

Abstract
About one-third of all proteins are associated with a metal. Metalloproteomics is defined as the structural and functional characterization of metalloproteins on a genome-wide scale. The methodologies utilized in metalloproteomics, including both forward (bottom-up) and reverse (top-down) technologies, to provide information on the identity, quantity and function of metalloproteins are discussed. Important techniques frequently employed in metalloproteomics include classical proteomics tools such as mass spectrometry and 2-D gels, immobilized-metal affinity chromatography, bioinformatics sequence analysis and homology modeling, X-ray absorption spectroscopy and other synchrotron radiation based tools. Combinative applications of these techniques provide a powerful approach to understand the function of metalloproteins.
Keywords: metalloproteomics, metal, structural genomics, mass spectrometry, X-ray absorption spectroscopy, computational modeling
Approximately one-third of all proteins possess a bound metal. Examination of the Protein Data Bank (PDB, http://www.rcsb.org) reveals that Mg and Zn are the most abundant, while Ca, Mn, Fe, and Ni are also frequently observed [1]. Metalloproteins represent one of the most diverse classes of proteins, with the intrinsic metal atoms providing catalytic, regulatory, or structural roles critical to protein function [2]. For example, Zn, the most abundant metal in cells, plays a vital role in the function of more than 300 enzyme classes, in stabilizing the DNA double helix and in control of gene expression [3].
Metalloproteomics is a relatively new field addressing genome-scale identification, structural characterization, and functional annotation of metal-associated proteins [4–6]. Several major experimental approaches have been successfully employed in metalloproteomics, including both forward (bottom-up) and reverse (top-down) technologies. Reverse (top-down) technologies follow classical biochemical approaches, wherein samples are fractionated, by sequential chromatographic or 2-D gel separations, and “fractions” are subjected to metal and protein identification techniques, such as atomic absorption and mass spectrometry (LC-MS and/or ICP-MS) to identify the metal and protein associated with specific fractions [6–11]. Advantage of this approach includes isolation of proteins from a native cell or tissue environment; potential disadvantages are potential loss of native metal and difficulties in generating sufficient quantities of the protein of interest for high signal-to-noise analysis. Recent examples include a combined metal-based approach, reported by Tainer JA and Adams MWW et al. [12], to identify metals and metalloproteins in three microorganisms (Pyrococcus furiosus, Escherichia coli and Sulfolobus solfataricus) on a genome-wide scale. 343 metal peaks, in chromatography fractions and likely associated with specific metalloproteins, were analyzed using ICP-MS and the most abundance metals from Pyrococcus furiosus were identified as iron (Fe), Zinc (Zn), tungsten (W) and nickel (Ni) and cobalt (Co). Other metals by inadvertent assimilation of the analyzed sample include lead (Pb), titanium (Ti) and uranium (U). Several novel Nickel- and molybdenum-containing proteins were identified through further purification steps and matrix-assisted laser desorption/ionization (MALDI)-MS.
Immobilized-metal affinity chromatography (IMAC) is the key technique commonly used for fractionation of metalloproteins dependent on their differential binding affinities of the surface exposed amino acids towards immobilized metal ion [13,14]. Metal depleted samples are loaded on an IMAC column/chip saturated with the metal of interest, and proteins with affinity to the metal are recovered and can be analyzed by any of the classical proteomics methods, for examples, 2-D gels [8,15–17] and surface enhanced laser desorption ionization (SELDI) MS [18]. In addition to protein identification and quantitative analysis, post-translation modifications of proteins are also targeted as many metalloproteins including metallo-enzymes, metal transporters and metallo-chaperones are involved in metal-dependent catalysis and regulatory functions related to phosphorylation and de-phosphorylation. Detection of phosphopeptides by MS can be integrated with IMAC enrichment technique [19–21]. Combination of IMAC and the hybrid LTQ-obitrap MS with high sensitivity and high resolution was used to identified phosphoproteome in Streptococcus pneumonia [22]. IMAC provides information on the presence of metal-binding sites in proteins but it does not detect the metal-protein complexes present. The other drawback of the IMAC technique is that metalloproteins with a high metal affinity site will very likely pass through the column/chip undetected as the metal sites are already occupied.
The forward (bottom-up) approach is also derived from classic methods, and involves cloning and expression of the genes of interest followed by analysis of metal content and function of metalloproteins. Advantages include the ability to optimize the expression and amount of the protein of interest; disadvantages include non-native metal incorporation or loss of native metal in protein expression and purification steps, although strategies for metal exchange to optimize spectroscopic analysis are well known and extremely valuable [1,23,24].
Both the reverse (top-down) and the forward (bottom-up) experimental approaches suffer from potential mis-annotation of a metalloprotein in two distinct ways. First, when native metals are lost in purification a bound metal may not be associated with the protein (false negative). Second, non-native metals may bind in the place of native metals, which may mislead the investigator with respect to the native and functionally active metalloprotein species. Such challenges have been well understood in the metalloprotein field for many years, as confirmation of the role of a metal in a protein’s native function requires careful experimental work. By combining a range of computational approaches with HT-experimental annotation, using either forward or reverse methods, these factors are under active exploration by many research groups [1,4,12].
Computational approaches can complement these experimental methods and explore wide ranges of sequence space for their potential connections to metal binding and related enzyme functions [25]. Several bioinformatics approaches commonly used in metalloproteomics include prediction of metal-binding proteins based on known consensus sequences and prediction of a metal binding site based on a known 3-D structure. A bioinformatics approach to predict the metal binding ability of proteins to Zn, non-heme Fe and Cu in several organisms has been reported [25–27]. In this approach, metalloproteins are identified through the combined search of known metal binding domains and of local sequence similarity to known metal binding motif. The search can be applied to the whole genome sequences of any organism and provides an estimate of the complete ensemble of metalloproteins in the organism. A similar study using structural bioinformatics and whole genome sequences has been carried out to identify Fe, Zn, Mn and Co binding proteins [28]. Metal binding sites can also be predicted based on a known 3-D structure with reasonable accuracy because of the conserved nature of a metal binding site and its usual compact size [29,30]. The method is useful to identify metal binding sites when no metal binding sequence motifs are obvious.
Metalloproteomics involves the structural and functional characterization of metal-binding proteins. Synchrotron radiation sources provide a unique set of techniques including X-ray macromolecular crystallography (MX), X-ray solution scattering (SAXS) and X-ray absorption spectroscopy (XAS), well suited for the structural and functional studies of metalloproteins and metalloprotein complexes. Approximately one-third of all structures in PDB (http://www.rcsb.org) contain a metal, however, very few of these crystal structures achieve atomic resolution (better than 1.2 Å). The atomic resolution structures can reveal new features essential to the biological functions of the metalloproteins that are sometimes missed in medium-low resolution structures [31,32]. XAS is a complementary technique to MX in structural analysis of metalloproteins [33,34]. XAS can be used to determine aspects of metalloprotein active site structures at extremely high accuracy (±0.02 Å), comparable to structures at atomic resolution, and it can be applied to samples in very diverse conditions (solid, solution or gaseous state). It can be used independently or to supplement structural information derived from MX or nuclear magnetic resonance (NMR) spectroscopy [35]. This technique can provide element-specific structural information for reaction intermediates of protein samples that are not amenable to study as single crystals or on samples that are difficult to crystallize. SAXS is a relatively low resolution technique (~15 Å), however, it is very useful in determination of protein-protein complex structures to resolve the biological functions of the proteins [31,36,37].
A high-throughput forward approach leveraging purified proteins arising from the U.S. Protein Structure Initiative structural genomics effort has been reported [1]. In the metalloproteomics study, thousands of expression constructs and purified proteins are accessible and each protein was measured directly the metal content utilizing high-throughput X-ray absorption spectroscopy (HT-XAS). HT-XAS employs the physical principle of X-ray excitation of core electrons of metal atoms with detection of resulting X-ray fluorescence as in X-ray absorption spectroscopy [1]. Metal identification and quantification are based on the energy and intensities of the X-ray fluorescence signal emitted by intrinsic metals bound to the proteins, respectively. XAS data for hundreds of samples can be collected in a single run and finished in a few hours with a detection limit of approximately 100 μg per protein. HT-XAS in metalloproteomics so far has been closely associated with structural genomics projects [1,24].
One aim of the US structural genomics program has been to experimentally determine at least one protein structure from each protein family or subfamily that are 30% or less identical to any protein with a known 3-D structure, thus enabling structural analysis of the entire family or subfamily through the use of comparative modeling [38]. Targets are selected based on this overall strategy, and protein structures are determined by X-ray crystallography or NMR spectroscopy [39,40]. The success rate from cloning through purified protein to the final experimental 3-D structures by the large scale structural genomics centers in the NIH-funded Protein Structure Initiative (PSI) is ~10–14% [38,41,42], however, the set of purified proteins as a whole represents a useful resource for biochemical and biophysical characterization of the proteins to better understand function.
A HT-XAS study of a limited ensemble of proteins (a total of 654) from PSI Phase 1 (PSI-1) via quantitative analysis of six metals (Mn, Fe, Co, Cu, Ni, and Zn) was previously reported [1,23]. Protein samples were purified using the high-throughput protein production pipeline of the NYSGXRC [38,42]. Over 10% of the total samples showed the presence of transition metal atoms in stoichiometric amounts. The method was shown to be 94% accurate in predicting the presence or absence of a transition metal based on 50 crystal structures from the sample set. In addition, bioinformatics-based functional annotation was carried out for the metalloproteins identified by HT-XAS. In many cases, the metal binding information provided a distinct, new annotation for proteins of unknown function and improved annotation for proteins with poorly understood function.
The extension of the method to nearly 4000 purified proteins generated by the NYSGXRC during PSI Phase 2 (PSI-2) has been carried out. Automation of the method was improved to handle thousands of samples. Within the PSI structure determination pipeline, the results were used to assist in structure solution by X-ray crystallography, primarily in the map interpretation step where the metal identities are sometimes ambiguous. Proteins annotated as metal binding were analyzed by computational methods including PSI-BLAST [43] for detection of similar sequences and protein family identification via annotation transfer by homology, and by MetalDetector [44,45], a de novo method that predicts His and Cys residues that bind to transition metals using information derived from protein sequence. Structural models available from homology modeling of (often distant) templates deposited in MODBASE [46] or built utilizing SWISS-MODEL [47], were also used to identify putative metal binding sites and these models were evaluated in the context of experimental and bioinformatics results to provide a comprehensive understanding of the metalloprotein structure of the targets (Figure 1). Overall, the results show that the information from both experimental and bioinformatics approaches can be productively combined to improve the understanding of metal binding sites in metalloproteins.
Figure 1.

Experimental approaches to metalloproteomics.
Conclusion
The genome scale analysis of metalloprotein as a class is in its infancy. However, advances in high throughput technologies, both experimental and computational, are permitting novel annotations of sequence with metalloprotein function, which will lead to focused experimentation to conclusively identify the structure and activity of the newly annotated metalloproteins. Metalloproteomes from many species are still mostly uncharacterized. Combination of techniques and methods from multiple disciplines, including biochemistry, biophysics, engineering and computer sciences, are required in fully characterizing metalloproteomes and achieving a comprehensive understanding of the roles of metals in biology.
Figure 2.
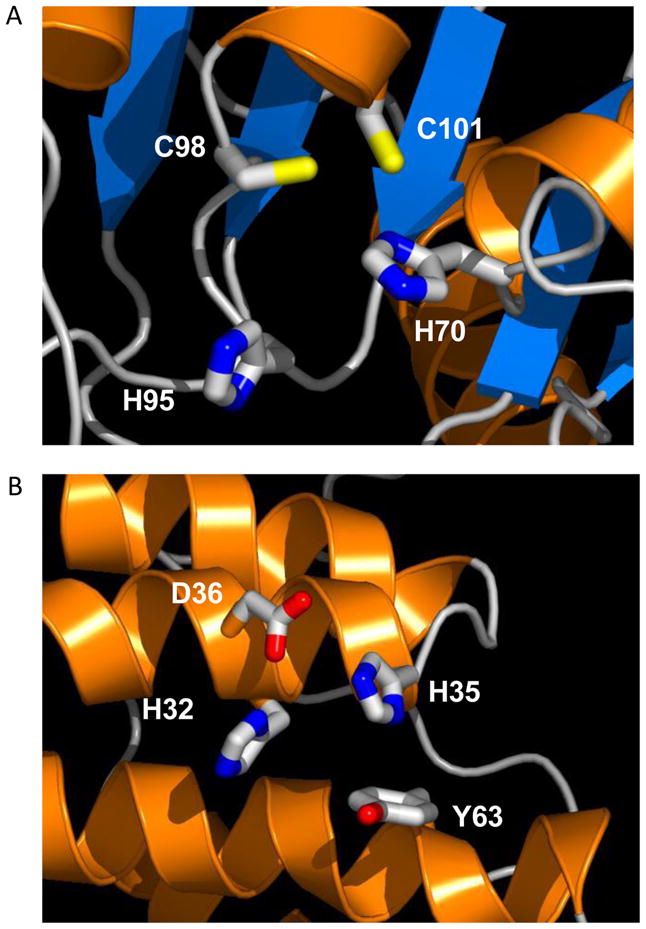
Examples of complementary use of HT-XAS, computational sequence analysis and homology modeling. A. Zn binding site for target NYSGXRC-892d (aka T812). HT-XAS showed that this protein contained Zn. MetalDetector [45] identified 3 residues, His70, Cys98, and Cys101, with high probabilities of 42%, 80% and 73%, respectively, to bind to a transition metal. Another His residue (His95), not identified by MetalDetector, is located nearby and likely contributes to metal binding. B. Ni/Zn binding site in the homology model of NYSGXRC-10060e. HT-XAS showed the protein contained Ni. MetalDetector identifies His35 with a probability of 21% to bind to a transition metal. MetalDetector only includes His and Cys in analysis. Based on the structure model, this protein mostly likely binds to a Zn. Ni detected in HT-XAS screening was probably trapped in the binding site in the purification step. Structure models were generated and retrieved from MODBASE [46].
References
- 1.Shi W, Zhan C, Ignatov A, Manjasetty BA, Marinkovic N, Sullivan M, Huang R, Chance MR. Metalloproteomics: high-throughput structural and functional annotation of proteins in structural genomics. Structure. 2005;13(10):1473–1486. doi: 10.1016/j.str.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Degtyarenko K. Bioinorganic motifs: towards functional classification of metalloproteins. Bioinformatics. 2000;16(10):851–864. doi: 10.1093/bioinformatics/16.10.851. [DOI] [PubMed] [Google Scholar]
- 3.Andreini C, Banci L, Bertini I, Rosato A. Zinc through the three domains of life. J Proteome Res. 2006;5(11):3173–3178. doi: 10.1021/pr0603699. [DOI] [PubMed] [Google Scholar]
- 4.Bertini I, Cavallaro G. Metals in the “omics” world: copper homeostasis and cytochrome c oxidase assembly in a new light. J Biol Inorg Chem. 2008;13(1):3–14. doi: 10.1007/s00775-007-0316-9. [DOI] [PubMed] [Google Scholar]
- 5.Shi W, Chance MR. Metallomics and metalloproteomics. Cell Mol Life Sci. 2008;65 (19):3040–3048. doi: 10.1007/s00018-008-8189-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szpunar J. Advances in analytical methodology for bioinorganic speciation analysis: metallomics, metalloproteomics and heteroatom-tagged proteomics and metabolomics. Analyst. 2005;130(4):442–465. doi: 10.1039/b418265k. [DOI] [PubMed] [Google Scholar]
- 7.Bettmer J. Metalloproteomics: a challenge for analytical chemists. Anal Bioanal Chem. 2005;383(3):370–371. doi: 10.1007/s00216-005-3404-0. [DOI] [PubMed] [Google Scholar]
- 8.Kulkarni PP, She YM, Smith SD, Roberts EA, Sarkar B. Proteomics of metal transport and metal-associated diseases. Chemistry. 2006;12(9):2410–2422. doi: 10.1002/chem.200500664. [DOI] [PubMed] [Google Scholar]
- 9.Bartel J, Charkiewicz E, Bartz T, Schmidt D, Grbavac I, Kyriakopoulos A. Metalloproteome of the prostate: carcinoma cell line DU-145 in comparison to healthy rat tissue. Cancer Genomics Proteomics. 2010;7(2):81–86. [PubMed] [Google Scholar]
- 10.Manley SA, Byrns S, Lyon AW, Brown P, Gailer J. Simultaneous Cu-, Fe-, and Zn-specific detection of metalloproteins contained in rabbit plasma by size-exclusion chromatography-inductively coupled plasma atomic emission spectroscopy. J Biol Inorg Chem. 2009;14(1):61–74. doi: 10.1007/s00775-008-0424-1. [DOI] [PubMed] [Google Scholar]
- 11 ·.Mounicou S, Szpunar J, Lobinski R. Metallomics: the concept and methodology. Chem Soc Rev. 2009;38(4):1119–1138. doi: 10.1039/b713633c. This review summarizes the various proteomics tools which can be used in metallomics and metalloproteomics. [DOI] [PubMed] [Google Scholar]
- 12 ··.Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole FL, 2nd, Jenney FE, Jr, Lancaster WA, Praissman JL, Shanmukh S, Vaccaro BJ, Trauger SA, et al. Microbial metalloproteomes are largely uncharacterized. Nature. 2010;466(7307):779–782. doi: 10.1038/nature09265. Using a combine metal-based approach, this paper reports the characterization of metalloproteomes from three microorganisms. High-throughput MS/MS and ICP-MS are used to identify cytoplamic metalloproteins from these species. [DOI] [PubMed] [Google Scholar]
- 13.Sun X, Chiu JF, He QY. Fractionation of proteins by immobilized metal affinity chromatography. Methods Mol Biol. 2008;424:205–212. doi: 10.1007/978-1-60327-064-9_17. [DOI] [PubMed] [Google Scholar]
- 14.Porath J, Carlsson J, Olsson I, Belfrage G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 1975;258(5536):598–599. doi: 10.1038/258598a0. [DOI] [PubMed] [Google Scholar]
- 15.She YM, Narindrasorasak S, Yang S, Spitale N, Roberts EA, Sarkar B. Identification of metal-binding proteins in human hepatoma lines by immobilized metal affinity chromatography and mass spectrometry. Mol Cell Proteomics. 2003;2(12):1306–1318. doi: 10.1074/mcp.M300080-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Smith SD, She YM, Roberts EA, Sarkar B. Using immobilized metal affinity chromatography, two-dimensional electrophoresis and mass spectrometry to identify hepatocellular proteins with copper-binding ability. J Proteome Res. 2004;3 (4):834–840. doi: 10.1021/pr049941r. [DOI] [PubMed] [Google Scholar]
- 17.Sun X, Ge R, Chiu JF, Sun H, He QY. Identification of Proteins Related to Nickel Homeostasis in Helicobater pylori by Immobilized Metal Affinity Chromatography and Two-Dimensional Gel Electrophoresis. Met Based Drugs. 2008;2008:289490. doi: 10.1155/2008/289490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roelofsen H, Balgobind R, Vonk RJ. Proteomic analyzes of copper metabolism in an in vitro model of Wilson disease using surface enhanced laser desorption/ionization-time of flight-mass spectrometry. J Cell Biochem. 2004;93(4):732–740. doi: 10.1002/jcb.20226. [DOI] [PubMed] [Google Scholar]
- 19.Thingholm TE, Jensen ON. Enrichment and characterization of phosphopeptides by immobilized metal affinity chromatography (IMAC) and mass spectrometry. Methods Mol Biol. 2009;527:47–56. xi. doi: 10.1007/978-1-60327-834-8_4. [DOI] [PubMed] [Google Scholar]
- 20.Thingholm TE, Jensen ON, Larsen MR. Enrichment and separation of mono- and multiply phosphorylated peptides using sequential elution from IMAC prior to mass spectrometric analysis. Methods Mol Biol. 2009;527:67–78. xi. doi: 10.1007/978-1-60327-834-8_6. [DOI] [PubMed] [Google Scholar]
- 21.Ye J, Zhang X, Young C, Zhao X, Hao Q, Cheng L, Jensen ON. Optimized IMAC-IMAC protocol for phosphopeptide recovery from complex biological samples. J Proteome Res. 2010;9(7):3561–3573. doi: 10.1021/pr100075x. [DOI] [PubMed] [Google Scholar]
- 22.Sun X, Ge F, Xiao CL, Yin XF, Ge R, Zhang LH, He QY. Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J Proteome Res. 2010;9(1):275–282. doi: 10.1021/pr900612v. [DOI] [PubMed] [Google Scholar]
- 23.Chance MR, Fiser A, Sali A, Pieper U, Eswar N, Xu G, Fajardo JE, Radhakannan T, Marinkovic N. High-throughput computational and experimental techniques in structural genomics. Genome Res. 2004;14(10B):2145–2154. doi: 10.1101/gr.2537904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott RA, Shokes JE, Cosper NJ, Jenney FE, Adams MW. Bottlenecks and roadblocks in high-throughput XAS for structural genomics. J Synchrotron Radiat. 2005;12(Pt 1):19–22. doi: 10.1107/S0909049504028791. [DOI] [PubMed] [Google Scholar]
- 25 ·.Andreini C, Bertini I, Rosato A. Metalloproteomes: a bioinformatic approach. Acc Chem Res. 2009;42(10):1471–1479. doi: 10.1021/ar900015x. The paper discusses the current progress in the development of bioinformatics methods for the prediction of metalloproteins based on protein sequences. It also includes a case study in prediction of Zn, Fe and Cu-proteins in a representative set of organisms. [DOI] [PubMed] [Google Scholar]
- 26.Andreini C, Bertini I, Rosato A. A hint to search for metalloproteins in gene banks. Bioinformatics. 2004;20(9):1373–1380. doi: 10.1093/bioinformatics/bth095. [DOI] [PubMed] [Google Scholar]
- 27.Andreini C, Banci L, Bertini I, Rosato A. Occurrence of copper proteins through the three domains of life: a bioinformatic approach. J Proteome Res. 2008;7(1):209–216. doi: 10.1021/pr070480u. [DOI] [PubMed] [Google Scholar]
- 28.Dupont CL, Yang S, Palenik B, Bourne PE. Modern proteomes contain putative imprints of ancient shifts in trace metal geochemistry. Proc Natl Acad Sci U S A. 2006;103(47):17822–17827. doi: 10.1073/pnas.0605798103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schymkowitz JW, Rousseau F, Martins IC, Ferkinghoff-Borg J, Stricher F, Serrano L. Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc Natl Acad Sci U S A. 2005;102(29):10147–10152. doi: 10.1073/pnas.0501980102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sodhi JS, Bryson K, McGuffin LJ, Ward JJ, Wernisch L, Jones DT. Predicting metal-binding site residues in low-resolution structural models. J Mol Biol. 2004;342(1):307–320. doi: 10.1016/j.jmb.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 31.Hasnain SS. Synchrotron techniques for metalloproteins and human disease in post genome era. J Synchrotron Radiat. 2004;11(Pt 1):7–11. doi: 10.1107/s0909049503024166. [DOI] [PubMed] [Google Scholar]
- 32.Einsle O, Tezcan FA, Andrade SL, Schmid B, Yoshida M, Howard JB, Rees DC. Nitrogenase MoFe-protein at 1.16 A resolution: a central ligand in the FeMo-cofactor. Science. 2002;297(5587):1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 33.Ascone I, Fourme R, Hasnain S, Hodgson K. Metallogenomics and biological X-ray absorption spectroscopy. J Synchrotron Radiat. 2005;12(Pt 1):1–3. doi: 10.1107/S0909049504033412. [DOI] [PubMed] [Google Scholar]
- 34.Ascone I, Strange R. Biological X-ray absorption spectroscopy and metalloproteomics. J Synchrotron Radiat. 2009;16(Pt 3):413–421. doi: 10.1107/S0909049509010425. [DOI] [PubMed] [Google Scholar]
- 35 ·.Hasnain SS, Strange RW. Marriage of XAFS and crystallography for structure-function studies of metalloproteins. J Synchrotron Radiat. 2003;10(Pt 1):9–15. doi: 10.1107/s0909049502022446. Recent progresses in biological X-ray absorption spectroscopy are reported in this review. New developments required to enable XAS a high-throughput method in metalloproteomics are also discussed. [DOI] [PubMed] [Google Scholar]
- 36.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6(8):606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40(3):191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 38.Bonanno JB, Almo SC, Bresnick A, Chance MR, Fiser A, Swaminathan S, Jiang J, Studier FW, Shapiro L, Lima CD, Gaasterland TM, et al. New York-Structural GenomiX Research Consortium (NYSGXRC): a large scale center for the protein structure initiative. J Struct Funct Genomics. 2005;6(2–3):225–232. doi: 10.1007/s10969-005-6827-0. [DOI] [PubMed] [Google Scholar]
- 39.Shi W, Chance MR. Structural Genomics: High-Throughput Structure Determination of Protein Domains. In: Triggle JBTaDJ., editor. Comprehensive Medicinal Chemistry. Vol. 3. Elsevier; Armsterdam, the Netherlands: 2006. [Google Scholar]
- 40.Manjasetty BA, Shi W, Zhan C, Fiser A, Chance MR. A high-throughput approach to protein structure analysis. Genet Eng (N Y) 2007;28:105–128. doi: 10.1007/978-0-387-34504-8_7. [DOI] [PubMed] [Google Scholar]
- 41.Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, et al. Protein production and purification. Nat Methods. 2008;5(2):135–146. doi: 10.1038/nmeth.f.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauder JM, et al. High throughput protein production and crystallization at NYSGXRC. In: Kobe BMG, Huber T, editors. Methods in Molecular Biology: Structural Proteomics: High-throughput methods. Chapter 37. Vol. 426. Humana Press; Totowa, NJ: 2008. [DOI] [PubMed] [Google Scholar]
- 43.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lippi M, Passerini A, Punta M, Rost B, Frasconi P. MetalDetector: a web server for predicting metal-binding sites and disulfide bridges in proteins from sequence. Bioinformatics. 2008;24(18):2094–2095. doi: 10.1093/bioinformatics/btn371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Passerini A, Punta M, Ceroni A, Rost B, Frasconi P. Identifying cysteines and histidines in transition-metal-binding sites using support vector machines and neural networks. Proteins. 2006;65(2):305–316. doi: 10.1002/prot.21135. [DOI] [PubMed] [Google Scholar]
- 46.Pieper U, Eswar N, Braberg H, Madhusudhan MS, Davis FP, Stuart AC, Mirkovic N, Rossi A, Marti-Renom MA, Fiser A, Webb B, et al. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004;32(Database issue):D217–222. doi: 10.1093/nar/gkh095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37(Database issue):D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]