

Abstract
Painful peripheral neuropathies produced by nerve trauma are accompanied by substantial axonal degeneration and by a response in spinal cord microglia that is characterized by hypertrophy and increased expression of several intracellular and cell-surface markers, including ionizing calcium-binding adapter molecule 1 (Iba1) and Cd11b (a complement receptor 3 antigen recognized by the OX42 antibody). The microglia response has been hypothesized to be essential for the pathogenesis of the neuropathic pain state. In contrast, the painful peripheral neuropathies produced by low doses of cancer chemotherapeutics do not produce degeneration of axons in the peripheral nerve, although they do cause partial degeneration of the sensory axons’ distal-most tips, i.e. the intraepidermal nerve fibers that form the axons’ terminal receptor arbors. The question thus arises as to whether the relatively minor and distal axonal injury characterizing the chemotherapy-evoked neuropathies is sufficient to evoke the microglial response that is seen after traumatic nerve injury. We examined the lumbar spinal cord of rats with painful peripheral neuropathies due to the anti-neoplastic agents, paclitaxel, vincristine, and oxaliplatin, and the anti-retroviral agent, 2′,3′-dideoxycytidine (ddC), and compared them to rats with a complete sciatic nerve transection and the partial sciatic nerve injury produced in the chronic constriction injury model (CCI). As expected, microglia hypertrophy and increased expression of Iba1 were pronounced in the nerve transection and CCI animals. However, there was no microglia hypertrophy or increased Iba1 staining in the animals treated with paclitaxel, vincristine, oxaliplatin, or ddC. These results suggest that the mechanisms that produce neuropathic pain after exposure to chemotherapeutics may be fundamentally different than those operating after nerve trauma.
Keywords: 2′,3′-dideoxycytidine; microglial hypertrophy; neuropathic pain; oxaliplatin; paclitaxel; vincristine
Transection of a peripheral nerve evokes dramatic changes in spinal cord microglia. The changes include an alteration of cell shape (hypertrophy), increased expression of cell surface and intracellular proteins, proliferation of resident microglia, and the in-migration of hematogenous microglia precursors [reviewed in (Inoue and Tsuda, 2009; McMahon and Malcangio, 2009; Milligan and Watkins, 2009)]. These changes are subsumed under the term “activation” following del Rio-Hortega and Penfield’s description (1927) of the microglial response to a stab wound in the cerebral cortex. For the response of spinal cord microglia to peripheral nerve injury, activation is generally detected immunocytochemically using antisera directed against the cell surface marker, Cd11b (a complement receptor 3 antigen recognized by the OX42 antibody), or the intracellular marker, ionizing calcium-binding adapter molecule 1 (Iba1); both markers demonstrate increased staining levels and hypertrophy. Following transection of the sciatic nerve, responding spinal microglia are found in the dorsal horn territory where the transected sensory axons terminate, while in the ventral horn the responding microglia are restricted to the lateral motor neuron pools that contain the cells whose axons have been cut (Beggs and Salter, 2007).
Changes in spinal cord microglia after a complete sciatic nerve transection were first reported over 45 years ago [for review see (Gehrmann et al., 1991)], but interest in the phenomenon was greatly increased by reports showing that microglia also responded after partial nerve injuries that evoked neuropathic pain. Such nerve injuries are accompanied by an up-regulation of pro-inflammatory cytokines and chemokines. Microglia are immune cells and they have thus been proposed as a likely source of these substances (Coyle, 1998; DeLeo et al., 1997). Consistent with this idea, microglial inhibitors (e.g., minocycline, propentofylline, and ibudilast (AV411)) block the neuropathic pain state produced by partial nerve injury. These observations have led to the hypothesis that the microglial response is an important, and perhaps a necessary, factor in the pathogenesis of neuropathic pain (Inoue and Tsuda, 2009; McMahon and Malcangio, 2009; Milligan and Watkins, 2009).
We have investigated the painful peripheral neuropathies produced by cytotoxins; specifically, the cancer chemotherapeutic agents, paclitaxel, oxaliplatin, and vincristine, and the anti-retroviral agent, 2′,3′-dideoxycytidine (ddC). With the low-dose protocols that we use, these agents produce neuropathic pain but do not produce degeneration of axons in the peripheral nerve. However, they do produce a statistically significant partial degeneration of the intraepidermal nerve fibers (IENF) that form the terminal receptor arbors of the sensory axons that innervate the epidermis (Jin et al., 2008; Siau et al., 2006; Xiao and Bennett, 2010; Xiao et al., 2009).
The experiments reported here investigate the question of whether the relatively minor and very distal degeneration seen in chemotherapy-evoked peripheral neuropathies causes the microglial response that is seen after traumatic nerve injury.
EXPERIMENTAL PROCEDURES
These experiments conformed to the ethics guidelines of the International Association for the Study of Pain (Zimmermann, 1983), the National Institutes of Health (USA), and the Canadian Institutes of Health Research. All experimental protocols were approved by the Animal Care Committee of the Faculty of Medicine, McGill University, in accordance with the regulations of the Canadian Council on Animal Care.
Animals
Adult male Sprague-Dawley rats (200–300g, Harlan Inc., Indianapolis, IN; Frederick, MD breeding colony) were housed in groups of 3–4 on sawdust bedding in plastic cages. Artificial lighting was provided on a fixed 12 hour light-dark cycle with food and water available ad libitum.
Drug administration and surgery
Paclitaxel (Taxol™; Biolyse Pharma Corp.; St. Catherines, ON, Canada) was administered via our standard protocol: the stock solution (in Cremophor/EL) was diluted with saline to a concentration of 2 mg/ml and injected IP at 2 mg/kg on four alternate days (D0, D2, D4 and D6) (Flatters and Bennett, 2004; Polomano et al., 2001).
Vincristine sulfate (Novopharm Ltd.; Toronto, ON, Canada) was given by diluting the stock solution with saline to a concentration of 50 μg/ml and injecting IP daily at 50 μg/kg for 10 consecutive days (D0–D9) (Siau and Bennett, 2006; Siau et al. 2006).
Oxaliplatin (Eloxatin™; Sanofi-aventis; Laval, QC, Canada) was given according to the protocol of Xiao and Bennett (2010): the stock solution was diluted with 5% dextrose in distilled water to a concentration of 2 mg/ml and injected IP daily for five consecutive days at 2 mg/kg.
2′,3′-dideoxycytidine (ddC; Sigma-Aldrich; Oakville, ON, Canada)) was administered as a single bolus via the tail vein at 50 mg/ml/kg (Joseph et al., 2004; Siau and Bennett, 2006).
For comparisons to the three paclitaxel groups, we used a control group consisting of animals that had received four IP vehicle injections (two rats sacrificed on D7 and two on D14). For comparisons to the vincristine, oxaliplatin, and ddC groups, we used another control group consisting of animals that had received five IP injections of 5% dextrose in water (1 ml/kg) on five consecutive days and then sacrificed on D35.
Rats with a complete unilateral nerve transection were prepared by cutting the common sciatic nerve at mid-thigh level under isoflurane anesthesia. Rats were prepared with the chronic constriction injury (CCI) as described previously (Bennett and Xie, 1988). The side contralateral to transection or CCI was used as the control comparison. It is known that the contralateral side in such cases may not be normal; however, all published studies of the effects of traumatic nerve injury on microglia activation have used this as the control. All vehicle control, drug, and surgery groups were n = 4.
Pain assays
The rats were habituated to the testing apparatus on two daily sessions. As per our standard procedure (Flatters and Bennett, 2004), 4 g and 15 g von Frey hairs (VFH) were applied to the plantar hind paw five times on each side for each hair. For each hair, the responses from both sides were summed and expressed as a percent response. Normal rats rarely or never respond to the 4 g VFH, indicating that it is an innocuous stimulus. Increased response frequency to this stimulus is thus indicative of mechano-allodynia. Normal rats respond to the 15 g VFH about 10–20% of the time, suggesting that it is a barely painful stimulus (it evokes a mild stinging pain when applied to our volar wrist). An increase in response frequency to the 15 g VFH is thus indicative of mechano-hyperalgesia.
Paclitaxel-treated rats were tested prior to drug exposure and on D7, D14, or D27. We have shown that paclitaxel-evoked mechano-allodynia and mechano-hyperalgesia begin with a distinct delay after the last injection of paclitaxel. Pain symptoms are absent on D7 (one day after the last paclitaxel injection), first appear 14–18 days after the last injection, and reach peak severity by D27 (Bennett et al. 2010; Flatters and Bennett, 2004, 2006; Xiao et al., 2009).
Immunocytochemistry
Sciatic nerve transection rats were sacrificed 14 after surgery. CCI rats were sacrificed 8 days after surgery, which is the time of onset of the plateau phase of peak pain severity (Bennett and Xie, 1988). Chemotherapy-treated rats were sacrificed immediately after behavioral testing: paclitaxel-treated rats on D7, D14, or D27; vincristine-treated rats on D16, oxaliplatin-treated rats on D35, and ddC-treated rats on D8. For the vincristine and ddC dosing protocols used here, robust mechano-allodynia and mechano-hyperalgesia are known to be present at the selected time points (Joseph et al., 2004; Siau and Bennett, 2006; Siau et al., 2006). A complete time-course of oxaliplatin-evoked pain with the dosing protocol used here shows that the selected time point is near the beginning of the phase of peak pain severity (Xiao and Bennett, 2010).
The rats were over-dosed with sodium pentobarbital (100 mg/kg; IP) and perfused transcardially with a vascular rinse (phosphate buffered saline (PBS) containing 0.05% sodium bicarbonate and 0.1% sodium nitrite) for 1 min; followed by freshly prepared 4% paraformaldehyde in 0.1M phosphate buffer, pH 7.4. The lumbosacral vertebral column was removed and post-fixed overnight, after which the L4/L5 segments of the spinal cord were exposed via laminectomy and identified by tracing the dorsal roots from their respective dorsal root ganglia (DRG). For the animals with CCI and sciatic nerve transection, a notch was made in the ventral side of the spinal cord contralateral to the side of nerve injury. Spinal cords were cryoprotected in 30% sucrose solution at 4º C overnight and then and stored at −80° C.
Cryostat sections (30 μm) sections were collected in PBS. Following one hour incubation in PBS containing 0.2% Triton-X 100 (PBS+T) and 10% normal donkey serum (Jackson ImmunoResearch Laboratories; Mississauga, ON, Canada) at room temperature, sections were incubated for 24 h at 4°C in PBS+T containing rabbit primary antisera diluted 1:1000 and 5% NDS. The primary antibody was directed against Iba1 (Wako Chemicals; Richmond, VA). After rinsing in PBS + T, sections were incubated in donkey anti-rabbit IgG secondary antibody labeled with Cy3 (Jackson ImmunoReseach) diluted 1:200 for 1.5 h. Negative control sections (no exposure to the primary antisera) were processed concurrently for each rat.
Quantification of microglial staining
Each section was photographed twice - first with bright-field and then with epifluorescence illumination. Care was taken to ensure that the fluorescence images from all animals were obtained in exactly the same way.
Using Image-Pro software (version 6.2; MediaCybernetics; Bethesda, MD), two areas-of-interest (AOI) were delimited on each side of the bright-field images. The first AOI captured the dorsal horn (DH; laminae I–III/IV) and was delimited medially, dorsally and laterally by the white matter-gray matter border, and ventrally by a horizontal line originating at the ventrolateral termination of laminae I–II, i.e., where the laminae curve around the lateral edge of the dorsal horn (see Fig. 1). The second AOI captured that part of the ventral horn (VH) that contains the lateral motor neurons, which innervate the distal musculature (calf and hind paw). It was delimited laterally and ventrally by the white matter-gray matter border, dorsally by a horizontal line originating at the level of the central canal and extending to the lateral gray matter-white matter border, and medially by a vertical line that bisected the aforesaid horizontal line (Fig. 1). The AOIs were subsequently superimposed on the fluorescence images. Setting AOI borders with the bright-field images has two advantages: the border between gray and white matter is clearly visible, and the observer is blind as to the signal to be measured (the fluorescent label). The fluorescence intensity values of all images were switched to gray scale values (0–255). For each animal, the negative control section (reacted concurrently but without exposure to primary antibody) was used to estimate non-specific staining. The distribution of pixel intensities from the entire gray matter was plotted and the highest value in this distribution was designated as the non-specific threshold intensity. This threshold value was then subtracted from all pixel intensity values in the AOIs from the section stained with primary antibody. We then obtained an “optical density” value by dividing the number of suprathreshold pixels by the number of pixels in the AOI. This method is equivalent to the “area percent” method described and validated by Blackbeard et al. (2007).
Figure 1.
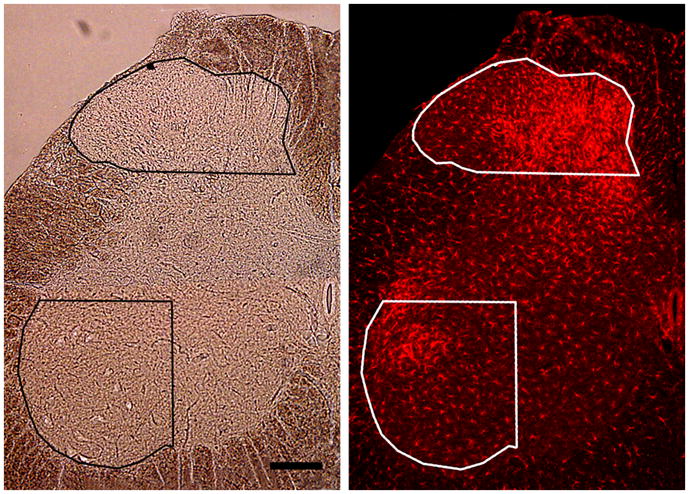
Method for assigning AOI borders. Left: Bright-field image of the lumbar spinal cord ipsilateral to a sciatic nerve transection showing the limits of the dorsal horn and ventral horn AOIs. Right: AOIs transferred to the epifluorescence image of the same section stained for Iba1. Scale bar: 200 μm.
For the drug-injected groups, in each rat we obtained a single optical density value for the DH and another for the VH by averaging the two sides and these values were compared to the homologous left-right average values from the vehicle-treated controls. For the nerve transection and CCI groups, we compared the DH and VH optical density values from the side of nerve injury with those on the opposite side.
Hypertrophy was assessed by inspection of at least three fields (60X objective) in the DH and VH per section.
RESULTS
Pain assays
Mechano-allodynia and mechano-hyperalgesia were present in the paclitaxel-treated rats on D27, but not on D7 or D14 (Fig. 2). Oxaliplatin, vincristine, and ddC treatment produced mechano-allodynia and mechano-hyperalgesia at the selected time points (Fig. 2).
Figure 2.
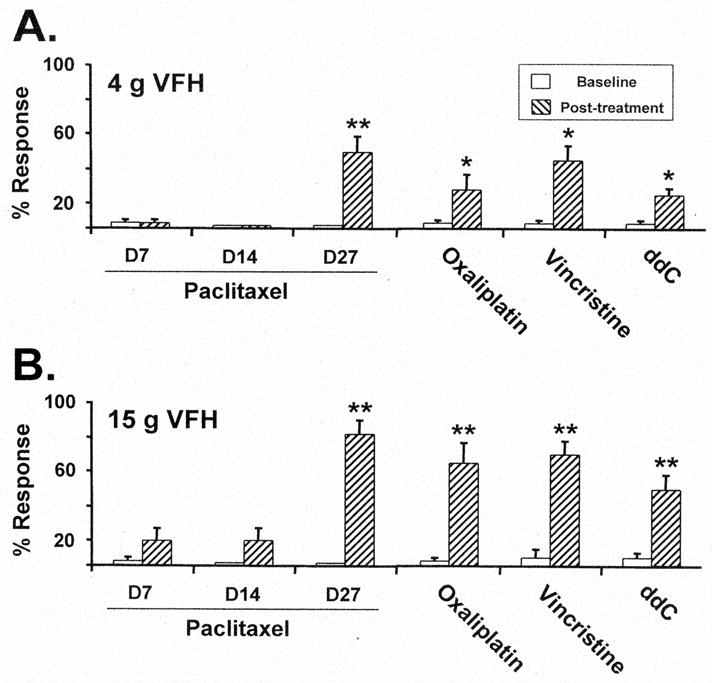
Chemotherapy-evoked neuropathic pain. Responses to (A) 4 g von Frey hair stimulus and (B) 15 g von Frey hair stimulus at baseline and after treatment with paclitaxel, oxaliplatin, vincristine, and ddC at the time points selected for analysis of microglial responses. Means ± SEM, n = 4/group. *, ** p < 0.05, 0.01, t-tests vs. pre-baseline).
Immunocytochemistry
The maximum gray scale intensity values for the negative control sections (no exposure to the primary antibody) from all the groups were very low and ranged from only 6 to 7, which is less than 3.0% of the scale (0–255).
The optical density values for the two control groups’ DH were nearly identical (mean ± SEM): paclitaxel control group: 76.7 ± 7.2%; vincristine, oxaliplatin and ddC control group: 76.3 ± 4.2%. The same was true for the optical density values for the VH: paclitaxel control group: 59.5 ± 10.4%; vincristine, oxaliplatin and ddC control group: 59.7 ± 5.3%. We saw no hypertrophic microglia in the DH and VH in the control groups (Fig. 3B).
Figure 3.

Microglial phenotypes in the medial dorsal horn. A: Microglia in a rat with a sciatic nerve transection. This is the hypertrophic phenotype, characterized by short, thickened dendrites. B: Normal microglia in a vehicle-treated control animal. This is the normal phenotype, characterized by long, thin, and highly ramified dendrites. C, D: Microglia in animals with paclitaxel-evoked neuropathy (C: D7, one day the last paclitaxel injection; D: D27, 21 days after the last paclitaxel and the time of peak pain severity). Microglia in animals treated with paclitaxel, vincristine, oxaliplatin or ddC always had the normal phenotype. Confocal z-stacks (100X objective, 16–22 0.8 μm thick optical slices). Scale bar: 10 μm.
Microglial after sciatic nerve transection
Axotomy produced the expected increased Iba1 staining in both the ipsilateral DH and VH (Figs. 1 and 5). For the DH, the optical density values were 94.7 ± 1.5 for the ipsilateral side and 68.0 ± 5.6 for the contralateral side. This is a statistically significant increase of 39.3% relative to the control. For the VH, the optical density values were 77.0 ± 7.1 ipsilaterally and 40.8 ± 9.0 contralaterally. This is a statistically significant increase of 88.6% relative to the control. The contralateral staining intensity was comparable to that seen in the vehicle-injected control group.
Figure 5.

Quantification of Iba1 labeling intensity. Percent control optical density values for (top) dorsal horn and (bottom) ventral horn AOIs. * p < 0.01 vs. own control group.
Hypertrophic microglia were seen throughout those regions of the ipsilateral DH and VH that expressed increased staining (Fig. 3A). No hypertrophy was seen in the contralateral DH and VH.
Microglia after CCI
Also as expected, CCI produced increased Iba1 staining in both the ipsilateral DH and VH (Figs. 4 and 5). For the DH, the optical density values were 86.1 ± 0.8 for the ipsilateral side and 56.3 ± 3.7 for the contralateral side. This is a statistically significant increase of 53.1% relative to the control (Fig. 5). For the VH, the optical density values were 79.9 ± 3.5 ipsilaterally and 31.6 ± 2.0 contralaterally. This is a statistically significant increase of 152.4% relative to the control. The increases seen in the DH and VH of CCI animals were greater than those seen after nerve transection; the significance of this is unknown. The contralateral DH and VH staining intensities were comparable to those seen in the vehicle-injected control group.
Figure 4.

Coronal sections from the L4/L5 spinal cord. (A) Section from a CCI animal. Traumatic nerve injury evokes a marked increase in Iba1 labeling in microglia in the ipsilateral dorsal and ventral horns. The side contralateral to nerve trauma lacks hypertrophic microglia and the staining intensity is indistinguishable from normal. (B) Section from a paclitaxel-treated animal sacrificed on D14. Neither paclitaxel nor any of the other agents produced any increase in Iba1 labeling in any region of the spinal cord grey matter. Scale bar: 1.0 mm.
As with transection, hypertrophic microglia were seen throughout those regions of the ipsilateral DH and VH that expressed increased staining. No hypertrophy was seen in the contralateral DH or VH.
Microglia after paclitaxel treatment
There was no significant change in Iba1 staining in the DH or VH of any of the three paclitaxel-treated groups (Figs. 4 and 5). For the DH, the optical density values were: control group: 76.7 ±7.2; paclitaxel D7: 80.0 ± 5.2; paclitaxel D14: 76.6 ± 2.6; paclitaxel D27: 70.8 ± 4.0. These are statistically non-significant changes relative to control: D7: 4.3%; D14: −0.1%; and D27: −7.7%. For the VH, the optical density values were: control group: 59.5 ±10.4; paclitaxel D7: 71.5 ± 9.3; paclitaxel D14: 57.0 ± 9.4; and paclitaxel D27: 55.8 ± 8.3. These are statistically non-significant changes relative to control: D7: 20.2%; D14: −4.2%; and D27: −6.3%.
Microglial hypertrophy was absent in the DH and VH of all the paclitaxel-treated groups (Fig. 3C, D).
Microglia after vincristine, oxaliplatin and ddC treatment
There was no significant change in Iba1 staining in the DH or VH of the groups treated with vincristine, oxaliplatin, or ddC (Fig. 5). For the DH, the optical density values were: control group: 76.3 ± 4.2; vincristine: 66.8 ± 3.6; oxaliplatin: 67.9 ± 4.9; and ddC: 81.8 ± 5.3. These are statistically non-significant changes relative to control: vincristine: −12.3%; oxaliplatin: −10.9%; and ddC: 7.3%. For the VH, the optical density values were: control group: 59.7 ± 5.3; vincristine: 55.8 ± 2.2; oxaliplatin: 55.3 ± 6.1; and ddC: 73.9 ± 5.9. These are statistically non-significant changes relative to control: vincristine: −6.4%; oxaliplatin: −7.3%; and ddC: 23.9%.
There was no microglia hypertrophy in the DH or VH in the vincristine, oxaliplatin, and ddC groups.
DISCUSSION
We found no evidence for increased microglia Iba1 staining or hypertrophy in the spinal cord DH in the painful peripheral neuropathies produced by our dosing protocols for paclitaxel, oxaliplatin, vincristine, and ddC. We also found no changes in the VH, but this is not surprising as these agents evoke peripheral neuropathies that are exclusively or predominately sensory rather than motor. Both nerve trauma models (transection and CCI) produced the expected increase in Iba1 staining and microglial hypertrophy in the ipsilateral DH and VH.
After traumatic nerve injury, microglial hypertrophy and Iba1 (and C11b) up-regulation begin and peak within a few days, but it can still be detected for at least seven weeks (Clark et al., 2007; Echeverry et al., 2008; Tawfik et al., 2007). Vincristine-treated and ddC-treated rats were examined at times when pain was present. Oxaliplatin-treated rats were also examined at a time when pain was present and we know from a time course study that the time point examined is early in the phase of peak pain severity (Xiao et al., 2010). Paclitaxel-treated rats were examined at three separate time points including the time of pre-pain (coasting) and a time early in the phase of peak pain severity. Thus, it is very unlikely that we missed microglia activation by looking at inappropriate times.
For the axotomy and CCI groups, including the entire mediolateral extent of the DH in the AOI diluted the estimate of microglia activation because there is no response in the lateral one-third of superficial laminae (Beggs and Salter, 2007), which is innervated by the uninjured posterior cutaneous nerve of the thigh (Swett and Woolf, 1985). However, for the toxins it is appropriate to include the entire mediolateral extent of the DH because the toxins would not be expected to spare any of the long nerves in the hind limb. The regions of microglial activation in the DH and VH are somatotopic (Beggs and Salter, 2007) and thus in the case of traumatic mononeuropathy they will vary with segmental level. In contrast, paclitaxel and the other chemotherapeutics evoke a polyneuropathy that involves all the long nerves innervating the hind paws and tail (Polomano et al., 2001). We carefully identified the fourth and fifth lumbar spinal segments that receive afferent axons from the hind paw and contain the motor neurons that innervate the muscles of the distal extremity. Thus we could not have missed a chemotherapeutic-evoked effect on microglia by examining the wrong spinal segments.
Iba1 up-regulation and hypertrophy as markers of microglia activation
Evidence suggests that different peripheral inflammatory pain conditions may evoke different microglia response programs with different temporal profiles [reviewed in (Fu et al., 2009; Hua et al., 2005)]. However, to our knowledge, the present study is the first demonstration of heterogeneity of the microglial response in different kinds of painful peripheral neuropathy.
Microglial response programs involve changes in the expression of very many proteins via activation of mitogen-activated protein kinases (MAPK) (Ji et al., 2009). It is therefore possible that the chemotherapy-evoked painful peripheral neuropathies evoke a microglial response program that does not involve Iba1 up-regulation (or hypertrophy). There is evidence that this is true in certain inflammatory pain conditions where, for example, microglial activation is instead marked by increased levels of phosphorylated p38 MAPK (Hua et al., 2005; Svensson et al., 2003). Thus, our data do not prove that the chemotherapy-evoked painful peripheral neuropathies examined here are completely without effect on spinal microglia. But our data do show that if a microglial response occurs, then it is clearly not the same as that evoked by nerve trauma.
Comparison to prior reports
Our results do not agree with those of two studies that reported increased microglial Iba1 or Cd11b staining with vincristine treatment (Kiguchi et al., 2008; Sweitzer et al., 2006) and one study that reported an increase in the staining levels of Cd11b with paclitaxel treatment (Ledeboer et al., 2007). The reason for the differences is not known. For the vincristine studies, several variables may be involved, including differences in the strain of rats used, a rat-mouse species difference, and small but nevertheless crucial differences due to dose and route of administration.
Ledeboer et al. (2007) used the same paclitaxel dosing protocol that we used and reported increased Cd11b staining but they did not report any quantification of staining intensity and they did not detect an increase in Cd11b mRNA levels. Kiguchi et al. (2008) examined vincristine in the mouse and reported increased staining but they also did not describe any method for quantification. Sweitzer et al. (2006) examined vincristine in the rat and reported a “mild” activation, based on a subjective rating scale. With a unilateral nerve injury, the contralateral side of the section offers an advantageous within-subject comparison that controls for the many variables that affect immunocytochemical staining intensity. But the toxic neuropathies are bilateral and comparisons must be between-subjects. Non-quantitative between-subject comparisons of immunocytochemically stained sections may be unreliable. Reports of paclitaxel-evoked (Ledeboer et al., 2007) and vincristine-evoked (Sweitzer et al., 2006) microglial activation in the ventral horn are difficult to understand as these agents produce a sensory neuropathy; motor effects are either very rare or non-existent. Lastly, we can not reject the possibility that some of the contradictory findings are due to the use of different markers of microglial activation. However this seems unlikely because (1) Iba1 and Cd11b are co-expressed in microglia from normal animals (Ito et al., 1998) and (2) following traumatic nerve injury, hypertrophic microglia showing increased levels of phospho-p38 MAPK also show increased levels of both Iba1 and Cd11b (Jin et al., 2003; Suter et al., 2009; Tsuda et al., 2004; Scholz et al., 2008; Wen et al., 2007; Zhou et al., 2010).
Low-dose vs. high-dose paclitaxel effects on microglia
Peters et al. (2007a, b) have described Cd11b up-regulation and hypertrophy in the DH of rats treated with a high-dose paclitaxel protocol (2 X 18 mg/kg, IV; a total dose that is 4.5X greater than what we used). Their dosing protocol produces massive degeneration of axons in the sciatic nerve and dorsal roots (Cliffer et al., 1988) and increases expression of activating transcription factor-3 (ATF-3) in the nuclei of DRG neurons (Jimenez-Andrade et al., 2006; Peters et al., 2007a, b). In contrast, our low-dose paclitaxel protocol does not cause degeneration of peripheral nerve axons and does not evoke the ATF-3 signal in DRG neurons (Flatters and Bennett, 2006).
It may be significant that microglia activation in both the nerve trauma and high-dose paclitaxel models is associated with extensive degeneration of peripheral nerve axons and the ATF-3 response in DRG neurons. Gene products associated with ATF-3 transcription may include the signal(s) that evokes increased Iba1 and CD11b staining and hypertrophy, and these genes may be activated only in the context of extensive axotomy. As with paclitaxel, the vincristine and oxaliplatin dosing protocols used here produce a partial degeneration of IENFs without degeneration of axons in the peripheral nerve (Siau et al., 2006; Tanner et al., 1998; Topp et al., 2000; Xiao and Bennett, 2010). The relatively minor and very distal degeneration of terminal arbors produced by the chemotherapeutics is thus not a sufficient stimulus for microglial hypertrophy and Iba1 up-regulation.
CONCLUSIONS
Our results show that the models of chemotherapy-evoked painful peripheral neuropathies that we used here do not evoke microglial hypertrophy or Iba1 up-regulation in the spinal cord dorsal horn. This is clearly different from the robust hypertrophy and Iba1 up-regulation seen with the painful peripheral neuropathy produced by a traumatic nerve injury (CCI) and clearly different from the microglial response to a complete nerve transection. Our results add to a growing body of evidence showing that chemotherapy-evoked painful peripheral neuropathies are mechanistically different from those evoked by nerve trauma [Bordet et al., 2008; Xiao and Bennett, 2008; Xiao et al., 2009].
The chemotherapy models used here produce neuropathic pain, but not Iba1 up-regulation or microglial hypertrophy. Prior reports have shown that there is no certain relationship between microglial hypertrophy and Iba1 or Cd11b up-regulation and neuropathic pain following various traumatic injuries to peripheral nerve and dorsal root (Clark et al., 2007; Colburn and DeLeo, 1999; Colburn et al., 1997; Colburn et al., 1999; Hashizume et al., 2000; Winkelstein and DeLeo, 2002).
Minocycline, propentofylline, and ibudilast inhibit microglial responses and have been shown to block the neuropathic pain produced by paclitaxel and vincristine [reviewed in (Inoue and Tsuda, 2009; McMahon and Malcangio, 2009; Milligan and Watkins, 2009)]. The data noted above are not necessarily inconsistent with these studies. The drugs may work on targets other than microglia or they may be blocking a microglial response program that does not include Iba1 (or Cd11b) up-regulation and hypertrophy.
Acknowledgments
This work was supported by the National Institutes of Health (R01-NS052255), the Canada Research Chairs Program, and the Canada Foundation for Infrastructure. GJB is a Canada Senior Research Chair. We thank Drs. J. DeLeo, S. Sweitzer, L. Watkins, and J. Zhang for discussions.
Abbreviations
- AOI
area of interest
- ATF-3
activating transcription factor 3
- CCI
chronic constriction injury
- ddC
2′, 3′-dideoxycytidine
- DH
spinal cord dorsal horn
- DRG
dorsal root ganglion
- IENF
intraepidermal nerve fiber
- Iba1
ionizing calcium-binding adaptor molecule 1
- MAPK
mitogen-activated protein kinases
- PBS
phosphate buffered saline
- PBS-T
PBS with Triton
- VFH
von Frey hair
- VH
spinal cord ventral horn
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beggs S, Salter MW. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav Immun. 2007;21:624–633. doi: 10.1016/j.bbi.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Blackbeard J, O’Dea KP, Wallace VCJ, Segerdahl A, Pheby T, Takata M, Field MJ, Rice ASC. Quantification of the rat spinal microglial response to peripheral nerve injury as revealed by immunohistochemical image analysis and flow cytometry. J Neurosci Meth. 2007;164:207–217. doi: 10.1016/j.jneumeth.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordet T, Buisson B, Michaud M, Abitbol JL, Marchand F, Grist J, Andriambeloson E, Malcangio M, Pruss RM. Specific antinociceptive activity of cholest-4-en-3-one, oxime ( TRO19622) in experimental models of painful diabetic and chemotherapy-induced neuropathy. J Pharmacol Exp Ther. 2008;326:623–632. doi: 10.1124/jpet.108.139410. [DOI] [PubMed] [Google Scholar]
- Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain. 2007;11:223–230. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Cliffer KD, Siuciak JA, Carson SR, Radley HE, Park JS, Lewis DR, Zlotchenko E, Nguyen T, Garcia K, Tonra JR, Stambler N, Cedarbaum JM, Bodine SC, Lindsay RM, DiStefano PS. Physiological characterization of Taxol-induced large-fiber sensory neuropathy in the rat. Ann Neurol. 1998;43:46–55. doi: 10.1002/ana.410430111. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA. The effect of perineural colchicine on nerve injury-induced spinal glial activation and neuropathic pain behavior. Brain Res Bull. 1999;49:419–427. doi: 10.1016/s0361-9230(99)00075-1. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79:163–175. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- Coyle DE. Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia. 1998;23:75–83. [PubMed] [Google Scholar]
- DeLeo JA, Colburn RW, Rickman AJ. Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res. 1997;759:50–57. doi: 10.1016/s0006-8993(97)00209-6. [DOI] [PubMed] [Google Scholar]
- del Rio-Hortega P, Penfield W. Cerebral cicatrix: the reaction of neuroglia and microglia to brain wounds. Johns Hopkins Hosp Bull. 1927;41:278–303. [Google Scholar]
- Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain. 2008;135:37–47. doi: 10.1016/j.pain.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Flatters SJL, Bennett GJ. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain. 2004;109:150–161. doi: 10.1016/j.pain.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Flatters SJL, Bennett GJ. studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: Evidence for mitochondrial dysfunction. Pain. 2006;122:245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu KY, Tan YH, Sung B, Mao J. Peripheral formalin injection induces unique spinal cord microglial phenotypic changes. Neurosci Lett. 2009;449:2340239. doi: 10.1016/j.neulet.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Monaco S, Kreutzberg GW. Spinal cord microglia and DRG satellite cells rapidly respond to transection of the rat sciatic nerve. Restor Neurol Neurosci. 1991;2:181–198. doi: 10.3233/RNN-1991-245605. [DOI] [PubMed] [Google Scholar]
- Hashizume H, DeLeo JA, Colburn RW, Weinstein JN. Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine. 2000;25:1206–1217. doi: 10.1097/00007632-200005150-00003. [DOI] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Inoue K, Tsuda M. Microglia and neuropathic pain. Glia. 2009;57:1469–1479. doi: 10.1002/glia.20871. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Peters CM, Mejia NA, Ghilardi JR, Kuskowski MA, Mantyh PW. Sensory neurons and their supporting cells located in the trigeminal, thoracic and lumbar ganglia differentially express markers of injury following intravenous administration of paclitaxel in the rat. Neurosci Lett. 2006;405:62–67. doi: 10.1016/j.neulet.2006.06.043. [DOI] [PubMed] [Google Scholar]
- Jin HW, Flatters SJL, Xiao WH, Mulhern HL, Bennett GJ. Prevention of paclitaxel-evoked painful peripheral neuropathy by acetyl-L-carnitine: Effects on axonal mitochondria, sensory nerve fiber terminal arbors, and cutaneous Langerhans cells. Exptl Neurol. 2008;210:229–237. doi: 10.1016/j.expneurol.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Khasar SG, Levine JD. Novel mechanism of enhanced nociception in a model of AIDS therapy-induced painful peripheral neuropathy in the rat. Pain. 2004;107:147–158. doi: 10.1016/j.pain.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Kiguchi N, Maeda T, Kobayashi Y, Kishioka S. Up-regulation of tumor necrosis factor-alpha in spinal cord contributes to vincristine-induced mechanical allodynia in mice. Neurosci Lett. 2008;445:140–143. doi: 10.1016/j.neulet.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, Martin D, Maier SF, Johnson KW, Leinwand LA, Chavez RA, Watkins LR. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. 2007;21:686–698. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Li K, Zhang FY, Zhang ZZ, Light AR, Fu KY. Dissociation of spinal microglia morphological activation and peripheral inflammation in inflammatory pain models. J Neuroimmunol. 2007;192:40–48. doi: 10.1016/j.jneuroim.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Rudin M, Kozlova EN. Glial cell proliferation in the spinal cord after dorsal rhizotomy or sciatic nerve transection in the adult rat. Exp Brain Res. 2000;131:64–73. doi: 10.1007/s002219900273. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CM, Jimenez-Andrade JM, Kuskowski MA, Ghilardi JR, Mantyh PW. An evolving cellular pathology occurs in dorsal root ganglia, peripheral nerve and spinal cord following intravenous administration of paclitaxel in the rat. Brain Res. 2007a;1168:46–59. doi: 10.1016/j.brainres.2007.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CM, Jimenez-Andrade JM, Jonas BM, Sevcik MA, Koewler NJ, Ghilardi JR, Wong GY, Mantyh PW. Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp Neurol. 2007b;203:42–54. doi: 10.1016/j.expneurol.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Polomano R, Clark U, Mannes AJ, Bennett GJ. A painful peripheral neuropathy in rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- Scholz J, Abele A, Marian C, Häussler A, Herbert TA, Woolf CJ, Tegeder I. Low-dose methotrexate reduces peripheral nerve injury-evoked spinal microglial activation and neuropathic pain behavior in rats. Pain. 2008;138:130–142. doi: 10.1016/j.pain.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siau C, Bennett GJ. Dysregulation of cellular calcium homeostasis in chemotherapy-evoked painful peripheral neuropathy. Anesth Analg. 2006;102:1485–1490. doi: 10.1213/01.ane.0000204318.35194.ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siau C, Xiao WH, Bennett GJ. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: loss of epidermal innervation and activation of Langerhans cells. Exptl Neurol. 2006;201:507–514. doi: 10.1016/j.expneurol.2006.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter MR, Berta T, Gao YJ, Decosterd I, Ji RR. Large A-fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resinifertoxin and bupivacaine on spinal microglial changes after spared nerve injury. Mol Pain. 2009;5:53. doi: 10.1186/1744-8069-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Pahl JL, DeLeo JA. Propentofylline attenuates vincristine-induced peripheral neuropathy in the rat. Neurosci Lett. 2006;400:258–261. doi: 10.1016/j.neulet.2006.02.058. [DOI] [PubMed] [Google Scholar]
- Swett JE, Woolf CJ. The somatotopic organization of primary afferent terminals in the superficial laminae of the dorsal horn of the rat spinal cord. J Comp Neurol. 1985;231:66–77. doi: 10.1002/cne.902310106. [DOI] [PubMed] [Google Scholar]
- Tanner KD, Levine JD, Topp KS. Microtubule disorientation and axonal swelling in unmyelinated sensory axons during vincristine induced painful neuropathy in rat. J Comp Neurol. 1998;395:481–492. [PubMed] [Google Scholar]
- Tawfik VL, Nutile-McMenemy N, Lacroix-Fralish ML, Deleo JA. Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain Behav Immun. 2007;21:238–246. doi: 10.1016/j.bbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Topp KS, Tanner KD, Levine JD. Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine induced painful peripheral neuropathy in the rat. J Comp Neurol. 2000;424:563–576. [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Wen YR, Suter MR, Kawasaki Y, Huang J, Pertin M, Kohno T, Berde CB, Decosterd I, Ji RR. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- Winkelstein BA, DeLeo JA. Nerve root injury severity differentially modulates spinal glial activation in a rat lumbar radiculopathy model: considerations for persistent pain. Brain Res. 2002;956:294–301. doi: 10.1016/s0006-8993(02)03560-6. [DOI] [PubMed] [Google Scholar]
- Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: Abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain. 2008;135:262–270. doi: 10.1016/j.pain.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao WH, Zheng H, Bennett GJ. Oxaliplatin-evoked painful peripheral neuropathy. 2010. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao WH, Zheng FY, Bennett GJ, Bordet T, Pruss RM. Olesoxime (cholest-4-en-3-one, oxime): Analgesic and neuroprotective effects in a rat model of painful peripheral neuropathy produced by the chemotherapeutic agent, paclitaxel. Pain. 2009;147:202–209. doi: 10.1016/j.pain.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LJ, Yang T, Wei X, Liu Y, Xin WJ, Chen Y, Pang RP, Zang Y, Li YY, Liu XG. Brain-derived neurotrophic factor contributes to spinal long-term potentiation and mechanical hypersensitivity by activation of spinal microglia in rat. Brain Behav Immun. 2010 doi: 10.1016/j.bbi.2010.09.025. In press. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]