


Abstract
Pyrimidine adducts in cellular DNA arise from modification of the pyrimidine 5,6-double bond by oxidation, reduction or hydration. The biological outcome includes increased mutation rate and potential lethality. A major DNA N-glycosylase responsible for the excision of modified pyrimidine bases is the base excision repair (BER) glycosylase endonuclease III, for which functional homologs have been identified and characterized in Escherichia coli, yeast and humans. So far, little is known about how hyperthermophilic Archaea cope with such pyrimidine damage. Here we report characterization of an endonuclease III homolog, PaNth, from the hyperthermophilic archaeon Pyrobaculum aerophilum, whose optimal growth temperature is 100°C. The predicted product of 223 amino acids shares significant sequence homology with several [4Fe-4S]-containing DNA N-glycosylases including E.coli endonuclease III (EcNth). The histidine-tagged recombinant protein was expressed in E.coli and purified. Under optimal conditions of 80–160 mM NaCl and 70°C, PaNth displays DNA glycosylase/β-lyase activity with the modified pyrimidine base 5,6-dihydrothymine (DHT). This activity is enhanced when DHT is paired with G. Our data, showing the structural and functional similarity between PaNth and EcNth, suggests that BER of modified pyrimidines may be a conserved repair mechanism in Archaea. Conserved amino acid residues are identified for five subfamilies of endonuclease III/UV endonuclease homologs clustered by phylogenetic analysis.
INTRODUCTION
Pyrimidine adducts in cellular DNA arise from modification of the pyrimidine 5,6-double bond by oxidation, reduction or hydration. So far, more than 20 stable pyrimidine adducts have been discovered (1,2). The biological consequences of some of these modified pyrimidine bases, including 5-hydroxycytosine, 5-hydroxyuracil, uracil glycol, thymine glycol and 5,6-dihydrothymine (DHT), have been studied in detail regarding miscoding potential and effect on DNA replication (1,3,4). With only a few exceptions, they all cause either miscoding or polymerase blocking. Both lead to increase in mutation rate or possible cell death.
Repair of pyrimidine adducts is mainly via the base excision repair (BER) process which is initiated by DNA N-glycosylases (1,5,6). In Escherichia coli, endonuclease III (EcNth) is responsible for the excision of most modified pyrimidine bases (7–14). It is bifunctional, having both DNA N-glycosylase activity that removes modified pyrimidine bases, and an intrinsic apurinic/apyrimidinic (AP) lyase activity that cleaves the phosphodiester backbone of DNA 3′ to AP sites. This generates 5′-deoxyribose-5-phosphate and a 3′-terminal unsaturated sugar derivative. The abnormal 3′-abasic residue is then removed by an AP endonuclease, another BER enzyme, to generate a 3′-OH nucleotide residue. Subsequently the gap is repaired by other BER components restoring the original genetic information. Functional endonuclease III homologs have been identified in organisms as diverse as yeast and humans (15–18), which suggests that BER of modified pyrimidines has been crucial for maintaining DNA stability during evolution.
Structurally, EcNth belongs to the HhH DNA N-glycosylase superfamily, which contains a helix–hairpin–helix (HhH) motif for DNA binding and a proline/glycine-rich loop (GPD motif). This GPD motif contains the catalytic aspartic acid residue corresponding to D138 in EcNth (19). Other members in this HhH superfamily are T/G and U/G mismatch-specific glycosylase (20–22), a few subgroups of methylpurine DNA glycosylases (23–25), UV endonuclease (26,27), adenine-DNA glycosylase (28–30) and 7,8-dihydro-8-oxoguanine DNA glycosylase (19,31–35). They all catalyze N-glycosyl bond cleavage of different unwanted bases from DNA. Many of the proteins that belong to the HhH DNA N-glycosylase superfamily also contain an additional conserved structural element—an iron–sulfur cluster loop (FCL motif). This motif contains four cysteine residues for binding of a [4Fe-4S] cluster (36). The crystal structures of members in the HhH superfamily reveal the nucleotide flipping mechanism for the base removal. The unwanted base is flipped out of the DNA helix into the glycosylase active site pocket and cleaved (37–42).
In thermophiles, whose optimal growth temperature is above 65°C, the rate of spontaneous deamination of cytosine and 5-methylcytosine is thermally enhanced, giving increased yields of U/G and T/G mismatches (20,21,43–46). Little is known about other types of pyrimidine lesions in thermophiles or their corresponding repair pathways. However, putative DNA N-glycosylase sequence homologs have been identified in thermophiles and hyperthermophiles whose genomes have been completely sequenced (47). This allows us to study, in vitro, the enzymatic activities of these putative homologs as a first step toward understanding the DNA repair pathways for pyrimidine lesions in these organisms. So far, the only published study for a thermophilic endonuclease III homolog is an NMR structure description of a homolog from the hyperthermophilic archaeon Archaeoglobus fulgidus (48). Here, we report cloning and characterization of a putative DNA N-glycosylase homolog from the hyperthermophilic archaeon, Pyrobaculum aerophilum, whose optimal growth temperature is 100°C (49). Using defined DNA substrates we demonstrate that this protein is an endonuclease III homolog (PaNth), which efficiently cleaves a modified pyrimidine, DHT, opposite G. Our data, showing the structural and functional similarity between PaNth and EcNth, suggests that BER of modified pyrimidines may be a conserved repair mechanism in Archaea. Conserved amino acid residues are identified for five subfamilies of endonuclease III/UV endonuclease homologs clustered by phylogenetic analysis.
MATERIALS AND METHODS
Identification of the candidate protein
The candidate protein coding region, pag5_880, was identified in the recently completed whole genome sequence of P.aerophilum (50; S.T.Fitz-Gibbon, H.Ladner, U.-J.Kim, K.O.Stetter, M.I.Simon and J.H.Miller, unpublished data) by analyzing sequence similarity to the public database using FASTA (51).
Expression and purification of PaNth
The pag5_880 DNA was amplified by polymerase chain reaction (PCR). The PCR product was cloned into pCR2.1-TOPO vector using TOPO TA Cloning kit (Invitrogen, Carlsbad, CA) and subsequently subcloned into the bacterial expression vector pQE30 (Qiagen, Chatsworth, CA) between the BamHI and HindIII sites. Transformants of E.coli CSH100 were grown at 37°C in 1.5 l of Luria–Bertani medium with ampicillin (200 µg/ml). The culture was induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when it reached optical density 1.5 at 600 nm, and allowed to continue growing overnight. The procedure for purification by Ni2+-NTA affinity chromatography was the same as previously described (21). The peak fractions that contained the olive color were combined and dialyzed overnight in Buffer B (50 mM Tris–HCl, pH 7.5, 30 mM NaCl, 1 mM EDTA, 1 mM DTT and 50% glycerol). A clear protein sample was obtained after centrifugation of the dialyzed sample and stored at –80°C. The protein concentration, determined by Bio-Rad Protein Assay (Bio-Rad, Hercules, CA), was 19 mg/ml. The method for determination of molecular mass by electron mass spectrometry was the same as previously described (21).
To prepare heat-treated PaNth, 50 µl of 10-fold-diluted purified recombinant PaNth (1.9 mg/ml) was incubated at 70 or 90°C for 10 min followed by centrifugation at 4°C for 15 min. The clear supernatant was collected and the protein concentration determined by Bio-Rad Protein Assay (Bio-Rad).
Oligonucleotide substrates
Sequences of two 96mer oligonucleotides, which were complementary to each other except at position 60, were previously described (21). The oligonucleotide that contains a single modified base, DHT at position 60, was synthesized and PAGE purified (DNAgency, Malvern, PA). The 96mer double-stranded DNA (dsDNA) substrates, containing a base pair X/Y at position 60 [X = A, C, G, T, U or DHT; Y = A, C, G, T or GO (7,8-dihydro-8-oxoguanine or 8-oxoG)], were prepared in an annealing buffer containing 70 mM Tris–HCl, pH 7.6, and 10 mM MgCl2, as previously described (21). Unless otherwise stated, the X-containing strand was 32P-labeled at the 5′-end.
Glycosylase activity assay
The glycosylase activity assay was essentially carried out as previously described (21). Briefly, the standard reaction mixture contained 20 mM Tris–HCl, pH 7.5, 1 mM DTT, 1 mM EDTA, 3% glycerol, 1 µl of 32P-labeled 96mer dsDNA substrate (final concentration of 1 nM) and 2 µl of the 90°C heat-treated recombinant PaNth in a total volume of 20 µl. Unless otherwise stated, the PaNth reaction mixture had 120 mM NaCl (a few cases contained 80 mM NaCl). The 2 µl of 90°C heat-treated recombinant PaNth was diluted to the desired concentrations using Buffer B (50 mM Tris–HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 30 mM NaCl and 50% glycerol). In the reactions with no PaNth, 2 µl Buffer B was used. The reaction was carried out for 15 min at 70°C (unless otherwise stated) and terminated by cooling to 4°C. Then 8 µl of loading buffer was added. The reaction mixture was heated to 94°C for 1 min and analyzed on a 15% polyacrylamide denaturing gel. Gels were dried and then visualized and quantified by PhosphorImager 445 SI (Sunnyville, CA) using the program Molecular Dynamics ImageQuaNT Version 4.2a. The percentage of cleaved product was determined by dividing the cleaved product intensity by the total intensity. Total intensity was the sum of intact DNA intensity and cleaved product intensity. All intensities were stated after subtracting background (a blank area of identical size).
EcNth (2 mg/ml) was a gift from Dr Richard M. Cunningham (Department of Biological Sciences, State University of New York at Albany, Albany, NY). For reactions containing EcNth, no NaCl was added to the glycosylase reaction buffer, unless otherwise stated. EcNth (2 µl) diluted in Buffer B to the desired concentration was used and the reactions were carried out at 37°C for 15 min. In the reactions with no EcNth, 2 µl Buffer B was used. The subsequent steps were similar to those described in the PaNth glycosylase activity assay (see above).
Reactions to determine the specific activity of the enzyme with DHT/G substrate were carried out using 1 nM DHT/G, 0.25 nM PaNth (at 70°C) or 5 nM EcNth (at 37°C) for 4 min. The reaction products were analyzed as described above.
AP lyase activity assay
A dsDNA substrate containing an AP site opposite G (AP/G) at position 60, was prepared from uracil-DNA glycosylase-treated U/G substrate as described previously (21). Increasing amounts of PaNth were incubated with 1 nM AP/G substrate at 70°C for 15 min followed by analysis on a 15% polyacrylamide denaturing gel as described in the glycosylase activity assay.
Escherichia coli endonuclease IV (endoIV, 0.157 mg/ml) was a gift from Dr Bruce Demple (Department of Cancer Cell Biology, Harvard School of Public Health, Boston, MA). The following experiments were carried out in the reaction buffer containing 20 mM Tris–HCl, pH 7.6, and 50 µg/ml bovine serum albumin (BSA). EcNth (10 nM) was incubated with 1 nM DHT/G substrate for 15 min at 37°C, followed by the addition of 5 nM endoIV and continued incubation for 15 min at 37°C. The reactions were analyzed on a 15% polyacrylamide denaturing gel as described in the glycosylase activity assay.
Oligonucleotide substrate containing 5,6-dihydrouracil (DHU) and activity assay
The 55mer dsDNA substrate, containing DHU paired with G at position 19, was prepared as described by Klungland et al. (52). Standard reaction mixtures (10 µl) contained 100 fmol of DNA substrate, 45 mM HEPES–KOH, pH 7.8, 70 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.4 mM EDTA, 2 mM ATP, 1 mM NAD, 20 µM each of dATP, dGTP, dCTP and dTTP, 3.6 µg BSA, 2% glycerol and either EcNth or PaNth. Prior to adding the glycosylases to the reactions, the proteins were heat-treated as indicated by incubating at 80°C for 20 min followed by centrifugation to remove any precipitate. The reactions were incubated at 37°C for 30 min and terminated by the addition of SDS and EDTA to final concentrations of 0.6% and 25 mM, respectively. The proteins were digested with proteinase K (500 µg/ml) at 37°C for 1 h. The DNA was purified and resolved by electrophoresis on a 20% polyacrylamide denaturing gel. The reaction products were visualized by autoradiography.
UV endonuclease activity assay
The UV-damaged DNA substrates were produced by irradiating plasmid pBluescript KS+ DNA with 450 J/m2 UV light. Then both the UV-treated plasmid DNA and the non-damaged pHM14 plasmid DNA were treated with EcNth protein to remove pyrimidine hydrates. This was followed by repeated purification of closed circular forms of plasmid DNA as described (53). The UV-damaged DNA was mixed with an equal amount of non-damaged closed circular pHM14 plasmid DNA. The repair reactions (25 µl) containing either PaNth or T4 endoV (as a positive control) and 500 ng DNA mixture (±UV) were incubated in buffer containing 40 mM HEPES–KOH, pH 7.8, 0.5 mM DTT, 4 mM EDTA, 70 mM KCl and 0.36 mg/ml of BSA at either 37°C for 30 min or 70°C for 15 min. The DNA was purified by first treating with proteinase K, phenol–chloroform and precipitating the DNA in absolute ethanol in the presence of 10 µg yeast tRNA. After centrifugation, the DNA pellet was washed with 70% ethanol and dried under vacuum. The DNA was resuspended in TE and incubated at 37°C for 15 min. RNase A (80 µg/ml) was added to the DNA to digest the carrier yeast tRNA prior to loading the DNA on a 1% agarose gel containing 0.25 µg/ml ethidium bromide. After electrophoresis, the gel was viewed under UV light.
Preparation of crude extract from P.aerophilum
Pyrobaculum aerophilum cells (400 ml) were grown in BSY medium (49) at 90°C for 3 days to optical density 0.2 at 600 nm. Cells were harvested by centrifugation. The cell pellet was washed once with Buffer P (10 mM NaPO4, pH 7.0, 50 mM NaCl), centrifuged and then resuspended in 4 ml Buffer P and sonicated. The crude extract was obtained following centrifugation. The protein concentration of the crude extract was determined by Bio-Rad Protein Assay (Bio-Rad).
Adduct formation in the presence of NaBH4
The DNA-trapping reaction was carried out using 120 mM NaBH4 (instead of 120 mM NaCl). After incubation at 70°C for 10 min the reaction mixture was denatured at 90°C for 3 min in standard SDS–PAGE loading buffer and was then separated by electrophoresis on a 10% SDS–polyacrylamide gel. The gel was dried and analyzed using a phosphoimager. In the case of EcNth, 25 mM NaBH4 was used and the reaction was carried out at 37°C for 10 min. The subsequent steps were similar to those described above.
Phylogenetic analysis
Distance analysis was performed using neighbor joining in the PAUP program (54). A multiple sequence alignment of the ‘HhH–GPD’ domain was retrieved from the Pfam Web site (http://pfam.wustl.edu; 55–57). Sequences containing a lysine residue in the HhH motif corresponding to Lys120 in EcNth were selected from the Pfam alignment. Sixteen additional sequences were manually added (Aa4, Ap1, Buch1, Cj1, Dr1, Dr2, Dr3, MjOgg, Nm1, Pab1, Pae1, PaNth, Tm1, Tm2, Vc1 and Xf1; see Fig. 9 legend).
Figure 9.
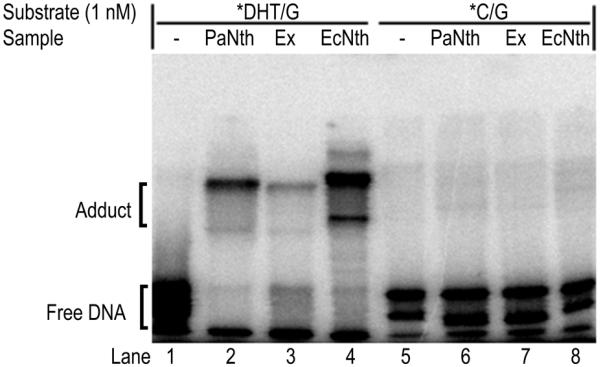
Adduct formation by DNA-trapping. The reaction was carried out for 10 min with 1 nM *DHT/G or *C/G in the presence of NaBH4. The asterisk indicates the 5′-end-labeled strand. The samples and the temperatures used for the reactions are as follows: 10 nM PaNth and 2.24 µg P.aerophilum crude extract (Ex) at 70°C, or 10 nM EcNth at 37°C. The reaction mixture was analyzed on a 10% SDS–polyacrylamide gel and a phosphoimage of the reactions is shown. The adduct and free DNA are indicated on the left.
RESULTS
Homology between PaNth and other DNA N-glycosylase homologs
Open reading frame (ORF), pag5_880, from the hyperthermophilic archaeon P.aerophilum, was identified as encoding a putative DNA N-glycosylase by FASTA (51). The amino acid alignments show that it contains all the structural attributes of a [4Fe-4S]-containing HhH DNA N-glycosylase (Fig. 1): a HhH motif, a strictly conserved aspartic acid residue at position 148 (GPD motif) and four cysteine residues for binding of a [4Fe-4S] cluster (FCL motif). Overall, amino acid sequence similarity was seen when comparing this putative DNA N-glycosylase to other characterized [4Fe-4S]-containing HhH DNA N-glycosylases using Bestfit (58): EcNth (37.4% identity), Micrococcus luteus UV endonuclease (MlUVendo, 37.6% identity), Thermotoga maritima methylpurine glycosylase II (TmMpgII, 36.5% identity), P.aerophilum T/G and U/G mismatch glycosylase (PaMig, 33.3% identity) and E.coli adenine-DNA glycosylase (EcMutY, 30.4% identity over 100 amino acid residues). The biochemical data that follows demonstrates that the purified recombinant protein can catalyze the base removal of a modified pyrimidine, a substrate for endonuclease III, but lacks detectable activity for substrates of UV endonuclease, T/G and U/G mismatch glycosylase, and adenine-DNA glycosylase. Therefore, we designate it as P.aerophilum endonuclease III homolog (PaNth).
Figure 1.

Sequence alignment of PaNth and other [4Fe-4S]-containing DNA glycosylase homologs using the program ClustalW (68). The homologs are as follows (protein code and GenBank accession number in parentheses): E.coli endonuclease III (EcNth, P20625), M.luteus UV endonuclease (MlUVendo, P46303), P.aerophilum endonuclease III homolog (PaNth, AF222334), T.maritima MpgII (TmMpgII, AAD35467), P.aerophilum T/G and U/G mismatch-specific glycosylase (PaMig, AF222335) and E.coli adenine DNA-glycosylase (EcMutY, P17802). The conserved amino acid residues are shaded as follows: black, strictly conserved identical amino acid; dark grey, conserved identical amino acid residues; light grey, conserved similar amino acid residues. The HhH motif is indicated. The cysteine residues for binding of [4Fe-4S] cluster are marked with asterisks. The strictly conserved aspartic acid residue is marked with a dot. The conserved lysine residue within the endonuclease III family is marked with an arrowhead. The regions A and B are indicated for sequence alignment in Figure 10.
The amino acid alignment shown in Figure 1 reveals that PaNth contains a lysine residue at position 131 (arrowhead indicates position). It appears that this is a strictly conserved residue in the endonuclease III family and is also present in the UV endonuclease from M.luteus (26). In contrast, at the corresponding position, methylpurine glycosylase homologs have a strictly conserved glutamic acid residue (24), and the T/G and U/G mismatch glycosylase/adenine-DNA glycosylase homologs have a conserved tyrosine or a serine residue (21,29). In addition, PaNth lacks the adenine-DNA glycosylase-specific C-terminal domain (59).
Expression and purification of the recombinant PaNth
The histidine-tagged recombinant PaNth was expressed in E.coli and was purified to near homogeneity by affinity chromatography on a Ni2+-NTA column (Fig. 2, lane 3). Although the apparent molecular mass, estimated from the SDS gel, was ∼30 kDa (Fig. 2), the mass spectrometry study showed the recombinant PaNth to be 26 624 Da (data not shown). This is in close agreement with the size of 26 594 Da predicted from the histidine-tagged pag5_880 ORF sequence. The majority of the purified recombinant PaNth remained soluble after heat treatment at either 70 or 90°C (Fig. 2, lanes 4 and 5) suggesting that it is heat stable. Therefore, 90°C heat treatment was used as a second step purification to further reduce the amount of contaminants from E.coli that precipitated at this temperature. The purified PaNth displays a distinct olive color due to the presence of [4Fe-4S] clusters with a corresponding absorption peak at 410 nm (data not shown).
Figure 2.
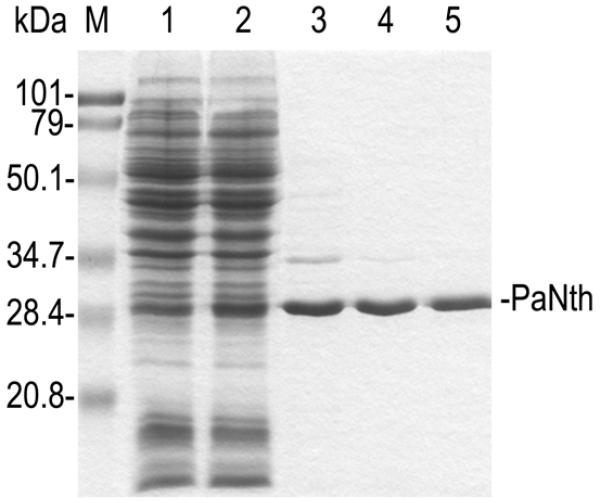
Expression and purification of PaNth protein. The 10% SDS–polyacrylamide gel contains the following samples: lysate of CSH100/pQE30 cells (lane 1), lysate of CSH100/pQE30PaNth after IPTG induction (lane 2), PaNth protein eluted from Ni2+-NTA column (lane 3), 70°C heat-treated PaNth (lane 4) and 90°C heat-treated PaNth (lane 5). The gel was visualized by Coomassie Blue staining. PaNth protein is indicated on the right. Lane M contains molecular mass standards (Bio-Rad) as indicated on the left.
PaNth glycosylase activity
Defined oligonucleotide substrates were used for the characterization of PaNth glycosylase activity. Endonuclease III substrates containing a single modified pyrimidine (DHT) paired with each of the four normal DNA bases, were first tested (Fig. 3). Efficient removal of DHT opposite G (Fig. 3A, lane 8), along with weak activity for removal of DHT opposite A, C and T, was observed (Fig. 3A, lanes 7, 9 and 10). The concentration dependence curve of PaNth with DHT/G substrate is shown in Figure 3B and its specific activity for DHT/G is 11 fmol/µg/min at 70°C. EcNth was used as a positive control and shows a similar opposite base preference on DHT substrates (Fig. 3C and D). Its specific activity for DHT/G is 0.6 fmol/µg/min at 37°C and is apparently much less efficient than PaNth.
Figure 3.

PaNth and EcNth glycosylase activity on DHT-containing substrates. The glycosylase reaction was carried out for 15 min with 1 nM *DHT/Y (Y = A, G, C or T) or *T/A, where the asterisk indicates the 5′-end-labeled strand. Each reaction mixture was then analyzed on a 15% denaturing polyacrylamide gel. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left. (A) Phosphoimage of the reaction in the presence (+) or absence (–) of 5 nM PaNth at 70°C. (B) Plot of the percentage cleaved versus PaNth concentration. Each individual point represented with error bars is as an average of data from two to five experiments. (C) Phosphoimage of the reaction with increasing amounts of EcNth at 37°C. (D) Plot of the percentage cleaved versus EcNth concentration.
Optimal conditions were studied for PaNth activity with DHT/G. The temperature dependence of the PaNth glycosylase activity with DHT/G substrate is shown in Figure 4A. While weak PaNth activity was detected at temperatures below 37°C (Fig. 4A, lanes 9–11), maximum cleavage was observed at temperatures between 60 and 70°C (Fig. 4A, lanes 13 and 14). At temperatures above 70°C, the activity of PaNth decreases (Fig. 4A, lanes 15 and 16).
Figure 4.
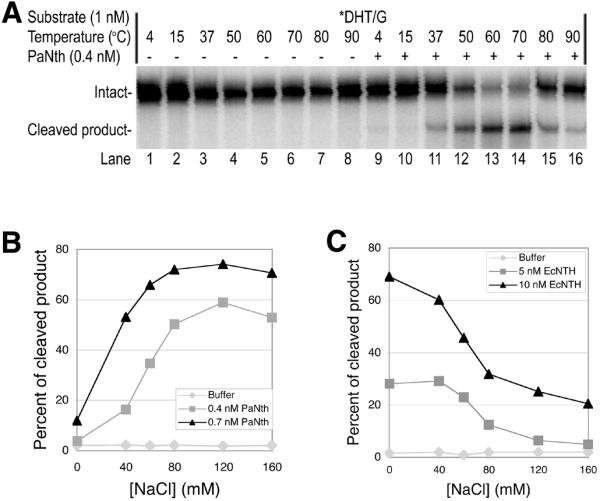
Optimal temperature and NaCl concentrations for PaNth activity. The glycosylase reaction was carried out for 15 min with 1 nM *DHT/G, where the asterisk indicates the 5′-end-labeled strand. Each reaction was then analyzed on a 15% denaturing polyacrylamide gel. (A) Phosphoimage of the reactions in the presence (+) or absence (–) of 0.4 nM PaNth over increasing temperatures. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left. (B) Plot of the percentage cleaved versus NaCl concentration with 0.4 or 0.7 nM PaNth or buffer at 70°C. (C) Plot of the percentage cleaved versus NaCl concentration with 5 or 10 nM EcNth or buffer at 37°C.
Figure 4B shows the salt dependence of PaNth with DHT/G substrate. The optimal NaCl concentration for PaNth is between 80 and 160 mM, while little activity was observed in the absence of NaCl (Fig. 4B). In contrast, EcNth prefers low NaCl conditions and even performs best in the absence of NaCl (Fig. 4C).
PaNth AP lyase activity
AP lyase activity is present in DNA N-glycosylases that have a conserved lysine residue in the HhH motif (Fig. 1, arrowhead indicates position). This conserved lysine residue is also present in PaNth suggesting that it may have AP lyase activity. Indeed, in the presence of suitable substrate, PaNth was able to generate a cleaved DNA product rather than just an AP product. To confirm the AP lyase activity, we looked at PaNth’s ability to cleave AP containing DNA derived from UDG-treated U/G substrate. We found that PaNth can efficiently cleave an existing AP site (Fig. 5A). So far, three types of glycosylase/lyase actions have been identified: β-elimination, 3′-OH hydrolysis and β,δ-elimination. The products of these three actions can be distinguished on a polyacrylamide gel due to different mobility: the β,δ-elimination product is fastest followed by the 3′-OH product and finally the β-elimination product (60). To define PaNth’s glycosylase/lyase action, the size of the PaNth product was compared with the β-elimination product generated by EcNth. The 3′-OH product, generated by the combination of EcNth and endoIV (61), was also included as a size marker. The PaNth product is the same size as the EcNth β-elimination product (Fig. 5B, lanes 2 and 3), and both migrated more slowly than the EcNth/endoIV 3′-OH product (Fig. 5B, lane 4). Therefore, we conclude that PaNth’s glycosylase/lyase action is likely via β-elimination.
Figure 5.
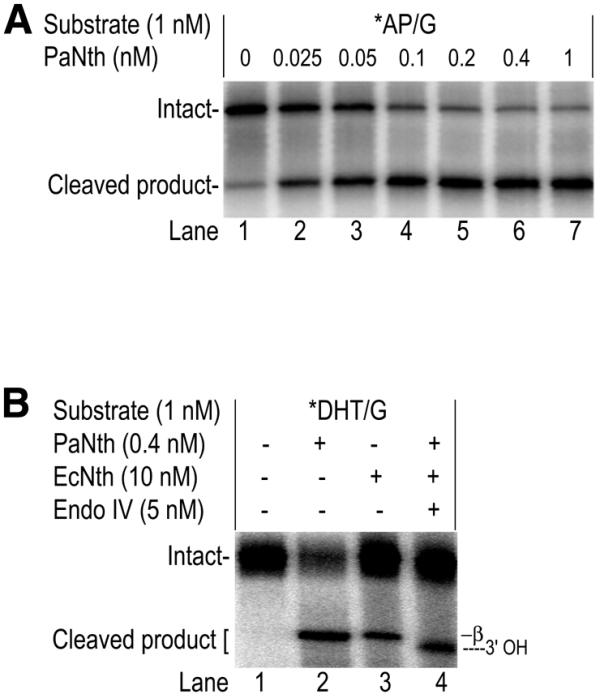
AP lyase activity of PaNth. The glycosylase reaction was carried out for 15 min. Each reaction mixture was then analyzed on a 15% denaturing polyacrylamide gel. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left. Asterisk indicates the 5′-end-labeled strand. (A) 1 nM *AP/G and increasing amounts of PaNth at 70°C. (B) 1 nM *DHT/G with: 0.4 nM PaNth (at 70°C), 10 nM EcNth (at 37°C) or 10 nM EcNth/5 nM Endo IV (at 37°C).
Other substrates for PaNth
Another known endonuclease III substrate is dsDNA containing DHU. We used a 55mer containing DHU paired with G at position 19 to test for PaNth cleavage (Fig. 6). The reactions were carried out at 37°C using both non-heat-treated and heat-treated PaNth samples to evaluate whether the observed PaNth activity was heat resistant. In the non-heat-treated PaNth samples as little as 2 ng PaNth was able to nick the DNA at the lesion site to generate a 19mer product (Fig. 6, lane 4). In addition, nuclease activity was observed when using a 100-fold excess of non-heat-treated PaNth, which caused DNA degradation (Fig. 6, lane 2). In the heat-treated PaNth samples the glycosylase/lyase activity was retained (Fig. 6, lanes 5–7), but the nuclease activity was not (Fig. 6, lane 5) indicating that the latter activity was from an E.coli contaminant. EcNth was used as a positive control and also generated the 19mer product (Fig. 6, lane 8), and its activity was heat labile (Fig. 6, lane 9).
Figure 6.
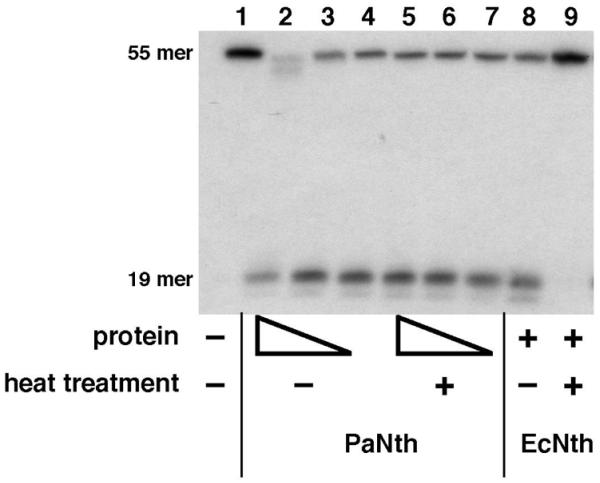
PaNth and EcNth activity on DHU/G. In each reaction, different amounts of PaNth (200 ng, lanes 2 and 5; 20 ng, lanes 3 and 6; 2 ng, lanes 4 and 7) were incubated with 100 fmol of 5′ end-labeled 55mer DNA substrate containing DHU. EcNth was present in reactions shown in lanes 8 and 9. The heat treated PaNth and EcNth were present in lanes 5–7 and 9, respectively. A control reaction without any protein is shown in lane 1. The intact 55mer DNA substrate and the nicked 19mer are indicated.
Since PaNth shares similar amino acid identity with both EcNth (37.4%) and M1UVendo (37.6%), UV-damaged DNA containing cyclobutane pyrimidine dimers was also tested as a substrate. PaNth did not show any nicking activity towards the DNA containing cyclobutane pyrimidine dimers (data not shown).
T/G and U/G mismatch glycosylase substrates were tested. Only minor cleavage of T/G substrate by PaNth was detected (Fig. 7A, lane 2). The adenine-DNA glycosylase substrate, A/GO, was also tested for possible cleavage by PaNth (Fig. 7B). While only minor activity was observed with the T/GO substrate (Fig. 7B, lane 8), no activity was detected with G/GO, C/GO, A/GO or U/GO substrates (Fig. 7B, lanes 2, 4, 6 and 10). These results are consistent with the structural analysis described earlier, where PaNth lacks some amino acid residues that are conserved either in the T/G and U/G mismatch glycosylases or in the adenine-DNA glycosylases.
Figure 7.
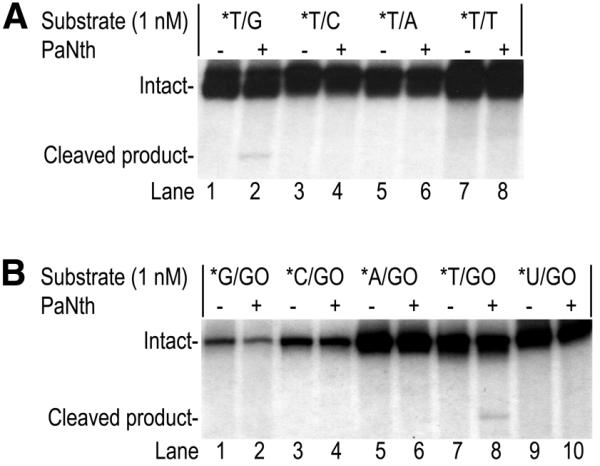
PaNth activity on T/G and T/GO. The glycosylase reaction was carried out at 70°C for 15 min with (A) 1 nM *T/Y (Y = G, C, A and T) or (B) 1 nM *X/GO (X = G, C, A, T or U), where the asterisk indicates the 5′-end-labeled strand. Phosphoimages of the reactions on 15% polyacrylamide gels are shown with (+) or without (–) 50 nM PaNth. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left.
We also looked at 8-oxoG DNA glycosylase substrate C/GO (where GO is labeled) and other unmodified base pair mismatches (A/A, A/C, A/G, C/A, C/C, C/T, G/A, G/G, G/T, T/C and T/T). No activity was observed with any of these substrates (data not shown).
DHT/G cleavage in crude cell extract of P.aerophilum
A crude extract of P.aerophilum cells was prepared and tested for activity with DHT substrates (Fig. 8). As expected, a similar opposite base preference was observed: DHT opposite G was efficiently removed (Fig. 8, lane 3) but not DHT opposite A, C or T (Fig. 8, lanes 2, 4 and 5). To identify the size of the glycosylase in the crude extract responsible for this cleavage, we utilized the method of DNA adduct formation (also known as DNA-trapping) by bifunctional N-glycosylase/β-lyases in the presence of NaBH4 (62–64). The DNA-trapping experiment was carried out and the sizes of adducts formed with purified recombinant PaNth versus crude extract were compared (Fig. 9). EcNth was used as a positive control. While no adducts were formed with control C/G DNA (Fig. 9, lanes 6, 7 and 8), multiple adducts with DHT/G were observed in the presence of PaNth, crude extract and EcNth (Fig. 9, lanes 2, 3 and 4). The pattern of adducts formed in the crude extract was similar to the pattern formed with purified PaNth suggesting that the major DHT cleaving glycosylase in the crude extract is PaNth or possibly a glycosylase of similar molecular weight.
Figure 8.
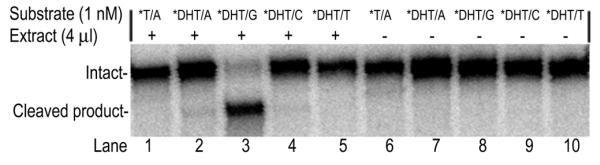
Cleavage of DHT substrates by P.aerophilum crude extract. The glycosylase reaction was carried out at 70°C for 15 min with 1 nM *DHT/G in the presence (+) or absence (–) of 2.24 µg P.aerophilum crude extract. The asterisk indicates the 5′ end-labeled strand. Each reaction mixture was analyzed on a 15% denaturing polyacrylamide gel. A phosphoimage of the reactions is shown. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left.
Phylogenetic study of endonuclease III homologs
DNA N-glycosylase homologs, which contain a conserved lysine residue in the HhH motif corresponding to Lys120 in EcNth (Fig. 1, arrowhead indicates position), were used to construct a phylogenetic tree (Fig. 10A). So far, among 34 completed archaeal and bacterial genomes, it appears that only three lack endonuclease III homologs: Mycoplasma genitalium, Mycoplasma pneumoniae and Ureaplasma irealyticum. All three are obligate parasitic bacteria. Notably, characterized DNA N-glycosylases that have different substrate preference (i.e. endonuclease III homologs and 8-oxoG DNA glycosylase homologs) cluster separately on the tree. When we look only at the endonuclease III/UV endonuclease homologs, we find that they cluster into five distinct groups. One of these contains PaNth and two other putative DNA N-glycosylase homologs from Aeropyrum pernix (Ap, 43.5% identity) and from Sulfolobus solfataricus (Ss, 41.8% identity).
Figure 10.


(A) Phylogenetic analysis of members of the DNA glycosylase superfamily that have a lysine residue in the HhH motif corresponding to Lys120 in EcNth (protein code and GenBank accession number in parentheses). Aeropyrum pernix (Ap1, BAA79061), Aquifex aeolicus (Aa1 AAC06594; Aa2, AAC06742; Aa4, AAC06576), A.fulgidus (Af1, AAB89556; Af2, AAB90876), Bacillus subtilis (Bs1, P39788), Borrelia burgdorferi (Bb1, AAC67089), Buchnera sp. APS (Buch1, BAB12837), Caenorhabditis elegans (Ce1, P54137), Campylobacter jejuni (Cj1, CAB75231), Chlamydia pneumoniae (Cp1, AAD18975), Chlamydia trachomatis (Ct1, AAC68292), Deinococcus radiodurans (Dr1, B75537; Dr2, D75275; Dr3 C75459) E.coli (EcNth, P20625), Haemophilus influenzae (Hi1, P44319), Helicobacter pylori (Hp1, AAD07651), Homo sapiens (hNTH1, AAC34209; hOGG1, CAA10351), Methanobacterium thermoautotrophicum (Mth1, AAB85267; Mth4, AAB85820), Methanococcus jannaschii (Mj1, AAB98606; MjOgg, Q58134), M.luteus (Ml UVendo, P46303), Mus musculus (mNth1, BAA22080; mOgg1, AAB61289), Mycobacterium tuberculosis (TB1,CAA17996), Neisseria meningitidis MC58 (Nm1, AAF40962), Pseudomonas aeruginosa (Pae1, AAG06883), Pyrobaculum aerophilum (PaNth, AAF37269), Pyrococcus abyssi (Pab1, A75109), Pyrococcus horikoshii (Ph1, BBA30606), Rattus norvegicus (ratOgg1, AAC77525), Rickettsia prowazekii (Rp1, CAA72458), Saccharomyces cerevisiae (ScNtg1, AAC04942; ScNtg2, CAA99045; ScOgg1, AAC49312), Schizosaccharomyces pombe (SpNth, CAA91893), Streptomyces coelicolor (Strep1, T36554), Sulfolobus solfataricus (Ss1, CAA69576), Synechocystis sp. (Syn1, P73715), Thermotoga maritima (Tm1, Q9WYK0; Tm2, Q9X2E1), Treponema pallidum (Tp1, AAC65744), Vibrio cholerae (Vc1, AAF94172), Xylella fastidiosa (Xf1, AAF83457). The bar scale stands for number of substitutions per site. Bold labels designate proteins with biochemically determined functions. The asterisk marks the endonuclease III homolog from A.fulgidus (Af1, AAB89556) which has been studied only by NMR structure (48). To help differentiate cluster groups, we use lines of different shades; dotted lines indicate homologs, which apparently do not fall into any of these existing groups. (B) Sequence alignment of regions A and B for phylogenetic groups of endonuclease III/UV endonuclease homologs using the program ClustalW Alignments in Color (68; http://www2.ebi.ac.uk/clustalW). Putative secondary structure assignments, according to the crystal structure of E.coli endonuclease III (37), are listed at the top. The program colors amino acid residues as follows: red, AVFPMILW (small and hydrophobic); blue, DE (acidic); magenta, RK (basic); green, STYHCNGQ (hydroxyl and amine). The dots mark amino acid residues occurring >80% of the time within a single group and outside the group only once or not at all. The arrows mark amino acid residues corresponding to residues in E.coli AlkA and human hOGG1, which are involved in protein–DNA interactions (41,42). Bold labels designate proteins with biochemically determined functions. For positions of regions A and B, with respect to the whole protein sequence, see Figure 1.
Figure 10B shows amino acid alignments of two regions for the five endonuclease III/UV endonuclease groups identified in Figure 10A. The two regions have been shown, through crystal structure analysis of hOGG1 and E.coli AlkA, to contain amino acid residues involved in DNA minor-groove reading (region A) and base binding (region B) (41,42). When we perform the phylogenetic analysis of region A, we obtained the same branching pattern seen in Figure 10A. However the tree for the base binding region, region B, fails to discriminate the five groups (data not shown), apparently due to both less divergence in this region among these groups and the shorter length of region B in comparison with region A.
DISCUSSION
In this study, we report characterization of a putative DNA N-glycosylase homolog from a hyperthermophilic archaeon P.aerophilum, whose optimal growth temperature is 100°C. We designated the protein as an endonuclease III homolog from P.aerophilum (PaNth) because it displays glycosylase/lyase activity on a modified pyrimidine, DHT. At optimal NaCl concentration, between 80 and 160 mM, PaNth can remove DHT. Efficient removal is achieved when the lesion is specifically paired with G. A similar opposite base preference for DHT substrates was also observed with EcNth at its optimal low NaCl concentrations, although the efficiency was less than PaNth. While this manuscript was in preparation, Asagoshi et al. published a paper showing significant activity of EcNth and a murine endonuclease III homolog (mNth1) when DHT was paired with G (65). These findings are consistent with our observations for EcNth. The fact that DHT/G preference is observed for endonuclease III homologs from organisms in each of the three kingdoms (Bacteria, Archaea and Eucarya) suggests that DHT/G preference is a common endonuclease III characteristic. Apparently DHT/G mispairs are not found under physiological conditions and therefore what we may be seeing is DHT’s resemblance to another pyrimidine lesion that pairs with G. It may be this unknown paired lesion that is biologically relevant. It is most likely generated by direct modification of an original cytosine base or one of its derivatives (such as 5-methylcytosine), or by a replication error which inserts this unknown lesion opposite G. The observed high activity of PaNth could reflect a more efficient repair of such lesions in P.aerophilum, and may have evolved due to the additional load of DNA damage caused by an extreme environment, such as high temperature.
In E.coli, two other DNA glycosylases, endonuclease VIII and MutM (formamidopyrimidine DNA glycosylase or Fpg), have substrate spectra that overlap with that of EcNth (66,67). We searched the P.aerophilum genome for putative sequence homologs of E.coli endonuclease VIII and Fpg, but found none. Pyrobaculum aerophilum has a U/G and T/G specific DNA glycosylase (PaMig; 21) which has extensive shared amino acid sequence identity (33.3%) to PaNth. It seems PaMig is quite specific for U or T opposite G but has no detectable activity on DHT/G (data not shown). PaNth, on the other hand, is active with DHT/G and only minor activity was detected with T/G and T/GO substrates. The observed activity of PaNth on T/G and T/GO substrates could be due to a contaminating T/G glycosylase activity from E.coli or, a more interesting possibility, that PaNth might recognize T/G and T/GO mispairs. The difference in substrate spectrum of PaNth and PaMig suggests that each protects DNA from different sources of pyrimidine lesions in P.aerophilum. We have also found that P.aerophilum contains two uncharacterized DNA glycosylases of similar molecular weight to PaNth. Further work is needed to clarify whether they contribute to DHT/G repair. The definitive solution for finding a backup system for PaNth in P.aerophilum would be to construct a PaNth knockout strain and look for any residual activity with DHT/G substrate.
Our phylogenetic study, using completely sequenced whole genomes, shows that all except three obligate parasitic bacteria contain an endonuclease III/UV endonuclease sequence homolog, underlining the importance of BER in maintaining DNA stability in both mesophilic and thermophilic organisms. Endonuclease III/UV endonuclease homologs are clustered into five major groups, each with unique conserved amino acid residues. One example of this is seen in region A, which is presumably involved in DNA–protein interactions. Since PaNth is not clustered with any other characterized endonuclease III homologs and contains its own unique amino acid residues in regions A, our work helps for future annotation of putative DNA N-glycosylase sequence homologs.
At this point, the nature of these multiple endonuclease III/UV endonuclease homolog clusters, identified by phylogenetic analysis, is not clear but many possibilities exist. One possibility is that these clusters may be subfamilies of endonuclease III/UV endonuclease, each having a different function in terms of substrate preference. A possible example of this clustering by enzymatic function is the branch containing the single characterized UV endonuclease. Despite the fact that UV endonuclease and endonuclease III share close sequence homology, they catalyze different glycosylase reactions. In our phylogenetic analysis six putative endonuclease III homologs cluster with M.luteus UV endonuclease. They also contain some amino acid residues in region A that are unique to this cluster. The next logical step is to see whether these homologs have similar catalytic function to UV endonuclease. Another possibility for the multiple endonuclease III/UV endonuclease homolog clusters is that the pattern of clustering reflects amino acid sequence differences that occur either due to interaction with other protein (52) or simply due to independent evolution along the organism lineage. Further characterization of BER pathways and all their related cofactors will help clarify any biochemical basis for the clustering of endonuclease III/UV endonuclease homologs.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Richard M. Cunningham for providing EcNth and Dr Bruce Demple for providing E.coli endonuclease IV. We also thank Dr Arne Klungland (Department of Molecular Biology, Institute of Medical Microbiology, University of Oslo, The National Hospital, Norway) for providing DHU substrate. We thank Jennifer H. Tai for technical assistance on the project. The phosphoimaging analysis was done in the UCLA-DOE Biochemistry Instrumentation facility. Mass spectrometry was done in the UCLA Pasarow Mass Spectrometry Laboratory by Kym F. Faull and was financially supported by the W.M. Keck Foundation. This work was supported by USHHS Institutional National Research Service Award T32 CA09056 (to H.Y.) and by National Institutes of Health Grant GM 57917 (to J.H.M.). M.K.K.S. and R.D.W. were supported by the Imperial Cancer Research Fund, UK.
DDBJ/EMBL/GenBank accession no. AF222334
References
- 1.Wallace S.S. (1998) Enzymatic processing of radiation-induced free radical damage in DNA. Radiat. Res., 150 (5 Suppl.), S60–S79. [PubMed] [Google Scholar]
- 2.Dizdaroglu M., Bauche,C., Rodriguez,H. and Laval,J. (2000) Novel substrates of Escherichia coli nth protein and its kinetics for excision of modified bases from DNA damaged by free radicals. Biochemistry, 39, 5586–5592. [DOI] [PubMed] [Google Scholar]
- 3.Wang D., Kreutzer,D.A. and Essigmann,J.M. (1998) Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat. Res., 400, 99–115. [DOI] [PubMed] [Google Scholar]
- 4.Gasparutto D., Bourdat,A.G., D’Ham,C., Duarte,V., Romieu,A. and Cadet,J. (2000) Repair and replication of oxidized DNA bases using modified oligodeoxyribonucleotides. Biochimie, 82, 19–24. [DOI] [PubMed] [Google Scholar]
- 5.Laval J., Jurado,J., Saparbaev,M. and Sidorkina,O. (1998) Antimutagenic role of base-excision repair enzymes upon free radical-induced DNA damage. Mutat. Res., 402, 93–102. [DOI] [PubMed] [Google Scholar]
- 6.Cadet J., Bourdat,A.G., D’Ham,C., Duarte,V., Gasparutto,D., Romieu,A. and Ravanat,J.L. (2000) Oxidative base damage to DNA: specificity of base excision repair enzymes. Mutat. Res., 462, 121–128. [DOI] [PubMed] [Google Scholar]
- 7.Demple B. and Linn,S. (1982) On the recognition and cleavage mechanism of Escherichia coli endodeoxyribonuclease V, a possible DNA repair enzyme. J. Biol. Chem., 257, 2848–2855. [PubMed] [Google Scholar]
- 8.Katcher H.L. and Wallace,S.S. (1983) Characterization of the Escherichia coli X-ray endonuclease, endonuclease III. Biochemistry, 22, 4071–4081. [DOI] [PubMed] [Google Scholar]
- 9.Breimer L.H. and Lindahl,T. (1985) Thymine lesions produced by ionizing radiation in double-stranded DNA. Biochemistry, 24, 4018–4022. [DOI] [PubMed] [Google Scholar]
- 10.Boorstein R.J., Hilbert,T.P., Cadet,J., Cunningham,R.P. and Teebor,G.W. (1989) UV-induced pyrimidine hydrates in DNA are repaired by bacterial and mammalian DNA glycosylase activities. Biochemistry, 28, 6164–6170. [DOI] [PubMed] [Google Scholar]
- 11.Dizdaroglu M., Laval,J. and Boiteux,S. (1993) Substrate specificity of the Escherichia coli endonuclease III: excision of thymine- and cytosine-derived lesions in DNA produced by radiation-generated free radicals. Biochemistry, 32, 12105–12111. [DOI] [PubMed] [Google Scholar]
- 12.Wang D. and Essigmann,J.M. (1997) Kinetics of oxidized cytosine repair by endonuclease III of Escherichia coli. Biochemistry, 36, 8628–8633. [DOI] [PubMed] [Google Scholar]
- 13.D’Ham C., Romieu,A., Jaquinod,M., Gasparutto,D. and Cadet,J. (1999) Excision of 5,6-dihydroxy-5,6-dihydrothymine, 5,6-dihydrothymine and 5-hydroxycytosine from defined sequence oligonucleotides by Escherichia coli endonuclease III and Fpg proteins: kinetic and mechanistic aspects. Biochemistry, 38, 3335–3344. [DOI] [PubMed] [Google Scholar]
- 14.Mazumder A., Gerlt,J.A., Absalon,M.J., Stubbe,J., Cunningham,R.P., Withka,J. and Bolton,P.H. (1991) Stereochemical studies of the beta-elimination reactions at aldehydic abasic sites in DNA: endonuclease III from Escherichia coli, sodium hydroxide and Lys-Trp-Lys. Biochemistry, 30, 1119–1126. [DOI] [PubMed] [Google Scholar]
- 15.Eide L., Bjørås,M., Pirovano,M., Alseth,I., Berdal,K.G. and Seeberg,E. (1996) Base excision of oxidative purine and pyrimidine DNA damage in Saccharomyces cerevisiae by a DNA glycosylase with sequence similarity to endonuclease III from Escherichia coli. Proc. Natl Acad. Sci. USA, 93, 10735–10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aspinwall R., Rothwell,D.G., Roldan-Arjona,T., Anselmino,C., Ward,C.J., Cheadle,J.P., Sampson,J.R., Lindahl,T., Harris,P.C. and Hickson,I.D. (1997) Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl Acad. Sci. USA, 94, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilbert T.P., Chaung,W., Boorstein,R.J., Cunningham,R.P. and Teebor,G.W. (1997) Cloning and expression of the cDNA encoding the human homologue of the DNA repair enzyme, Escherichia coli endonuclease III. J. Biol. Chem., 272, 6733–6740. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda S., Biswas,T., Roy,R., Izumi,T., Boldogh,I., Kurosky,A., Sarker,A.H., Seki,S. and Mitra,S. (1998) Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem., 273, 21585–21593. [DOI] [PubMed] [Google Scholar]
- 19.Nash H.M., Bruner,S.D., Schärer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 20.Horst J.P. and Fritz,H.J. (1996) Counteracting the mutagenic effect of hydrolytic deamination of DNA 5-methylcytosine residues at high temperature: DNA mismatch N-glycosylase Mig.Mth of the thermophilic archaeon Methanobacterium thermoautotrophicum THF. EMBO J., 15, 5459–5469. [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H., Fitz-Gibbon,S., Marcotte,E.M., Tai,J.H., Hyman,E.C. and Miller,J.H. (2000) Characterization of a thermostable DNA glycosylase specific for U/G and T/G mismatches from the hyperthermophilic archaeon Pyrobaculum aerophilum. J. Bacteriol., 182, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendrich B., Hardeland,U., Ng,H.H., Jiricny,J. and Bird,A. (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- 23.Lindahl T., Sedgwick,B., Sekiguchi,M. and Nakabeppu,Y. (1988) Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem., 57, 133–157. [DOI] [PubMed] [Google Scholar]
- 24.Begley T.J., Haas,B.J., Noel,J., Shekhtman,A., Williams,W.A. and Cunningham,R.P. (1999) A new member of the endonuclease III family of DNA repair enzymes that removes methylated purines from DNA. Curr. Biol., 9, 653–656. [DOI] [PubMed] [Google Scholar]
- 25.O’Rourke E.J., Chevalier,C., Boiteux,S., Labigne,A., Ielpi,L. and Radicella,J.P. (2000) A novel 3-methyladenine DNA glycosylase from Helicobacter pylori defines a new class within the endonuclease III family of base excision repair glycosylases. J. Biol. Chem., 275, 20077–20083. [DOI] [PubMed] [Google Scholar]
- 26.Piersen C.E., Prince,M.A., Augustine,M.L., Dodson,M.L. and Lloyd,R.S. (1995) Purification and cloning of Micrococcus luteus ultraviolet endonuclease, an N-glycosylase/abasic lyase that proceeds via an imino enzyme-DNA intermediate. J. Biol. Chem., 270, 23475–23484. [DOI] [PubMed] [Google Scholar]
- 27.Nyaga S.G. and Lloyd,R.S. (2000) Two Glycosylase/Abasic Lyases from Neisseria mucosa that Initiate DNA Repair at Sites of UV-induced Photoproducts. J. Biol. Chem., 275, 23569–23576. [DOI] [PubMed] [Google Scholar]
- 28.Michaels M.L., Pham,L., Nghiem,Y., Cruz,C. and Miller,J.H. (1990) MutY, an adenine glycosylase active on G-A mispairs, has homology to endonuclease III. Nucleic Acids Res., 18, 3841–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu A.L. and Fawcett,W.P. (1998) Characterization of the recombinant MutY homolog, an adenine DNA glycosylase, from yeast Schizosaccharomyces pombe. J. Biol. Chem., 273, 25098–25105. [DOI] [PubMed] [Google Scholar]
- 30.Slupska M.M., Baikalov,C., Luther,W.M., Chiang,J.-H., Wei,Y.-F. and Miller,J.H. (1996) Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol., 178, 3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bjørås M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Rognes,T. and Seeberg,E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radicella J.P., Dherin,C., Desmaze,C., Fox,M.S. and Boiteux,S. (1997) Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 8010–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arai K., Morishita,K., Shinmura,K., Kohno,T., Kim,S.R., Nohmi,T., Taniwaki,M., Ohwada,S. and Yokota,J. (1997) Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene, 14, 2857–2861. [DOI] [PubMed] [Google Scholar]
- 35.Roldán-Arjona T., Wei,Y.F., Carter,K.C., Klungland,A., Anselmino,C., Wang,R.P., Augustus,M. and Lindahl,T. (1997) Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 8016–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham R.P., Asahara,H., Bank,J.F., Scholes,C.P., Salerno,J.C., Surerus,K., Münck,E., McCracken,J., Peisach,J. and Emptage,M.H. (1989) Endonuclease III is an iron-sulfur protein. Biochemistry, 28, 4450–4455. [DOI] [PubMed] [Google Scholar]
- 37.Thayer M.M., Ahern,H., Xing,D., Cunningham,R.P. and Tainer,J.A. (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J., 14, 4108–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Labahn J., Schärer,O.D., Long,A., Ezaz-Nikpay,K., Verdine,G.L. and Ellenberger,T.E. (1996) Structural basis for the excision repair of alkylation-damaged DNA. Cell, 86, 321–329. [DOI] [PubMed] [Google Scholar]
- 39.Yamagata Y., Kato,M., Odawara,K., Tokuno,Y., Nakashima,Y., Matsushima,N., Yasumura,K., Tomita,K., Ihara,K., Fujii,Y., Nakabeppu,Y., Sekiguchi,M. and Fujii,S. (1996) Three-dimensional structure of a DNA repair enzyme, 3-methyladenine DNA glycosylase II, from Escherichia coli. Cell, 86, 311–319. [DOI] [PubMed] [Google Scholar]
- 40.Guan Y., Manuel,P.C., Arvai,A.S., Parikh,S.S., Mol,C.D., Miller,J.H., Lloyd,R.S. and Tainer,J.A. (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature Struct. Biol., 5, 1058–1064. [DOI] [PubMed] [Google Scholar]
- 41.Hollis T., Ichikawa,Y. and Ellenberger,T. (2000) DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J., 19, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruner S.D., Norman,D.P. and Verdine,G.L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature, 403, 859–866. [DOI] [PubMed] [Google Scholar]
- 43.Lindahl T. and Nyberg,B. (1974) Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry, 13, 3405–3410. [DOI] [PubMed] [Google Scholar]
- 44.Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 45.Sandigursky M. and Franklin,W.A. (1999) Thermostable uracil-DNA glycosylase from Thermotoga maritima a member of a novel class of DNA repair enzymes. Curr. Biol., 9, 531–534. [DOI] [PubMed] [Google Scholar]
- 46.Sandigursky M. and Franklin,W.A. (2000) Uracil-DNA glycosylase in the extreme thermophile Archaeoglobus fulgidus. J. Biol. Chem., 275, 19146–19149. [DOI] [PubMed] [Google Scholar]
- 47.Eisen J.A. and Hanawalt,P.C. (1999) A phylogenomic study of DNA repair genes, proteins and processes. Mutat. Res., 435, 171–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shekhtman A., McNaughton,L., Cunningham,R.P. and Baxter,S.M. (1999) Identification of the Archaeoglobus fulgidus endonuclease III DNA interaction surface using heteronuclear NMR methods. Struct. Fold. Des., 7, 919–930. [DOI] [PubMed] [Google Scholar]
- 49.Völkl P., Huber,R., Drobner,E., Rachel,R., Burggraf,S., Trincone,A. and Stetter,K.O. (1993) Pyrobaculum aerophilum sp. nov., a novel nitrate-reducing hyperthermophilic archaeum. Appl. Environ. Microbiol., 59, 2918–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fitz-Gibbon S., Choi,A.J., Miller,J.H., Stetter,K.O., Simon,M.I., Swanson,R. and Kim,U.J. (1997) A fosmid-based genomic map and identification of 474 genes of the hyperthermophilic archaeon Pyrobaculum aerophilum. Extremophiles, 1, 36–51. [DOI] [PubMed] [Google Scholar]
- 51.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klungland A., Höss,M., Gunz,D., Constantinou,A., Clarkson,S.G., Doetsch,P.W., Bolton,P.H., Wood,R.D. and Lindahl,T. (1999) Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell, 3, 33–42. [DOI] [PubMed] [Google Scholar]
- 53.Biggerstaff M. and Wood,R.D. (1999) Assay for nucleotide excision repair protein activity using fractionated cell extracts and UV-damaged plasmid DNA. Methods Mol. Biol., 113, 357–372. [DOI] [PubMed] [Google Scholar]
- 54.Swofford D.L. (1999) PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4. Sinauer Associates, Sunderland, MA.
- 55.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L. and Sonnhammer,E.L. (2000) The Pfam protein families database. Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Finn,R.D. and Sonnhammer,E.L. (1999) Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res., 27, 260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonnhammer E.L., Eddy,S.R., Birney,E., Bateman,A. and Durbin,R. (1998) Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res., 26, 320–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wisconsin package 9.1 (1997) Genetics Computer Group (GCG), Madison, WI.
- 59.Noll D.M., Gogos,A., Granek,J.A. and Clarke,N.D. (1999) The C-terminal domain of the adenine-DNA glycosylase MutY confers specificity for 8-oxoguanine.adenine mispairs and may have evolved from MutT, an 8-oxo-dGTPase. Biochemistry, 38, 6374–6379. [DOI] [PubMed] [Google Scholar]
- 60.Jiang D., Hatahet,Z., Melamede,R.J., Kow,Y.W. and Wallace,S.S. (1997) Characterization of Escherichia coli Endonuclease VIII. J. Biol. Chem., 272, 32230–32239. [DOI] [PubMed] [Google Scholar]
- 61.Warner H.R., Demple,B.F., Deutsch,W.A., Kane,C.M. and Linn,S. (1980) Apurinic/apyrimidinic endonucleases in repair of pyrimidine dimers and other lesions in DNA. Proc. Natl Acad. Sci. USA, 77, 4602–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dodson M.L., Michaels,M.L. and Lloyd,R.S. (1994) Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem., 269, 32709–32712. [PubMed] [Google Scholar]
- 63.Sun B., Latham,K.A., Dodson,M.L. and Lloyd,R.S. (1995) Studies on the catalytic mechanism of five DNA glycosylases. Probing for enzyme-DNA imino intermediates. J. Biol. Chem., 270, 19501–19508. [DOI] [PubMed] [Google Scholar]
- 64.Tchou J. and Grollman,A.P. (1995) The catalytic mechanism of Fpg protein. Evidence for a Schiff base intermediate and amino terminus localization of the catalytic site. J. Biol. Chem., 270, 11671–11677. [DOI] [PubMed] [Google Scholar]
- 65.Asagoshi K., Odawara,H., Nakano,H., Miyano,T., Terato,H., Ohyama,Y., Seki,S. and Ide,H. (2000) Comparison of substrate specificities of Escherichia coli endonuclease III and its mouse homologue (mNTH1) using defined oligonucleotide substrates. Biochemistry, 39, 11389–11398. [DOI] [PubMed] [Google Scholar]
- 66.Blaisdell J.O., Hatahet,Z. and Wallace,S.S. (1999) A novel role for Escherichia coli endonuclease VIII in prevention of spontaneous G→T transversions. J. Bacteriol ., 181, 6396–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q.M., Miyabe,I., Matsumoto,Y., Kino,K., Sugiyama,H. and Yonei,S. (2000) Identification of repair enzymes for 5-formyluracil in DNA: Nth, Nei and MutM proteins of Escherichia coli. J. Biol. Chem., Aug 23 [epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 68.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]