has a structural role independent from transcription in establishing chromatin contacts.
Human MHC-II: Cytokine-induced loops for co-regulated gene activation
Boss and colleagues report the first evidence for CTCF-mediated long-range interactions at a subset of genes within the human major histocompatibility complex class II (MHC-II) locus. Specifically, they focus their analysis on a region containing two divergently expressed MHC-II genes (HLA-DRB1 and HLA-DQA1) co-regulated by an intergenic element termed XL9 (Figure 3). CTCF binds to XL9, which shows EB activity in transgene assays, suggesting it may be a functional insulator in vivo (Majumder et al., 2006). This locus is an excellent model for the study of developmentally regulated transcription because MHCII genes are constitutively expressed in B lymphocytes, macrophages, and dendritic cells, while treatment with the cytokine interferon-γ (IFNγ) can induce transcription in non-expressing cell types.
Figure 3. An inducible chromatin loop.
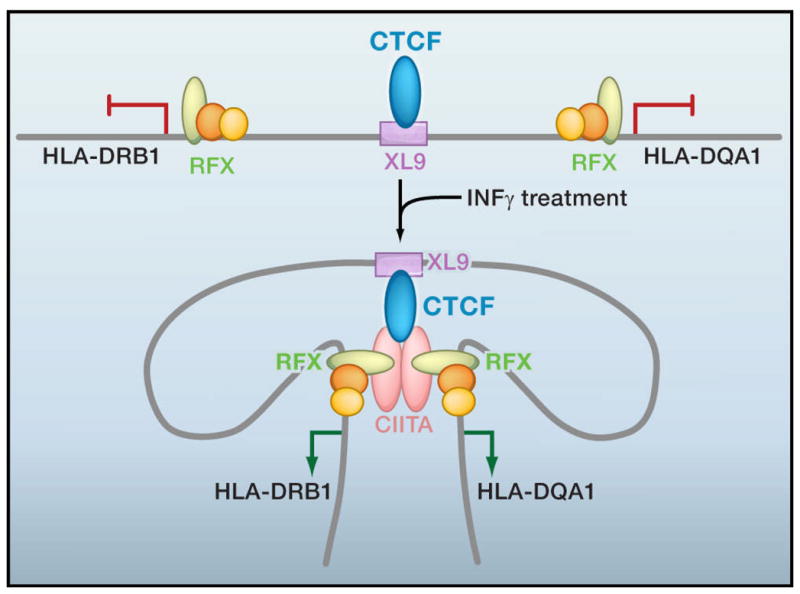
Model for cytokine-induced loop formation at the human major histocompatibility complex class II (MHC-II) locus. CTCF binds to the XL9 enhancer element between two co-regulated genes, HLA-DRB1 and HLA-DQA1, driven by divergent promoters. Transcription factors RFX, CREB, and NF-Y bind to regulatory sequences within the proximal promoter of MHCII genes when they are transcriptionally inactive. Interferon γ (IFNγ) treatment induces transcription in non-expressing cell types by upregulating the non-DNA binding co-activator CIITA, which subsequently forms a heteromultimer with RFX-CREB-NF-Y-bound promoters and the CTCF-bound enhancer in parallel with HLA-DRB1 and HLA-DQA1 gene activation.
3C analysis demonstrates physical interactions between the CTCF-bound XL9 intergenic enhancer and two divergent promoters upstream of HLA-DRB1 and HLA-DQA1 genes (Majumder et al., 2008). RNAi-mediated CTCF knockdown markedly reduces XL9-promoter interactions and also decreases expression of both genes, suggesting that CTCF-based loops may be essential for co-regulated gene activation at this particular locus. To our knowledge, this study is the first to report two new CTCF-mediated phenomena, namely the ability to form loops via hetero-multimerization and the ability to form transcriptionally-functional loops in response to cytokine treatment (Figure 3). Promoter-XL9 loop formation is dependent on a complex formed by CTCF, the co-activator CIITA, and the RFX transcription factor bound to a protein complex (containing CREB, NF-Y, and RFX) assembled at the proximal-promoter. Knockdown of any of these three factors (e.g. CIITA, RFX, or CTCF) abolishes long-range interactions. In order to study the interplay between loops and transcription, the authors use non-MHC-expressing epithelial cells as a model system in which the CIITA transactivating factor is not expressed and the HLA-DRB1/DQA1 regulatory region is in a relatively linear conformation and transcriptionally silent. Upon stimulation with IFNγ, kinetic experiments indicate that CIITA is expressed prior to the concurrent formation of CTCF-based contact with divergent gene promoters and initiation of HLA-DRB1 and HLA-DQA1 gene expression. Genetic studies will be necessary to determine if this interaction is a cause or a consequence of transcriptional activation.
The unique nature of these contacts, between an intergenic enhancer and a promoter, suggests that CTCF may not be functioning as a canonical EB insulator at this locus in vivo. Nevertheless, we cannot rule out the possibility that CTCF is blocking inappropriate regulatory elements contained within the larger 4 Mb MHC-II locus. A full characterization of all possible enhancer sequences, CTCF binding sites, and physical contacts throughout this region will be necessary to determine the structure and role(s) for these physical interactions. On the basis of multiple CTCF binding sites identified by genome-wide studies, it is tempting to speculate that the entire MHC-II domain assembles into an active chromatin hub reminiscent of the β-globin locus.
Overall, data from these three developmentally regulated genes suggest that CTCF may predominantly function in spatial organization of chromatin topology via loop formation, with insulation and/or downstream effects on transcription a secondary consequence of the genomic context of the endogenous locus. We note that the models described here are limited by their two-dimensional representation and do not reflect the possible topological configurations adopted within the three-dimensional space of the nucleus. Nonetheless, this evidence supports the hypothesis that the sequence of the CTCF binding site and the spatial positioning of each consensus with respect to genes and other regulatory elements would dictate the types of CTCF-based chromatin loops structures formed (Figure 4a–d). Mechanistic models to explain how looping between CTCF insulators mediates downstream effects on transcription are an active area of investigation and have been reviewed elsewhere (Gaszner and Felsenfeld, 2006).
Figure 4. Potential classes of CTCF-mediated contacts.
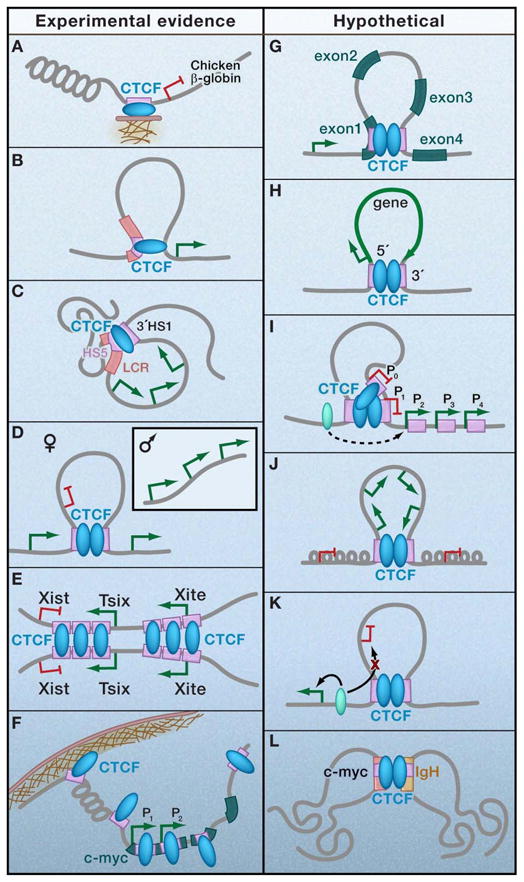
Experimental evidence for certain sub-classes of CTCF loops exists (a–f), whereas others can be hypothesized based on genome-wide distribution patterns (g–l). (a) Anchoring via direct attachment to subnuclear structures such as the nucleolus and/or nuclear matrix; (b) Transcriptional regulation via contact between intergenic locus control region and promoter-proximal regulatory element; (c) Active chromatin hub around multiple co-regulated genes via contact between multiple distal CTCF binding sites; (d) Mono-allelic gene expression via allele-specific contacts between multiple imprinted regulatory elements; (e) X-chromosome inactivation or mono-allelic gene expression via interchromosomal contacts between regulatory elements in trans; (f) Global nuclear organization via demarcation of lamina-associated domains (LADs; (g) RNA polymerase II pausing and/or termination via intragenic contacts between introns and exons; (h) RNA processing or transcriptional re-initiation via a single gene 5′-3′ loop; (i) Alternative promoter selection via contact between two insulator elements demarcating transitions in chromatin structure; (j) Boundary/barrier loops to demarcate independently regulated chromatin domains containing a co-regulated gene-dense cluster via contact between two insulator elements; (k) Enhancer blocking loops that topologically separate inappropriate enhancer-promoter interactions via contact between two insulator elements; (l) Interchromosomal translocations via contacts between two regulatory elements in trans.
More than Loops…a Nuclear Web?
Recent evidence supports a much larger role for chromosome intermingling between territories than previously thought and it may not be a coincidence that CTCF binding sites have been implicated in many of the interchromosomal contacts identified to date. Ohlsson and colleagues used a strategy termed “circular chromosome conformation capture” (4C) that leverages the principles of 3C in combination with high-throughput sequencing in order to query genome-wide loci in close spatial proximity with the H19/Igf2 ICR (Zhao et al., 2006). This analysis detected >100 interactions in cells derived from neonatal mouse liver tissue samples. A critical feature of this 4C library is that it contains a significant number of sequences representing potential interchromosomal contacts in addition to the expected overrepresentation of cis-acting intrachromosomal interactions. Notably, in some cases sequences derived from as many as 4 separate chromosomes are detected within a single 4C clone, suggesting the potential for multiple simultaneous trans interactions. By contrast, Hoffman and colleagues used a conceptually similar technique and identified only 3 genome-wide interactions, one in cis and two in trans, with the CTCF-bound maternal H19/Igf2 ICR in mouse bone marrow-derived fibroblasts (Ling et al., 2006). The marked discrepancy between these two studies may be due to cell type or technique-dependent differences and the likely possibility that neither approach exhaustively identifies all interactions.
What is the evidence that CTCF is necessary for these interchromosomal contacts? By using transgenic mice that allow the two alleles to be distinguished, Zhao et al. discovered a high percentage of the 4C-identified interchromosomal interactions are specific for the maternal (CTCF bound) H19 ICR allele, and the majority of these contacts are lost upon genetic deletion of 3 out of 4 CTCF binding sites. Evidence that at least some of these interactions are functional is provided by specific examples in which gene expression was deregulated and the physical juxtaposition of two loci are lost as a result of global CTCF knock-down (Ling et al., 2006) or mutation of the CTCF consensus to abrogate binding (Zhao et al., 2006). It is not clear whether binding on both sequences by CTCF is essential in every case for interchromosomal interactions or if ChIP data detects indirect CTCF-DNA interactions via formation of a multi-component bridging complex.
Additional evidence supporting the role for CTCF in functional interchromosomal interactions comes from studies of X-chromosome inactivation (XCI) in mammals. In order to equalize dosage of X-linked genes between females and males, one female X chromosome is selected for silencing in a random manner in cells originating from the post-implantation epiblast. This process requires counting, choice, and mutually-exclusive silencing, and is controlled by a genomic locus termed the ‘X inactivation center’ (Xic) that contains multiple non-coding genes including Xist, Tsix, and Xite (reviewed in detail (Payer and Lee, 2008)). By using embryoid body-induced differentiation of mouse ES cells, a model that recapitulates the early stages of random XCI, it has been recently discovered that homologous X chromosomes come into close spatial proximity in a significant fraction of nuclei within a cell population (Bacher et al., 2006; Xu et al., 2006). This interchromosomal pairing event is transient, occurs in parallel with the onset of counting/choice, and is an important prerequisite for proper initiation of molecular events involved in XCI.
Several lines of evidence link CTCF to interchromosomal contacts involved in XCI (Figure 4e). Specific regions within the Xic sufficient for interchromosomal interactions, including sub-fragments within a 3.7 kb domain around Tsix as well as 5.6 kb of Xite, contain numerous CTCF binding sites (Xu et al., 2007; Xu et al., 2006). Insertion of transgenes representing these sequences into autosomes results in ectopic X-autosome (X-A) interactions and disruption of the normal pairing event between homologous wild type X chromosomes (Xu et al., 2007). RNAi-mediated knockdown of CTCF markedly reduces the frequency of wild type X-X pairing, as well as X-A pairing mediated by Tsix or Xite transgenes. Interestingly, intrachromosomal loops have also been detected between Tsix and Xite specifically on the inactive X chromosome during the time frame for counting/choice (Tsai et al., 2008). Although direct evidence for involvement of CTCF in these loops has not yet been reported, it is interesting to note that the genomic domains responsible for these interactions contain CTCF binding sites and are also the same fragments that mediate transient interchromosomal interactions.
What is the functional role for CTCF-mediated contacts during random XCI? CTCF depletion results in Tsix downregulation (Donohoe et al., 2007), as well as a markedly deregulated Xist accumulation (Donohoe et al., 2007; Xu et al., 2007), providing evidence, albeit indirect, that CTCF-mediated chromatin structures are important for proper expression of noncoding genes essential for the early stages of XCI. Another intriguing possibility [proposed in (Tsai et al., 2008)] is that intrachromosomal loops between Tsix/Xite could also be favorable for the transient interchromosomal interactions mediated by these fragments via “bundling” of CTCF molecules into a high affinity bridging complex. Does pairing require interactions between the same CTCF binding sites on each homologous Xic sub-fragment? Alternatively, can CTCF more promiscuously associate with any of the CTCF binding sites within the Xic on the opposite chromosome? Is CTCF homodimerization a necessary or sufficient component of the protein bridge? If so, what additional factors and regulatory mechanisms enable the transient and specific nature of this interaction? As answers to these questions emerge they may reveal principles more broadly applicable to other imprinted loci.
Overall, although these studies provide support for the notion that CTCF mediates interchromosomal interactions, they only represent the first step in this largely unexplored area. Are these interchromosomal interactions directed and functional, or just a coincidence of the need to share transcriptional machinery? To what extent are they cell type-specific? How dynamic are they and is the transient nature of these interactions absorbed into the averaged data? Are they specific to imprinted genes requiring mono-allelic expression? Interestingly, the high percentage of interchromosomal interactions with known or candidate imprinted domains suggests the intriguing possibility that, at least at the H19 ICR, CTCF-mediated interchromosomal contacts may function to control epigenetic information in trans (Zhao et al., 2006). Current evidence indicates that the majority of long-range chromatin interactions appear to be in cis (Simonis et al., 2006; Zhao et al., 2006), suggesting that only a small (but intriguing) proportion of CTCF-mediated contacts will turn out to be functional interchromosomal interactions.
The Audacity of Loops: Potential Classes and Roles
Chromatin loops are a ubiquitous structural element involved in many hierarchical levels of nuclear organization. Accumulating evidence (discussed above) has revealed several general subclasses of CTCF-mediated chromatin contacts involved in transcription (Figure 4a–e). What additional hypothetical classes of these interactions could be functionally important in a broader range of genomic regulatory processes?
van Steensel and colleagues recently identified >1300 sharply-defined, large (0.1–10 Mb) genomic regions in human lung-derived fibroblasts that interact with LaminB1 and, in principle, are generally localized to the nuclear periphery (Guelen et al., 2008). These ‘Lamina-associated domains’ (LADs) correlate with low gene density, promoter depletion of RNA polymerase II, and markedly decreased gene expression compared to the rest of the human genome. By aligning LADs with genome-wide CTCF binding sites, ~10–15% (365/2,688) of LAD borders are found adjacent (within 5–10 kb) to a CTCF binding site, revealing a potentially new role for CTCF in the demarcation of LAD boundaries. The ubiquitous distribution of ~15,000 CTCF binding sites and the potential for LADs to represent up to 40% of the fibroblast genome raises the possibility that this correlation is coincidental. More specifically, at least with the CTCF library generated in IMR90 fibroblasts, at most 3% of CTCF binding sites appear to demarcate LADs, while ~19% bind within LADs, leaving the majority (~80%) of CTCF binding sites unassociated with these domains (Pagie, L., van Steensel, B., personal communication). Nevertheless, we note that the enrichment of CTCF around LAD borders is significantly higher than can be expected by chance, indicating that this localization is non-random and unlikely to be merely due to the high overall density of genome-wide CTCF binding sites. What is the functional relevance of CTCF-mediated LAD demarcation? In one example, a LAD border maps to a CTCF binding site ~10kb upstream of the human c-myc gene (Figure 4f), where a dynamic rosette-like structure localized to the nuclear periphery can be envisioned given the number of well-characterized CTCF sites throughout the locus (Gombert et al., 2003). Future experiments in which CTCF is knocked down globally or binding is abrogated genetically at specific LADs will be highly informative toward establishing a causal mechanism for LAD demarcation.
Genome-wide CTCF distribution patterns provide insight into additional putative classes of CTCF loops. The high percentage of binding sites localized to the 5′UTR, exons, introns, and the 3′UTR suggest that intragenic and/or single-gene loops may have functional roles (Figure 4g, h). We speculate that putative loops formed between the 5′UTR and intron- or exon-localized CTCF binding sites could regulate transcription by interfering with a processive activating signal (e.g. RNA Polymerase II, histone modification) (Figure 4g). Although it may be difficult to fathom that one CTCF molecule could block Pol II, it is less difficult to imagine that a multimeric protein aggregate may serve as a physical barrier to a tracking signal. In support of this notion, in the specific case of the mammalian c-myc gene, a CTCF binding site maps precisely within a conserved +5→+45 sequence downstream of the P2 promoter critical for Pol II pausing and release. It would be very interesting to determine how many CTCF consensus sequences map to sites of polymerase pausing genome-wide (Core et al., 2008). It is also plausible that loop formation between the proximal promoter and the 3′UTR or 3′ end of a single gene could facilitate the coordination of transcription re-initiation and RNA processing in a manner similar to the loops detected in S. cerevisiae (O’Sullivan et al., 2004) (Figure 4h).
Ren and colleagues recently identified two classes of large 2 Mb genomic domains containing higher- and lower-than-average CTCF binding densities (Kim et al., 2007). Insights from the characterization of these domains implicate CTCF loops in two additional mechanisms. CTCF-poor regions flanked by a pair of CTCF binding sites contain an average of 2.5, with as many as 56, genes/domain and tend to encompass multiple co-regulated or developmentally-related genes (such as olfactory receptor gene clusters or keratin associated protein gene clusters). By contrast, the vast majority (>80%) of CTCF-enriched regions contain genes expressed by multiple alternative promoters, with examples including the T cell receptor β locus, T cell receptor α/δ locus, and the Immunoglobulin λ light chain locus. Taken together, these results suggest that CTCF-loops could be involved in alternative promoter selection or sequestering clusters of co-regulated genes into separate, independently regulated chromatin domains (Figure 4i, j). CTCF’s ability to demarcate boundaries between active and repressive histone modifications has been implicated in both of these genomic processes (Barski et al., 2007).
Finally, as discussed above, enhancer blocking via loop formation between two endogenous CTCF-bound insulator sites remains to be demonstrated in vivo at a non-imprinted locus (Figure 4k). Furthermore, on the basis of recent CTCF binding maps throughout the Igh locus (Degner et al., 2009), coupled with known binding sites around c-myc, it is tempting to speculate a role for CTCF, albeit unproven, in mediating interchromosomal contacts that facilitate the frequent and preferential Igh/myc translocations observed in Burkitt’s lymphoma (Osborne et al., 2007)(Figure 4l).
Mechanisms of Regulation
If CTCF does indeed have an essential role in the establishment and maintenance of chromatin organization during development, it must be capable of dynamically responding to environmental and biological cues in a subset of binding sites, as well as remaining stable and not responding in the presence of the same signal within completely different genomic contexts. Evidence is increasing that CTCF-based intra-chromosomal contacts show cell type specificity and can be altered in response to cytokines (see Figures 2, 3). Furthermore, preliminary data also suggests that CTCF-mediated interchromosomal interactions can be developmentally-regulated, as the specific CTCF-dependent network of interacting partners with the H19/Igf2 ICR in ES cells changes significantly upon embryoid body-induced differentiation (Zhao et al., 2006). Accumulating data suggest that a complex regulatory network may exist to modulate CTCF’s diverse functions. Here we broadly categorize previously identified regulatory mechanisms into those that alter CTCF binding and those that may facilitate the de novo formation, maintenance, or stabilization of CTCF loops without altering CTCF interaction with its cognate binding site (Figure 5).
Figure 5. Possible mechanisms for developmentally-regulated loop formation.
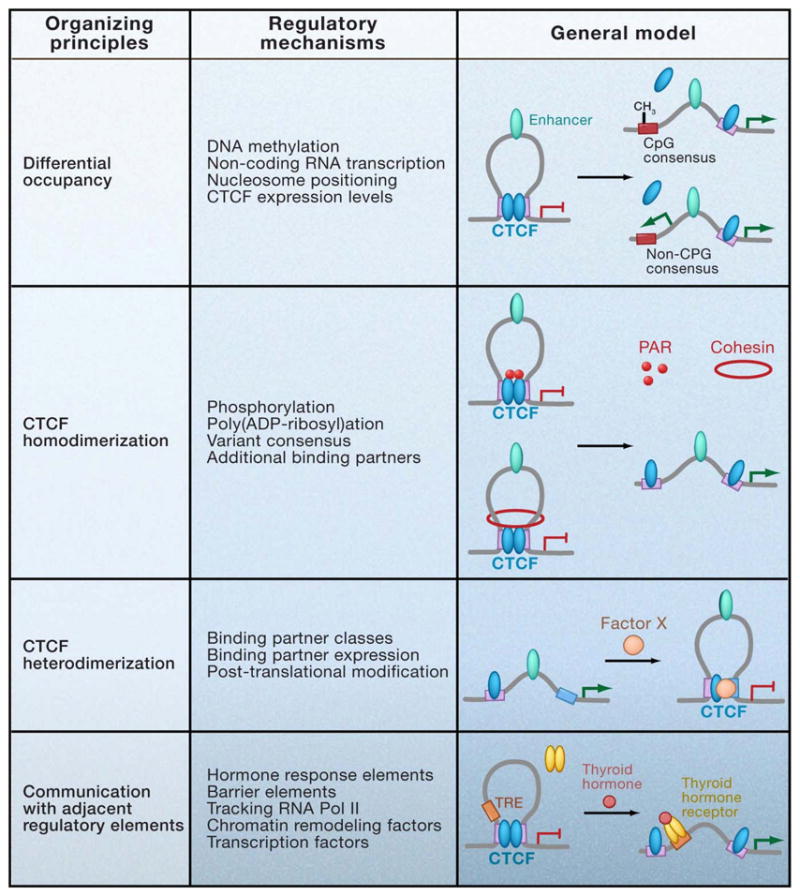
Previously identified mechanisms for mammalian insulator regulation are broadly categorized according to four general organizing principles, including those that alter CTCF occupancy and those that may facilitate the de novo formation, maintenance, or stabilization of CTCF loops without altering CTCF binding to its cognate consensus site. Models that generally illustrate specific regulatory mechanisms within each category are provided. -CH3, DNA methylation. Purple squares, CTCF consensus sites. Blue squares, thyroid hormone response element. Green ovals, enhancers. Blue ovals, CTCF. Red rings, cohesin. Pink dots, PARylation mark. Red ovals, Factor X representing any CTCF binding partner. Orange ovals, thyroid hormone receptor.
Regulation of CTCF Occupancy
Is there evidence on a genome-wide scale that CTCF can be regulated at the level of occupancy? A comparative analysis of ChIP-Seq data in HeLa, Jurkat, and CD4+ T cells reveals 40–60% overlap across cell types (Cuddapah et al., 2009). Furthermore, a smaller scale comparison using ChIP-chip data from ENCODE regions show at most 70% similarity between IMR90 fibroblasts and U937 erythroid progenitors (Kim et al., 2007). On the basis of these observations, it has been concluded that genome-wide CTCF binding is largely invariant between cell types. However, we suggest that the remaining ~25–50% may be functionally important for developmentally regulated gene expression. Gene ontology classification analysis of loci displaying differential binding will be necessary in order to elucidate the link between CTCF occupancy and tissue-specific chromatin interactions.
The best understood mechanism by which CTCF binding can be altered is via DNA methylation on CpG dinucleotides within and around the core consensus. For example, at the H19/Igf2 ICR all 4 CTCF binding sites are regulated by methylation to control allele-specific CTCF occupancy and subsequent loop formation. More recently, Pedone and colleagues used the chicken β-globin insulator to determine that methylation of only a single, specific CpG dinucleotide within the CTCF consensus principally affects binding of the protein (Renda et al., 2007). The general applicability of this finding to all sequence variations of the CTCF binding site has not yet been confirmed. CTCF regulation by methylation may not be only limited to imprinted genes where these marks are established early in embryogenesis. In principle, all CTCF binding sites with consensus variants containing CpG dinucleotides retain the potential for methylation-based regulation in response to biological or environmental signals.
At non-CpG binding sites, CTCF occupancy could be constitutive or regulated by alternative mechanisms. Bonifer and colleagues recently reported disruption of CTCF binding by transcription of an antisense non-coding RNA through an insulator element upstream of the chicken lysozyme gene (Lefevre et al., 2008). It is not yet known if abrogated CTCF binding is simply caused by tracking of transcriptional machinery during elongation or is due to non-coding RNA interactions with CTCF in trans. Data from this study also suggest that CTCF eviction is maintained by repositioning of nucleosomes over the binding site. This finding is corroborated by a recent genome-wide analysis indicating that nucleosome positioning may have a global role in regulating cell type-specific CTCF occupancy (Cuddapah et al., 2009).
Regulation of CTCF-based loops
Evidence that CTCF-mediated insulation can be altered in response to developmental signals without in vivo changes in binding indicates an additional layer of regulation with potential downstream effects on looping (Gombert et al., 2003). Because CTCF has an important role in protection against ectopic methylation, the ability to transiently alter chromatin loop structures without modifying CTCF binding would allow for more flexibility in gene regulation without permanently altering the epigenetic state of the cell. In principle, the potential for loop formation is directly linked to CTCF’s tertiary conformation, which is thought to be modulated in part by a combination of differential zinc finger binding to the underlying sequence and the surrounding genomic context. Assuming that CTCF homo- and/or hetero-dimerization is the organizing principle behind genome-wide chromatin contacts, the potential models for regulating loops are illustrated in Figure 5. The role for these mechanisms in loop formation has not yet been directly proven via 3C. Therefore, we restrict the discussion to mechanisms with a functional effect on CTCF-mediated EB insulation and/or reporter gene expression without altering CTCF occupancy.
CTCF function can be regulated by post-translational modifications such as phosphorylation and poly(ADP-ribosyl)ation (PARylation). The poly(ADP-ribose) (PAR) mark, in particular, has been ChIPed at >100 insulator sequences genome-wide and specifically detected at the CTCF-bound maternal H19 ICR (Yu et al., 2004). General inhibition of PAR polymerases (PARPs) results in abrogation of CTCF-mediated insulation by the H19/Igf2 ICR in EB transgene assays, as well as de-repression of maternal Igf2 without changing CTCF binding. Importantly, this trend was recapitulated in a library of EB transgenes representing >100 candidate mouse insulator sequences, implying a more general genome-wide role for CTCF PARylation in functional insulation. The observation that PARP inhibition did not alter CTCF occupancy together with the known role for CTCF-based loops at the H19/Igf2 ICR led to the suggestion that PAR modification may stabilize CTCF-mediated chromatin contacts. It is unclear why general PARP inhibition does not abrogate binding at its cognate CpG-consensus binding sites within the H19/Igf2 ICR, particularly in light of the observation that PARP-1 is essential to maintain DNA hypomethylation in vivo (Guastafierro et al., 2008). Similarly, phosphorylation within a highly conserved, four serine residue motif in the C-terminus markedly influences reporter gene expression without altering DNA binding activity, indicating that this post-translational modification may also regulate CTCF-homodimerization or association with different binding partners during development (Klenova et al., 2001).
Numerous potential CTCF binding partners have been reported ((Filippova, 2008; Wallace and Felsenfeld, 2007)). For brevity’s sake, we focus on the considerably smaller subset of proteins that have been detected in vivo via ChIP at functional mammalian insulator sequences linked to chromatin loop formation. Among this list, the PcG repressor Suz12 is interesting because recruitment to the maternal Igf2 promoter requires CTCF binding to the H19/Igf2 ICR 90kb downstream (discussed above). Another factor, the SNF2-like chromodomain helicase protein (CHD8), has been detected at several insulator sites, including two (H19/Igf2 ICR and β-globin 5′HS5) with a direct role in CTCF-mediated intrachromosomal contacts (Ishihara et al., 2006). RNAi-mediated knockdown of CHD8 abrogates insulator activity of the H19/Igf2 ICR in both EB transgene assays and on the endogenous maternal allele without disrupting CTCF binding. A functional link between the known role for CHD8 in chromatin remodeling and its putative recruitment to and/or involvement in CTCF-mediated chromatin loops has not yet been established. Finally, recent ChIP-chip analysis of a representative sample of ~250 CTCF binding sites identifies a particularly small subset (<5%), including the H19/Igf2 ICR, associated with RNA Pol II (Chernukhin et al., 2007). Although direct Pol II recruitment to CTCF cannot be ruled out, an alternative interpretation is that these interactions (shown to be biochemically weak in vitro) are merely a consequence of the ability of CTCF loops to physically block a tracking polymerase.
For all proteins mentioned thus far, it is still unclear if these interactions are locus-specific or more generally found at a larger number of functional CTCF binding sites. A notable exception is the recent exciting discovery that cohesin proteins are enriched at thousands of CTCF-bound insulator sites. Several genome-wide analyses across multiple cell lines demonstrate a range of 65–90% cohesin sites overlapping CTCF and 55–80% CTCF sites overlapping cohesin, indicating that although a high proportion of sites are shared, a subset of cohesin-only and CTCF-only binding sites exist (Parelho et al., 2008; Wendt et al., 2008). Recruitment of cohesin to its chromosome locations appears to be mediated by CTCF because depletion of CTCF leads to absence of cohesin binding at specific shared insulator sites. What is the role for cohesin proteins at CTCF binding sites? Because the four subunits (Smc1, Smc3, Rad21/Scc1, and Scc3/SA1) are thought to coalesce into a ring-like structure that mediates sister chromatid cohesion during mitosis, it is tempting to speculate that a similar mechanism could facilitate the stabilization of loops. In support of this notion, data indicate that specific cohesin subunits are essential for functional insulation by the H19/Igf2 ICR in both EB transgene assays as well as on the endogenous maternal allele (Stedman et al., 2008; Wendt et al., 2008).
The possibility that cohesin plays a regulatory role in CTCF-mediated loop formation has also been recently illustrated by studies at the mouse Immunoglobulin heavy-chain (IgH) locus. This particular region adopts a strikingly compact 3-D topology during B cell differentiation that has been implicated in the highly regulated process of V(D)J recombination (Jhunjhunwala et al., 2008). The putative role for CTCF/cohesin in these intrachromosomal contacts is highlighted by the recent mapping of >50 CTCF binding sites throughout the extended Igh locus (Degner et al., 2009). Several of these sites are located close to the recombination signal sequence, suggesting that CTCF-mediated loops may be important in positioning distant genomic sites in close proximity to facilitate appropriate rearrangements within the locus. Interestingly, CTCF occupancy is constitutive and remains unchanged during B cell differentiation, whereas cohesin is progressively recruited to CTCF-bound sites in a cell type-specific manner that parallels conformational changes in locus topology. Taken together, these data provide a strong indication that cohesin and CTCF will act in concert at many genomic locations to facilitate the formation of developmentally-regulated long range interactions.
Evidence by Majumder et al. indicates that CTCF heterodimerization can also lead to loop formation, suggesting an additional layer of complexity regarding classes of intrachromosomal contacts (Majumder et al., 2008). Although CTCF is generally considered the sole mammalian insulator protein, it is possible that other factors will be identified with a primary role in genome-wide organization of nuclear architecture or demarcating the boundaries of independently regulated chromatin domains (Galande et al., 2007). One potentially exciting example of this possibility is provided by the discovery that the methyl-CpG-binding protein Kaiso binds in vivo to a methylated version of the CpG-containing consensus variant upstream of the Retinoblastoma tumor suppressor gene (De La Rosa-Velazquez et al., 2007). Kaiso specifically recognizes the motif 5′-CmGCmG-3′ that is found within many methylated consensus variants with abrogated CTCF binding (Defossez et al., 2005). These data coupled with evidence that Kaiso binds to the C-terminus of CTCF in vitro suggest that reciprocal binding between CTCF and Kaiso at the same consensus sequence may be a compelling new mechanism for both blockage of loop formation as well as the possibility of alternative loops formation via heterodimerization. This finding also offers an important mechanistic link between CTCF loops and epigenetic silencing via protection/maintenance against the spread of DNA methylation.
Finally, it should be pointed out that another class of factors that bind to sequences adjacent to the CTCF consensus, but may not directly interact with CTCF, might also affect loop formation. For example, numerous instances have been reported of composite elements containing a CTCF binding site adjacent to a thyroid hormone response element (TRE) that show enhancer blocking activity in transgene assays. Thyroid hormone treatment can alter CTCF-mediated EB activity without affecting occupancy, suggesting a potential way to rapidly and transiently alter insulation via loop formation (Lutz et al., 2003). In another example, multiple paired co-regulatory elements containing adjacent binding sites for CTCF and the zinc finger protein Yin Yang 1 (YY1) have been identified at a critical regulatory region at the 5′ end of Tsix within the mammalian X-inactivation center (Donohoe et al., 2007). YY1 and CTCF co-immunoprecipitate in vivo and both proteins are essential for appropriate expression of Tsix. We will be curious to see if YY1-mediated transactivation of Tsix is linked to a yet to be determined role for YY1 in regulating CTCF-based intrachromosomal interactions between regulatory regions involved in X-inactivation (Tsai et al., 2008). Future experiments should aim to clarify the causes and consequences of these binding partners on loop formation and the molecular mechanism(s) by which each protein modulates how the diverse functions of CTCF are made manifest throughout the genome.
Are CTCF-Mediated Chromatin Structures Heritable?
An ‘epigenetic’ mark is classically defined as any heritable change in genome function that does not involve alterations to the primary DNA sequence. A less stringent definition for the ‘epigenome’ has evolved in modern day reports to encompass a growing list of chromatin modifications (e.g. DNA methylation, chemical histone modifications, non-coding RNA, and DNAse I hypersensitive sites). Beyond semantics, however, it will be essential to evaluate heritability in order to establish a functional role for a specific subset of these so-called epigenetic marks during cellular memory and lineage commitment. More specifically, epigenetic inheritance would involve propagation of an individual mark through multiple cell divisions, as well as maintenance throughout developmental stages of the adult organism (and potentially even transmission on to progeny) in the absence of the original signal and/or developmental cue.
According to these criteria, could higher-order chromatin architectures mediated by CTCF carry intrinsic epigenetic information? Do these topologies play an essential role in regulating phenotype-specific gene expression patterns during development? Is there a subset of CTCF and CTCF-loops that are transmitted through cell division after the initial establishment signal has dissipated? Although no clear unified picture has yet emerged, enough data exist to support a working model for locus-specific epigenetic inheritance of CTCF-mediated chromatin structures. Data are consistent with the notion that CTCF-based chromatin structures may be a heritable component of the cell type-specific epigenome, as well as the possibility that CTCF itself may serve as a genome-wide ‘Epigenetic Shield’ to protect a specific subset of imprinted and developmentally controlled regulatory sequences (particularly those involved in looping) against the aberrant acquisition of DNA methylation. If proven, these two ‘epigenetic mechanisms’ may not be mutually exclusive.
If CTCF is an epigenetic mark then it must retain (or restore) its information content, presumably, but not necessarily, by remaining bound to DNA despite disruptions in chromatin caused by transcription, DNA replication, and chromatin compaction/decompaction during mitosis. Mitotic chromosomes show positive staining for CTCF antibodies in HeLa cells (Burke et al., 2005), but this observation appears to be dependent on the chromosome fixation technique and was not repeatable in an independent study (Wendt et al., 2008). In live HeLa cells, a CTCF-eGFP fusion protein associates with mitotic chromosomes in a manner dependent on the zinc finger DNA binding domain (Burke et al., 2005). At the molecular level, a recent ChIP-chip analysis with ENCODE regions indicates that CTCF remains bound to ~50% (70/147) of its cognate binding sites in both asynchronous and mitotic ally-arrested HBL100 cells (Rubio et al., 2008). Although further work is necessary to determine the general applicability of these trends across multiple cell types, these results suggest that a specific subset of CTCF, but likely not all, remains associated with chromosomes during mitosis.
The relationship between CTCF and DNA methylation provides important clues into how the subset of CTCF binding sites that remain associated with chromatin is determined. At the maternal H19/Igf2 ICR, for example, CTCF binding to CpG-containing variations of its consensus is essential for maintenance of the hypomethylated state during post-implantation development as well as protection from de novo methylation in oocytes (discussed above). This mechanism is likely not restricted to imprinted genes, but is also more generally applicable to genomic elements that undergo spatiotemporally-regulated methylation through development. For example, CTCF functions as a boundary element upstream of the Retinoblastoma gene by protecting both its cognate CpG consensus variant and the proximal CpG-island promoter from methylation and, subsequently, gene silencing (De La Rosa-Velazquez et al., 2007). Importantly, a small-scale comparison between pre-B and thymocyte cell lines indicates that sites with unchanged CTCF occupancy are generally unmethylated, whereas specific sites displaying differential binding between lineages acquire CpG methylation (Parelho et al., 2008). Taken together, these data indicate that there are three classes of CTCF binding sites: non-CpG-, unmethylated CpG-, and methylated CpG-containing consensus variants.
Preliminary evidence also suggests that these subclasses show different patterns of epigenetic inheritance. Specifically at the CpG-containing H19/Igf2 ICR, for example, ChIP experiments indicate high levels of CTCF binding in interphase and mitotic HeLa cells (Burke et al., 2005). By contrast, at a non-CpG human c-myc insulator, Komura et al. demonstrate that DNase I hypersensitivity and CTCF binding are markedly reduced to almost background levels during mitosis in the same cell type (Komura et al., 2007). We highlight these data with the caveat that both observations were not repeatable in independent ChIP experiments also conducted in HeLa cells (Burke et al., 2005; Wendt et al., 2008). It is not yet clear whether these observations truly reflect a CpG-consensus-dependent pattern in CTCF epigenetic inheritance or whether they are merely a result of technical issues that remain to be addressed, such as the heterogeneity of mitotic cell populations and the potential contribution of contaminating non-arrested cells to the detected signal. Further experiments are therefore needed to resolve procedural and cell type-specific discrepancies. Nevertheless, existing evidence is sufficient to support the possibility that CTCF binding at CpG-consensus variants during the cell cycle may be necessary to protect against ectopic methylation, whereas CTCF binding/protection may be dispensable during mitosis at non-CpG sites, a concept that would explain a general decrease in CTCF binding to mitotic chromosomes.
Beyond CTCF binding, is there evidence that CTCF-mediated chromatin interactions remain intact during mitosis? Physical contacts between the CpG-containing H19/Igf2 ICR and the upstream DMR1 region have been detected with 3C in both interphase and mitotic cells, whereas enhancer-promoter interactions are not detected (Burke et al., 2005). This observation supports the compelling possibility that if CTCF stays bound to CpG-consensus sites during cell division then the loops formed by these elements may also remain intact. This immediately raises two key questions: What enables loop stabilization? How is the subset of contacts that remain intact through the cell-cycle determined? A recent study suggesting that CTCF governs the crosstalk between PARP-1 and the maintenance methyltransferase DNMT1 provides an interesting clue (Guastafierro et al., 2008). Blocking global PARP activity results in aberrant DNA hypermethylation in vivo, suggesting a role for both CTCF and PARylation in protection against ectopic methylation. Notably, transient overexpression of CTCF induces PARylation of both PARP-1 and CTCF, as well as inhibition of DNMT1 and global DNA hypomethylation. These data, coupled with the direct interaction between PARP-1 and CTCF in vitro and in vivo (Guastafierro et al., 2008; Yusufzai et al., 2004), suggest that the PAR mark may be critical for maintaining CTCF-bound CpG-consensus sites in a hypomethylated state. According to this model, upon CTCF eviction at a specific genomic locus, PARP auto-PARylation would decrease, leading to an increase in DNMTI activity at the target site. It is not yet known if constitutive PAR modification is the default state or if CTCF is PARylated only at a subset of CpG-containing sites involved in long-range interactions. Although no direct evidence proving a role for PAR in stabilizing CTCF-mediated chromatin contacts has yet been reported, the marked effect of PAR on insulator function genome-wide provides strong support for this possibility.
Overall, these data support a critical role for CTCF in coordinating the complex relationship between DNA methylation, PARylation, and higher-order chromatin loops. Evidence is consistent with a model (Figure 6) in which a small subset of CpG consensus-containing CTCF binding sites remain associated with CTCF during mitosis to re-establish chromatin topologies that may be essential for propagating phenotype-specific transcriptional and epigenetic programs. At these sites, CTCF, and potentially CTCF-mediated higher-order chromatin structures, would serve as an ‘Epigenetic Shield’ that functions to protect specific regulatory sequences (particularly those involved in looping) against the aberrant acquisition of DNA methylation until the appropriate time in development. Although the role for these structures in lineage commitment is still up for debate, data also support the notion that the chromatin loops mediated by CTCF also contain intrinsic epigenetic information. Essential toward validating this model and assigning the terminology “epigenetic” to CTCF and/or CTCF-based loops will be future experiments proving that the original signal required for promoting both the initial binding event and subsequent chromatin loop formation is absent while these CTCF-mediated structures are maintained through multiple rounds of cell division. Another challenge critical to ascertaining epigenetic inheritance will be determining whether and how specific CTCF-mediated chromosome topologies are propagated or restored after the perturbations caused by the replication fork during S phase. Although heritablity of CTCF through the cell cycle is controversial, more extensive analyses at multiple binding sites within diverse genomic contexts across several cell types may reveal unifying principles and purposes for epigenetic inheritance of CTCF-mediated higher-order chromatin architecture.
Figure 6. Epigenetic inheritance of CTCF-mediated chromatin loops.
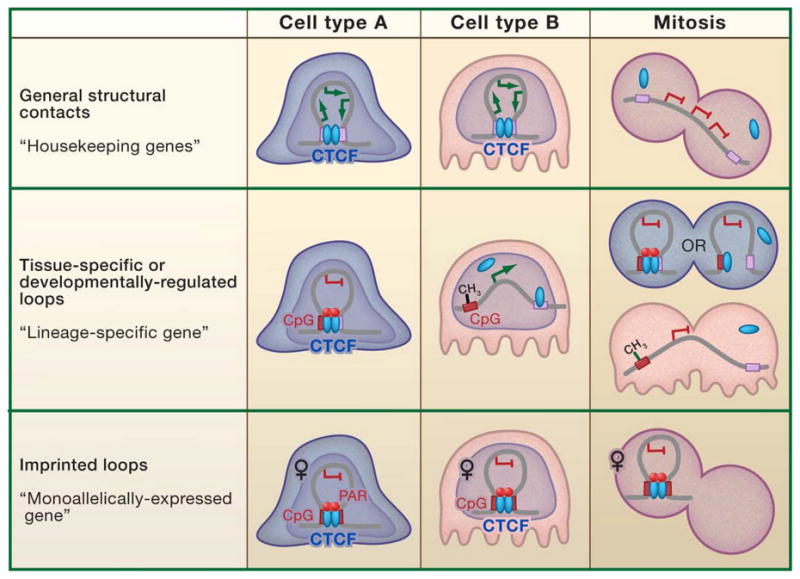
Data are consistent with a model centered on the interplay between DNA methylation, poly(ADP-ribosyl) ation, and higher-order chromatin loops. Three classes of CTCF consensus sites (non-CpG, unmethylated CpG-containing, and methylated CpG-containing) display differential patterns of CTCF binding and ‘heritability’ during cell division. In the case of general structural loops mediated by CTCF-bound non-CpG consensus sites (top panel), the majority would lose CTCF during mitosis. CTCF re-binding would generally be the default state and structural contacts across the cell would be re-established after every cell division. Conversely, a smaller subset of binding sites with unmethylated CpG-consensus variants or CpG dinucleotides in the surrounding 50–60 bp insulator element (middle, bottom panels) may retain CTCF binding through the cell cycle to protect against de novo methylation. Higher-order chromatin structures mediated by these particular elements would retain the potential for heritability through mitosis via cell type- and locus-specific post-translational modifications such as PAR or recruitment of protein binding partners such as cohesin. Among the putative classes of unmethylated CpG-consensus-mediated contacts, constitutive structures may be observed around key allele-specific imprinted genes (bottom panel), whereas acquisition of DNA methylation in response to developmental and/or environmental cues would result in abrogated CTCF binding and, consequently, potentially permanent disruption of contacts at a smaller subclass of loops encompassing developmentally-regulated loci (middle panel). Although only CpG-containing consensus sites retain the potential for heritable loop structures in this model, we cannot rule out the possibility of PAR-stabilized contacts between CpG- and non-CpG consensus sites or methylation-independent structures between non-CpG sites stabilized by PAR, cohesin, and/or additional unknown mechanism(s). PAR is shown only stabilizing loops between CpG consensus sites, but this has not yet been proven. Purple squares, non-CpG consensus sites. Red-hashed purple -squares, CpG-containing consensus sites. –CH3, DNA methylation. Red ovals, PARylation mark.
Perspectives and Future Questions
Evidence to date supports a genome-wide role for CTCF in the organization of developmentally regulated intra- and inter-chromosomal contacts. In light of the recent paradigm shift toward 3-D genome regulation, data are consistent with the notion that traditional regulatory functions of CTCF, including transcriptional activation, repression, insulation, and imprinting, may all be secondary effects of its primary, ubiquitous, and essential role as a genome-wide organizer of chromatin architecture. Although we favor this ‘CTCF-looping-centric’ viewpoint, it may be premature to rule out the possibility for instances where CTCF functions via looping-independent mechanisms by simply recruiting proteins involved in transcriptional activation.
Many important questions remain to be answered. Determination of the crystal structure of the zinc finger domain would lend significant understanding into how CTCF’s conformation and the specific zinc fingers associated with DNA change upon binding to divergent sequences. This knowledge will provide a reference point for inquiry into the poorly understood, and potentially locus-specific, relationship between CTCF conformation, post-translational modifications, association with additional co-regulatory proteins, and potential for chromatin contacts. Organizing principles for loop formation should be established, in particular an unambiguous conclusion regarding whether chromatin interactions involve CTCF homodimerization, heterodimerization, or if a single CTCF molecule can bring together multiple regulatory elements by serving as a substrate for proteins such as cohesin known to mediate chromatin contacts. Moreover, insight into the molecular mechanisms by which CTCF modulates chromatin structure and demarcates independent chromatin domains will be extremely important toward understanding if loops are a necessary and/or sufficient component of genome regulation. For example, a report by Yu et al. discovered that CTCF binding sites are generally localized in the chromatin linker region flanked by at least 20 symmetrically distributed nucleosomes, revealing a genome-wide role for CTCF in nucleosome positioning and a link to regulation of chromatin structure (Fu et al., 2008). Most recently, Merkenschlager, Fisher, and colleagues reported that cohesin is required for CTCF-mediated intrachromosomal contacts between specific sites around the interferon-γ locus (Hadjur et al., 2009). Determining whether cohesin recruitment is always essential for CTCF-mediated loop formation or if a subset of CTCF loops can form in a cohesin-independent manner will be important toward elucidating the mechanism by which CTCF promotes chromatin contacts.
The role for CTCF-mediated chromatin contacts as a heritable and functional component of the epigenome is still up for debate. Are CTCF-based structures established prior to and independent from lineage-specific transcription factor binding? Or, alternatively, is CTCF merely a generic looping device that is leveraged by the true master transcription factors to regulate gene expression across diverse genomic contexts? Recent development of technologies for high-throughput, unbiased identification of genome-wide chromatin interactions will enable the rigorous mapping of global chromatin architecture (Dostie et al., 2006). These maps will facilitate investigation into the dynamic nature of CTCF-based chromatin interactions on a genome-wide scale and how they are altered in a cell- and locus-specific manner. The role for higher-order chromatin structure as a potentially heritable carrier of epigenetic information and functional consequences of these structures on the establishment of lineage-specific gene expression profiles during development will be an exciting area of future inquiry. All information that we have at the moment indicates that CTCF is emerging as a (and perhaps the) master weaver of the mammalian genome.
Acknowledgments
The authors thank T. Misteli and J. M. Boss for valuable insights and critical review of the manuscript. We also acknowledge B. van Steensel and J. T. Lee, as well as past and present members of the Corces lab, in particular A. Bushey and E. Doorman, for helpful discussions. V.G.C. is funded by a U.S. Public Health Service Award (GM35463) from the NIH and J.E.P. is supported by an NIH NRSA postdoctoral fellowship.
References
- Apostolou E, Thanos D. Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell. 2008;134:85–96. doi: 10.1016/j.cell.2008.05.052. [DOI] [PubMed] [Google Scholar]
- Bacher CP, Guggiari M, Brors B, Augui S, Clerc P, Avner P, Eils R, Heard E. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat Cell Biol. 2006;8:293–299. doi: 10.1038/ncb1365. [DOI] [PubMed] [Google Scholar]
- Baniahmad A, Steiner C, Kohne AC, Renkawitz R. Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell. 1990;61:505–514. doi: 10.1016/0092-8674(90)90532-j. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- Bender MA, Byron R, Ragoczy T, Telling A, Bulger M, Groudine M. Flanking HS-62.5 and 3′ HS1, and regions upstream of the LCR, are not required for beta-globin transcription. Blood. 2006;108:1395–1401. doi: 10.1182/blood-2006-04-014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender MA, Reik A, Close J, Telling A, Epner E, Fiering S, Hardison R, Groudine M. Description and targeted deletion of 5′ hypersensitive site 5 and 6 of the mouse beta-globin locus control region. Blood. 1998;92:4394–4403. [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Boumil RM, Ogawa Y, Sun BK, Huynh KD, Lee JT. Differential methylation of Xite and CTCF sites in Tsix mirrors the pattern of X-inactivation choice in mice. Mol Cell Biol. 2006;26:2109–2117. doi: 10.1128/MCB.26.6.2109-2117.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Schubeler D, Bender MA, Hamilton J, Farrell CM, Hardison RC, Groudine M. A complex chromatin landscape revealed by patterns of nuclease sensitivity and histone modification within the mouse beta-globin locus. Mol Cell Biol. 2003;23:5234–5244. doi: 10.1128/MCB.23.15.5234-5244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burcin M, Arnold R, Lutz M, Kaiser B, Runge D, Lottspeich F, Filippova GN, Lobanenkov VV, Renkawitz R. Negative protein 1, which is required for function of the chicken lysozyme gene silencer in conjunction with hormone receptors, is identical to the multivalent zinc finger repressor CTCF. Mol Cell Biol. 1997;17:1281–1288. doi: 10.1128/mcb.17.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke LJ, Zhang R, Bartkuhn M, Tiwari VK, Tavoosidana G, Kurukuti S, Weth C, Leers J, Galjart N, Ohlsson R, et al. CTCF binding and higher order chromatin structure of the H19 locus are maintained in mitotic chromatin. EMBO J. 2005;24:3291–3300. doi: 10.1038/sj.emboj.7600793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Chernukhin I, Shamsuddin S, Kang SY, Bergstrom R, Kwon YW, Yu W, Whitehead J, Mukhopadhyay R, Docquier F, Farrar D, et al. CTCF interacts with and recruits the largest subunit of RNA polymerase II to CTCF target sites genome-wide. Mol Cell Biol. 2007;27:1631–1648. doi: 10.1128/MCB.01993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Rosa-Velazquez IA, Rincon-Arano H, Benitez-Bribiesca L, Recillas-Targa F. Epigenetic regulation of the human retinoblastoma tumor suppressor gene promoter by CTCF. Cancer Res. 2007;67:2577–2585. doi: 10.1158/0008-5472.CAN-06-2024. [DOI] [PubMed] [Google Scholar]
- Defossez PA, Kelly KF, Filion GJ, Perez-Torrado R, Magdinier F, Menoni H, Nordgaard CL, Daniel JM, Gilson E. The human enhancer blocker CTC-binding factor interacts with the transcription factor Kaiso. J Biol Chem. 2005;280:43017–43023. doi: 10.1074/jbc.M510802200. [DOI] [PubMed] [Google Scholar]
- Degner SC, Wong TP, Jankevicius G, Feeney AJ. Cutting edge: Developmental stage-specific recruitment of cohesin to CTCF sites throughout immunoglobulin loci during B lymphocyte development. J Immunol. 2009;182:44–48. doi: 10.4049/jimmunol.182.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat Methods. 2006;3:17–21. doi: 10.1038/nmeth823. [DOI] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell. 2007;25:43–56. doi: 10.1016/j.molcel.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel N, Thorvaldsen JL, Bartolomei MS. CTCF binding sites promote transcription initiation and prevent DNA methylation on the maternal allele at the imprinted H19/Igf2 locus. Hum Mol Genet. 2006;15:2945–2954. doi: 10.1093/hmg/ddl237. [DOI] [PubMed] [Google Scholar]
- Farrell CM, West AG, Felsenfeld G. Conserved CTCF insulator elements flank the mouse and human beta-globin loci. Mol Cell Biol. 2002;22:3820–3831. doi: 10.1128/MCB.22.11.3820-3831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw AM, Stein P, Svoboda P, Schultz RM, Bartolomei MS. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science. 2004;303:238–240. doi: 10.1126/science.1090934. [DOI] [PubMed] [Google Scholar]
- Filippova GN. Genetics and epigenetics of the multifunctional protein CTCF. Curr Top Dev Biol. 2008;80:337–360. doi: 10.1016/S0070-2153(07)80009-3. [DOI] [PubMed] [Google Scholar]
- Filippova GN, Cheng MK, Moore JM, Truong JP, Hu YJ, Nguyen DK, Tsuchiya KD, Disteche CM. Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev Cell. 2005;8:31–42. doi: 10.1016/j.devcel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16:2802–2813. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–417. doi: 10.1038/nature05916. [DOI] [PubMed] [Google Scholar]
- Fu Y, Sinha M, Peterson CL, Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss SH, Omura M, Mombaerts P. Local and cis effects of the H element on expression of odorant receptor genes in mouse. Cell. 2007;130:373–384. doi: 10.1016/j.cell.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Galande S, Purbey PK, Notani D, Kumar PP. The third dimension of gene regulation: organization of dynamic chromatin loopscape by SATB1. Curr Opin Genet Dev. 2007;17:408–414. doi: 10.1016/j.gde.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006;7:703–713. doi: 10.1038/nrg1925. [DOI] [PubMed] [Google Scholar]
- Gerasimova TI, Byrd K, Corces VG. A chromatin insulator determines the nuclear localization of DNA. Mol Cell. 2000;6:1025–1035. doi: 10.1016/s1097-2765(00)00101-5. [DOI] [PubMed] [Google Scholar]
- Gombert WM, Farris SD, Rubio ED, Morey-Rosler KM, Schubach WH, Krumm A. The c-myc insulator element and matrix attachment regions define the c-myc chromosomal domain. Mol Cell Biol. 2003;23:9338–9348. doi: 10.1128/MCB.23.24.9338-9348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guastafierro T, Cecchinelli B, Zampieri M, Reale A, Riggio G, Sthandier O, Zupi G, Calabrese L, Caiafa P. CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J Biol Chem. 2008;283:21873–21880. doi: 10.1074/jbc.M801170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009 doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Lee DH, Szabo PE. CTCF is the master organizer of domain-wide allele-specific chromatin at the H19/Igf2 imprinted region. Mol Cell Biol. 2008;28:1124–1135. doi: 10.1128/MCB.01361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- Heath H, de Almeida CR, Sleutels F, Dingjan G, van de Nobelen S, Jonkers I, Ling KW, Gribnau J, Renkawitz R, Grosveld F, et al. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J. 2008;27:2839–2850. doi: 10.1038/emboj.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C, Zhao H, Tanimoto K, Dean A. CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc Natl Acad Sci U S A. 2008;105:20398–20403. doi: 10.1073/pnas.0808506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Oshimura M, Nakao M. CTCF-dependent chromatin insulator is linked to epigenetic remodeling. Mol Cell. 2006;23:733–742. doi: 10.1016/j.molcel.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Jhunjhunwala S, van Zelm MC, Peak MM, Cutchin S, Riblet R, van Dongen JJ, Grosveld FG, Knoch TA, Murre C. The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions. Cell. 2008;133:265–279. doi: 10.1016/j.cell.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanduri C, Pant V, Loukinov D, Pugacheva E, Qi CF, Wolffe A, Ohlsson R, Lobanenkov VV. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol. 2000;10:853–856. doi: 10.1016/s0960-9822(00)00597-2. [DOI] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova EM, Chernukhin IV, El-Kady A, Lee RE, Pugacheva EM, Loukinov DI, Goodwin GH, Delgado D, Filippova GN, Leon J, et al. Functional phosphorylation sites in the C-terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol Cell Biol. 2001;21:2221–2234. doi: 10.1128/MCB.21.6.2221-2234.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol. 1993;13:7612–7624. doi: 10.1128/mcb.13.12.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohne AC, Baniahmad A, Renkawitz R. NeP1. A ubiquitous transcription factor synergizes with v-ERBA in transcriptional silencing. J Mol Biol. 1993;232:747–755. doi: 10.1006/jmbi.1993.1428. [DOI] [PubMed] [Google Scholar]
- Komura J, Ikehata H, Ono T. Chromatin fine structure of the c-MYC insulator element/DNase I-hypersensitive site I is not preserved during mitosis. Proc Natl Acad Sci U S A. 2007;104:15741–15746. doi: 10.1073/pnas.0702363104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, Lobanenkov V, Reik W, Ohlsson R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U S A. 2006;103:10684–10689. doi: 10.1073/pnas.0600326103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanctot C, Cheutin T, Cremer M, Cavalli G, Cremer T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet. 2007;8:104–115. doi: 10.1038/nrg2041. [DOI] [PubMed] [Google Scholar]
- Lefevre P, Witham J, Lacroix CE, Cockerill PN, Bonifer C. The LPS-induced transcriptional upregulation of the chicken lysozyme locus involves CTCF eviction and noncoding RNA transcription. Mol Cell. 2008;32:129–139. doi: 10.1016/j.molcel.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Hu JF, Qiu X, Ling J, Chen H, Wang S, Hou A, Vu TH, Hoffman AR. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol Cell Biol. 2008;28:6473–6482. doi: 10.1128/MCB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, Cherry AM, Hoffman AR. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–272. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene. 1990;5:1743–1753. [PubMed] [Google Scholar]
- Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R. Interchromosomal interactions and olfactory receptor choice. Cell. 2006;126:403–413. doi: 10.1016/j.cell.2006.06.035. [DOI] [PubMed] [Google Scholar]
- Lutz M, Burke LJ, LeFevre P, Myers FA, Thorne AW, Crane-Robinson C, Bonifer C, Filippova GN, Lobanenkov V, Renkawitz R. Thyroid hormone-regulated enhancer blocking: cooperation of CTCF and thyroid hormone receptor. EMBO J. 2003;22:1579–1587. doi: 10.1093/emboj/cdg147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder P, Gomez JA, Boss JM. The human major histocompatibility complex class II HLA-DRB1 and HLA-DQA1 genes are separated by a CTCF-binding enhancer-blocking element. J Biol Chem. 2006;281:18435–18443. doi: 10.1074/jbc.M601298200. [DOI] [PubMed] [Google Scholar]
- Majumder P, Gomez JA, Chadwick BP, Boss JM. The insulator factor CTCF controls MHC class II gene expression and is required for the formation of long-distance chromatin interactions. J Exp Med. 2008;205:785–798. doi: 10.1084/jem.20071843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- O’Sullivan JM, Tan-Wong SM, Morillon A, Lee B, Coles J, Mellor J, Proudfoot NJ. Gene loops juxtapose promoters and terminators in yeast. Nat Genet. 2004;36:1014–1018. doi: 10.1038/ng1411. [DOI] [PubMed] [Google Scholar]
- Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17:520–527. doi: 10.1016/s0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palstra RJ, Simonis M, Klous P, Brasset E, Eijkelkamp B, de Laat W. Maintenance of long-range DNA interactions after inhibition of ongoing RNA polymerase II transcription. PLoS ONE. 2008;3:e1661. doi: 10.1371/journal.pone.0001661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W. The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet. 2003;35:190–194. doi: 10.1038/ng1244. [DOI] [PubMed] [Google Scholar]
- Pant V, Kurukuti S, Pugacheva E, Shamsuddin S, Mariano P, Renkawitz R, Klenova E, Lobanenkov V, Ohlsson R. Mutation of a single CTCF target site within the H19 imprinting control region leads to loss of Igf2 imprinting and complex patterns of de novo methylation upon maternal inheritance. Mol Cell Biol. 2004;24:3497–3504. doi: 10.1128/MCB.24.8.3497-3504.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V, Mariano P, Kanduri C, Mattsson A, Lobanenkov V, Heuchel R, Ohlsson R. The nucleotides responsible for the direct physical contact between the chromatin insulator protein CTCF and the H19 imprinting control region manifest parent of origin-specific long-distance insulation and methylation-free domains. Genes Dev. 2003;17:586–590. doi: 10.1101/gad.254903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–772. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- Rand E, Ben-Porath I, Keshet I, Cedar H. CTCF elements direct allele-specific undermethylation at the imprinted H19 locus. Curr Biol. 2004;14:1007–1012. doi: 10.1016/j.cub.2004.05.041. [DOI] [PubMed] [Google Scholar]
- Recillas-Targa F, Pikaart MJ, Burgess-Beusse B, Bell AC, Litt MD, West AG, Gaszner M, Felsenfeld G. Position-effect protection and enhancer blocking by the chicken beta-globin insulator are separable activities. Proc Natl Acad Sci U S A. 2002;99:6883–6888. doi: 10.1073/pnas.102179399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renda M, Baglivo I, Burgess-Beusse B, Esposito S, Fattorusso R, Felsenfeld G, Pedone PV. Critical DNA binding interactions of the insulator protein CTCF: a small number of zinc fingers mediate strong binding, and a single finger-DNA interaction controls binding at imprinted loci. J Biol Chem. 2007;282:33336–33345. doi: 10.1074/jbc.M706213200. [DOI] [PubMed] [Google Scholar]
- Rubio ED, Reiss DJ, Welcsh PL, Disteche CM, Filippova GN, Baliga NS, Aebersold R, Ranish JA, Krumm A. CTCF physically links cohesin to chromatin. Proc Natl Acad Sci U S A. 2008;105:8309–8314. doi: 10.1073/pnas.0801273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh N, Bell AC, Recillas-Targa F, West AG, Simpson M, Pikaart M, Felsenfeld G. Structural and functional conservation at the boundaries of the chicken beta-globin domain. EMBO J. 2000;19:2315–2322. doi: 10.1093/emboj/19.10.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr CJ, Levorse JM, Tilghman SM. CTCF maintains differential methylation at the Igf2/H19 locus. Nat Genet. 2003;33:66–69. doi: 10.1038/ng1057. [DOI] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006;20:2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 2008;27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P, Tang SH, Rentsendorj A, Pfeifer GP, Mann JR. Maternal-specific footprints at putative CTCF sites in the H19 imprinting control region give evidence for insulator function. Curr Biol. 2000;10:607–610. doi: 10.1016/s0960-9822(00)00489-9. [DOI] [PubMed] [Google Scholar]
- Szabo PE, Tang SH, Silva FJ, Tsark WM, Mann JR. Role of CTCF binding sites in the Igf2/H19 imprinting control region. Mol Cell Biol. 2004;24:4791–4800. doi: 10.1128/MCB.24.11.4791-4800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell. 2002;10:1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- Torrano V, Chernukhin I, Docquier F, D’Arcy V, Leon J, Klenova E, Delgado MD. CTCF regulates growth and erythroid differentiation of human myeloid leukemia cells. J Biol Chem. 2005;280:28152–28161. doi: 10.1074/jbc.M501481200. [DOI] [PubMed] [Google Scholar]
- Tsai CL, Rowntree RK, Cohen DE, Lee JT. Higher order chromatin structure at the X-inactivation center via looping DNA. Dev Biol. 2008;319:416–425. doi: 10.1016/j.ydbio.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrov AA, Quitschke WW. The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta-protein precursor promoter. Evidence for a role in transcriptional activation. J Biol Chem. 1997;272:33353–33359. doi: 10.1074/jbc.272.52.33353. [DOI] [PubMed] [Google Scholar]
- Wallace JA, Felsenfeld G. We gather together: insulators and genome organization. Curr Opin Genet Dev. 2007;17:400–407. doi: 10.1016/j.gde.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan LB, Pan H, Hannenhalli S, Cheng Y, Ma J, Fedoriw A, Lobanenkov V, Latham KE, Schultz RM, Bartolomei MS. Maternal depletion of CTCF reveals multiple functions during oocyte and preimplantation embryo development. Development. 2008;135:2729–2738. doi: 10.1242/dev.024539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- Xie X, Mikkelsen TS, Gnirke A, Lindblad-Toh K, Kellis M, Lander ES. Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc Natl Acad Sci U S A. 2007;104:7145–7150. doi: 10.1073/pnas.0701811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Donohoe ME, Silva SS, Lee JT. Evidence that homologous X-chromosome pairing requires transcription and Ctcf protein. Nat Genet. 2007;39:1390–1396. doi: 10.1038/ng.2007.5. [DOI] [PubMed] [Google Scholar]
- Xu N, Tsai CL, Lee JT. Transient homologous chromosome pairing marks the onset of X inactivation. Science. 2006;311:1149–1152. doi: 10.1126/science.1122984. [DOI] [PubMed] [Google Scholar]
- Yoon YS, Jeong S, Rong Q, Park KY, Chung JH, Pfeifer K. Analysis of the H19ICR insulator. Mol Cell Biol. 2007;27:3499–3510. doi: 10.1128/MCB.02170-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Ginjala V, Pant V, Chernukhin I, Whitehead J, Docquier F, Farrar D, Tavoosidana G, Mukhopadhyay R, Kanduri C, et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat Genet. 2004;36:1105–1110. doi: 10.1038/ng1426. [DOI] [PubMed] [Google Scholar]
- Yusufzai TM, Tagami H, Nakatani Y, Felsenfeld G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol Cell. 2004;13:291–298. doi: 10.1016/s1097-2765(04)00029-2. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]