

Abstract
C5a is a proinflammatory mediator that has recently been shown to regulate adaptive immune responses. Here we demonstrate that C5a receptor (C5aR) signaling in dendritic cells (DC) affects the development of regulatory T cell (Treg) and Th17 cells. Genetic ablation or pharmacological targeting of the C5aR in spleen-derived DC results in increased production of TGF-β leading to de novo differentiation of Foxp3+ Treg within 12h after co-incubation with CD4+ T cells from DO11.10/RAG2-/- mice. Stimulation of C5aR-/- DC with ovalbumin and TLR2 ligand Pam3CSK4 increased TGF-β production and induced high levels of IL-6 and IL-23 but only minor amounts of IL-12 leading to differentiation of Th cells producing IL-17A and IL-21. Th17 differentiation was also found in vivo after adoptive transfer of CD4+ Th cell into C5aR-/- mice immunized with OVA and Pam3CSK4. The altered cytokine production of C5aR-/- DC was associated with low steady state MHC class II expression and an impaired ability to upregulate CD86 and CD40 in response to TLR2. Our data suggest critical roles for C5aR in Treg and Th17 cell differentiation through regulation of DC function.
Keywords: C5a, complement, dendritic cell, T cells, IL-17A
Introduction
Recognition of pathogens leads to activation of the complement system generating potent proinflammatory effector molecules, the so-called anaphylatoxins (AT) C3a and C5a [1]. C5a exerts its pro-inflammatory properties through G-protein-mediated signaling of the C5a receptor (C5aR), which is broadly expressed on myeloid as well as on tissue cells [2]. In addition to C5aR, C5a and its degradation product C5adesArg bind to C5L2, another seven-transmembrane receptor sharing the broad tissue distribution with C5aR [3]. The role of C5L2 in AT biology is controversial. Unlike C5aR, C5L2 is uncoupled from G-proteins, suggesting that C5L2 functions as a non-signaling decoy receptor [4,5]. In support of this view, C5L2-deficient mice exhibit exaggerated inflammation in an immune complex-mediated alveolitis model [6]. In contrast, recent studies suggest that C5a and C3a exert biologic functions via C5L2 in neutrophils and macrophages through MAPK activation [7].
In addition to their proinflammatory properties, AT regulate APC function and modify adaptive immune responses [1]. C5a down-regulates TLR4-induced expression of IL-12 via ERK and PI3K [8,9]. In contrast, C5-deficiency in macrophages or combined C5aR/C5L2 targeting in monocytes results in impaired IL-12 production stimulated by S. aureus [10]. Further, pulmonary C5aR targeting during allergen sensitization leads to a Th2-biased response in experimental allergic asthma [11]. Recently, it has been shown that local production of C3a and C5a by APC and T cells leads to bidirectional signaling at the APC-T cell interface and regulates MHC class II (MHC-II) and costimulatory molecule expression promoting T cell survival and Th1 differentiation [12]. In summary, these studies support a critical role for C5aR signaling in directing Th1 and Th2 cell polarization.
A lineage of IL-17-producing CD4+Th (Th17) cells has been recently discovered playing critical roles in autoimmunity, infection and allergy [13,14]. Several studies have demonstrated that in mice [15,16] and in humans [17,18] TGF-β and IL-6 are sufficient to drive Th17 cell differentiation from naïve Th cells. In addition to TGF-β and IL-6, IL-21 and IL-23 serve as Th17-promoting cytokines. IL-21 can act in an autocrine manner to induce Th17 differentiation in concert with TGF-β [19,20] whereas IL-23 mediates Th17 expansion and/or survival. In the presence of exogenous TGF-β, TLR4-stimulated DC gain the ability to promote Th17 differentiation [13]. However, the conditions that favor TGF-β production from APC remain elusive.
Here we show that C5aR but not C5L2 activation in spleen-derived DC (sDC) is an important signal for naïve CD4+ Th cells differentiate into either Th1 or Th17 effector cells and regulatory T cells (Treg). C5aR-deficient sDC drive the expansion of Treg, which is associated with upregulation of TGF-β. Such sDC also shifted TLR2-driven differentiation of naïve CD4+ T cells from IFN-γ+ Th1 cells toward IL-17A and IL-21-producing Th17 cells. Our data indicate that C5aR signaling in DC sets the threshold for Th cell differentiation into Th1, Treg and Th17 cells at the time of T cell priming.
Results
Increased production of Th17 promoting cytokines in the absence of C5aR signaling
To assess the contribution of C5aR and C5L2 signaling on cytokine production during initial T cell priming, we co-cultured WT, C5aR-/- and C5L2-/- sDC in the presence of OVA ± the triacyl-lipopeptide Pam3CSK4 (PAM) and OVA transgenic CD4+ T cells isolated from DO11.10 RAG2-/- mice [21]. In response to OVA treatment, we found some upregulation of IFN-γ in WT and C5L2-/- co-cultures as compared with unstimulated controls (Fig. 1A). Upon additional stimulation with PAM, we found substantial upregulation of IL-12p40, IL-12p70 and IFN-γ production and a moderate upregulation of IL-6 production. In contrast, we found only minor production of IL-12p70 and IFN-γ but strong production of the Th17 related cytokines IL-6, TGF-β, IL-23, IL-21 and IL-17A in C5aR-/- sDC/T cell co-cultures (Fig. 1A). The production of IL-10 (Fig. 1A) and IL-22 (data not shown) was not affected by the absence of AT receptor signaling. These data suggest that C5aR but not C5L2 signaling is critical for TLR2-driven Th1 differentiation of naïve CD4+ T cells.
Figure 1. TLR2-challenge of C5aR-/- sDC induces a cytokine milieu that drives Treg and Th17 differentiation.
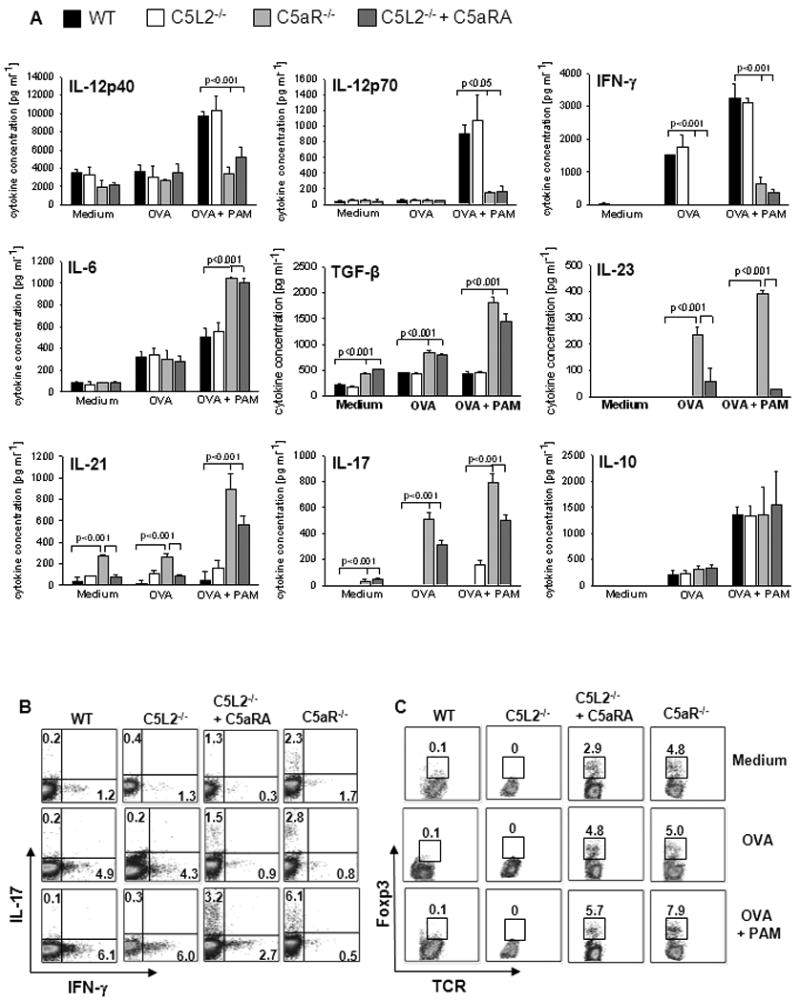
(A) sDC from WT, C5aR-/-, and C5L2-/- mice were co-cultured with naïve CD4+ T cells from OVA-TCR transgenic DO11.10/RAG2-/- mice for 4 days in the absence or the presence of OVA ± Pam3CSK4 (PAM). OVA was added at a concentration of 1 μM. C5aR antagonist A8Δ71-73 (C5aRA) was added to some cultures as indicated. Concentrations of IL-12, IFN-γ, IL-6, TGF-β, IL-23, IL-21, IL-17A and IL-10 in culture supernatants were determined by ELISA. Data show mean ± SD (n=3). The frequencies of (B) IL-17A+ and IFN-γ+ or (C) Foxp3+TCR+ among CD4+ cells were determined by intracellular staining and flow cytometry. Data are representative of 4 independent experiments.
We also blocked C5aR signaling in C5L2-/- DC using an antagonist (C5aRA). Similar to C5aR-/- DC, combined C5aR and C5L2 inhibition resulted in marked production of TGF-ß and IL-6 and substantial reduction of IL-12 and IFN-γ confirming the requirement of C5aR for the induction of a Th1-promoting microenvironment (Fig. 1A). However, the magnitude of the Th17 promoting effect was lower in response to pharmacological targeting than that obtained with C5aR-/- DC. Incomplete blockade of C5aR by the C5aRA may account for these differences.
The absence of C5aR signaling in sDC results in Treg and Th17 cell differentiation
The impact of C5aR signaling on the Th cell differentiating cytokine milieu was confirmed by intracellular staining of IFN-γ and IL-17A in CD4+ T cells. While antigen challenge ± PAM of WT and C5L2-/- cultures increased the frequency of IFN-γ+ Th1 cells, C5aR-/- sDC promoted the differentiation of IL-17A+ Th cells (Fig. 1B).
In addition to Th1 and Th17 polarization, we assessed the impact of C5aR and C5L2 on de novo CD25+Foxp3+ Treg differentiation. RAG2-/-DO11.10 mice are devoid of Treg in the periphery [22]. After 4d of WT or C5L2-/- sDC/T cell co-culture, no Treg were detectable under any conditions (Fig. 1C). Unexpectedly, we observed 3-8% of Treg when C5aR signaling in DC was absent, either by C5aR deficiency or C5aR blockade (Fig. 1C).
In addition we determined the ability of sDC to induce proliferation of CD4+ Th cells. The frequency of proliferating DO11.10 CD4+ T cells increased in response to OVA and was further enhanced by TLR2 ligation. Importantly, C5aR-/- sDC showed a significantly reduced ability to promote T cell proliferation in response to antigen ± PAM as compared with WT or C5L2 sDC (Supporting Information Fig. 1 and Table 1).
Taken together, these data suggest that C5aR-/- sDC differ in their ability to induce Th differentiation and proliferation as compared with WT and C5L2 sDC.
The extremely low levels of IL-12p70 produced by C5aR-/- sDC (Fig. 1A) prompted us to assess whether this is the cause for their defective ability to induce Th1 polarization. C5aR-/- sDC/T cell co-cultures performed in the presence of high IL-12 concentrations and additional blockade of IL-4 (“Th1 conditions”) still resulted in a dominant Th17 and Treg phenotype (Supporting Information Fig. 2), suggesting that the C5aR-mediated impact on Th17 and Treg differentiation is independent of IL-12.
Th17 cell differentiation in the absence of C5aR requires TGF-β and proinflammatory cytokines
As the addition of IL-12 was not sufficient to restrain C5aR-/- sDC ability in inducing Th17 responses, we assessed the importance of individual Th17 promoting cytokines in C5aR-dependent Th17 differentiation. We observed almost complete inhibition (∼80-90%) of Th17 cell population following anti-TGF-β treatment, which was associated with a concomitant increase in IFN-γ+ Th1 cells (Supporting Information Fig. 3A). Further, anti-TGF-β treatment resulted in complete absence of Treg whereas single inhibition of IL-6, IL-21 or IL-23 had no impact on Treg differentiation (Supporting Information Fig. 3B).
These findings were supported by ELISA data (Supporting Information Fig. 3C). TGF-ß inhibition reduced the production of Th17-related cytokines (IL-6, IL-23, IL-21 and IL-17A). Further, IL-6 inhibition had a negative regulatory impact on IL-23, IL-21 and IL-17A. Interestingly, IL-23 blockade moderately increased IL-12p70 and IFN-γ and shut down IL-21 and IL-17A production. IL-21 blockade reduced IL-17A production independent of IL-6, TGF-ß and IL-23. These data point toward a hierarchical rather than an equal contribution of Th-17 promoting cytokines in the order TGF-ß > IL-6 > IL-23 > IL-21.
Of note, the increase in IFN-γ production following TGF-ß inhibition was enhanced by additional inhibition of IL-6 and IL-23. In fact, this combination resulted in total abrogation of IL-17A production and marked IFN-γ secretion associated with strong IL-12p70 secretion from DC (Supporting Information Fig. 3C).
C5aR signaling regulates early development of Foxp3+ Treg and subsequent Th17 cells differentiation
Next, we determined the dynamics of Treg and Th17 cell development in C5aR-/- sDC/T cell co-cultures. We already found Foxp3+ Treg after 12h (Fig. 2A), which was associated with early production of TGF-β (Fig. 2C). The frequency of Foxp3+ cells was between 3.8 and 5.7%, did not change over time and was independent of OVA and TLR2 challenge (Fig. 2A). In contrast, IL-17A producing cells only appeared after 36h and the frequency of this population was strongly dependent on antigen, TLR and time (Fig. 2B). The ELISA data shown in Fig. 2C support this observation. We found sequential production of cytokines in the order TGF-β (12h), IL-23 (12-24h), IL-6 (24h) and IL-17A (36h). Of note, IL-23 levels reached their maximum after 24h and declined within the next 48h suggesting consumption by newly formed Th17 cells.
Figure 2. C5aR signaling regulates early development of Foxp3+ Treg and subsequent Th17 differentiation.
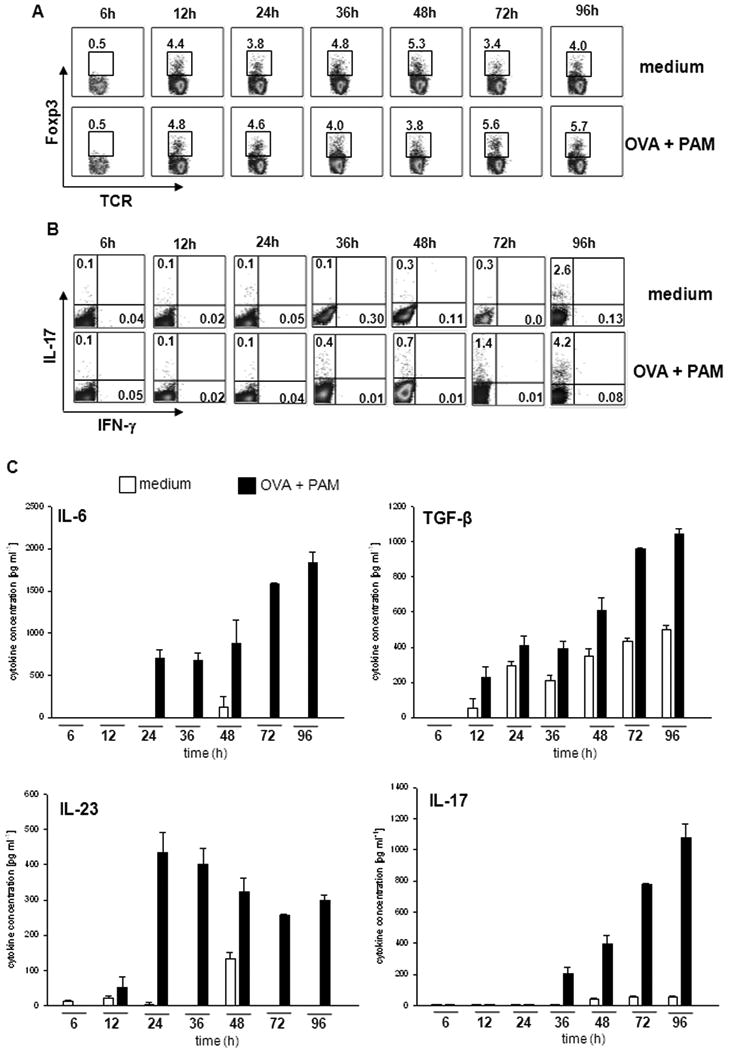
sDC from C5aR-/- mice were co-cultured with naïve CD4+ T cells from DO11.10/RAG2-/- mice in the absence or the presence of PAM. After the indicated incubation periods, the frequencies of (A) Foxp3+TCR+ T cells or (B) of IL-17A+IFN-γ+ cells among CD4+ T cells were determined by intracellular staining and flow cytometry. (C) Concentrations of IL-6, TGF-β, IL-23 and IL-17A in culture supernatants were determined by ELISA. Data show mean ± SD (n=3). Data are representative of 3 independent experiments.
IL-2 receptor targeting abrogates Th17 differentiation by C5aR-deficient DC
Several studies have shown that inhibition of IL-2 or blocking CD25 preferentially targets Treg survival [23]. Here, we found that anti-CD25 treatment not only prevented the differentiation of Treg (Fig. 3A) but also the differentiation of Th17 cells (Fig. 3B). Instead, the numbers of IFN-γ+ Th1 cells increased from 0.4% to 3.5% (Fig. 3B), which is similar to what we found in response to anti-TGF-ß treatment (Supporting information Fig. 3A).
Figure 3. IL-2 receptor alpha chain blockade abrogates C5aR-/--induced Th17 differentiation.
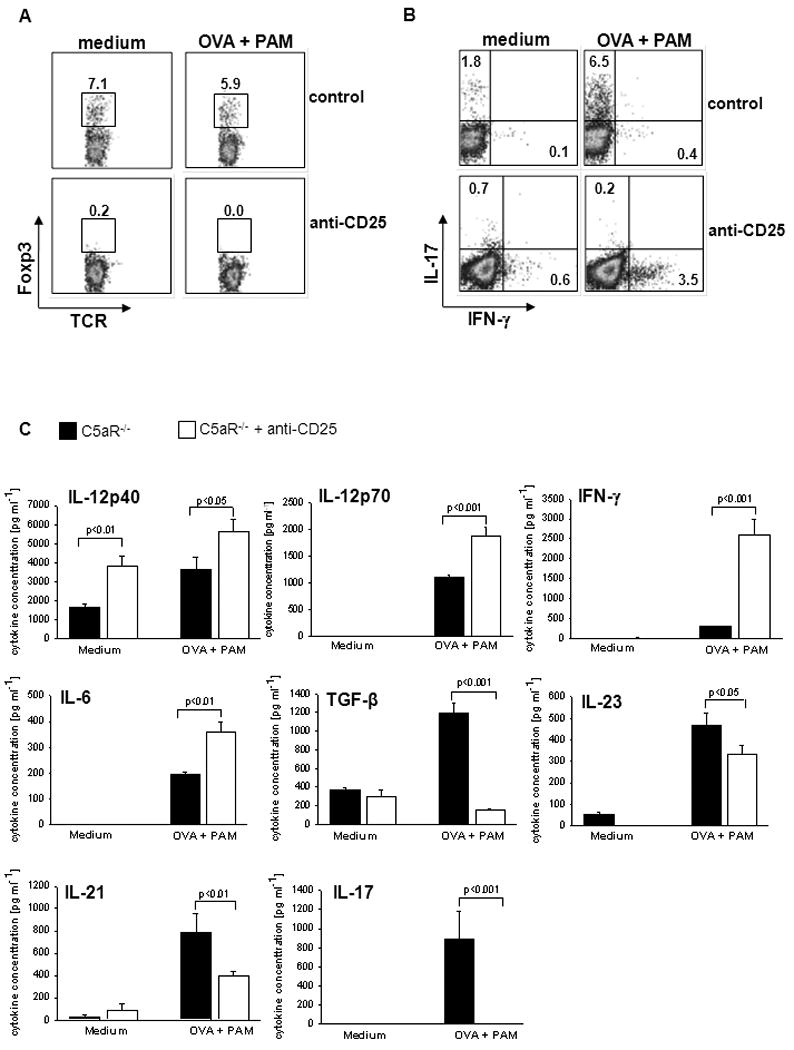
sDC from C5aR-/- mice were co-cultured with naïve CD4+ T cells from DO11.10/RAG2-/- mice. (A) Antibodies to CD25 or an appropriate isotype control (20 μg/ml) were added to the cell cultures and the frequencies of Foxp3+ (A) or IL-17A+/IFN-γ+ (B) CD4+ T cells were determined by intracellular staining using flow cytometry. (C) Concentrations of IL-12, IFN-γ, IL-6, TGF-β, IL-23, IL-21 and IL-17A in culture supernatants from (A) were determined by ELISA. Data show mean ± SD (n=4). Data are representative of 4 independent experiments.
The FACS data were confirmed by ELISA. Blockade of CD25 combined with TLR2 activation completely abolished IL-17A and TGF-ß secretion but promoted a substantial increase in IL-12p70 and IFN-γ production (Fig. 3C). IL-6 levels were also enhanced following anti-CD25 treatment. In contrast, IL-21 and IL-23 production was significantly reduced (Fig. 3C).
The lack of C5aR signaling promotes TLR2-driven Th17 differentiation in vivo
We then determined the impact of C5aR on Th differentiation in vivo. CD4+ cells from DO11.10 RAG2-/- mice were adoptively transferred into WT, C5aR-/- and C5L2-/- mice immunized i.p. with OVA and PAM.
Consistent with our in vitro findings, we observed a decreased frequency of OVA transgenic T cells in the spleen of C5aR-/- mice 4d after adoptive cell transfer, which indicates decreased proliferation in a C5aR-deficient environment (Fig. 4A). We further assessed the differentiation of OVA transgenic CD4+ T cells from spleens of injected mice after ex vivo re-stimulation with either medium or OVA plus PAM. DO11.10 CD4+ T cells activated in WT and C5L2-/- mice differentiated into IFN-γ-producing Th1 cells (Fig. 4B). In contrast, DO11.10 T cells from C5aR-/- mice were IL-17A+ and did not express IFN-γ (Fig. 4B).
Figure 4. Lack of C5aR in vivo promotes Th17 differentiation.
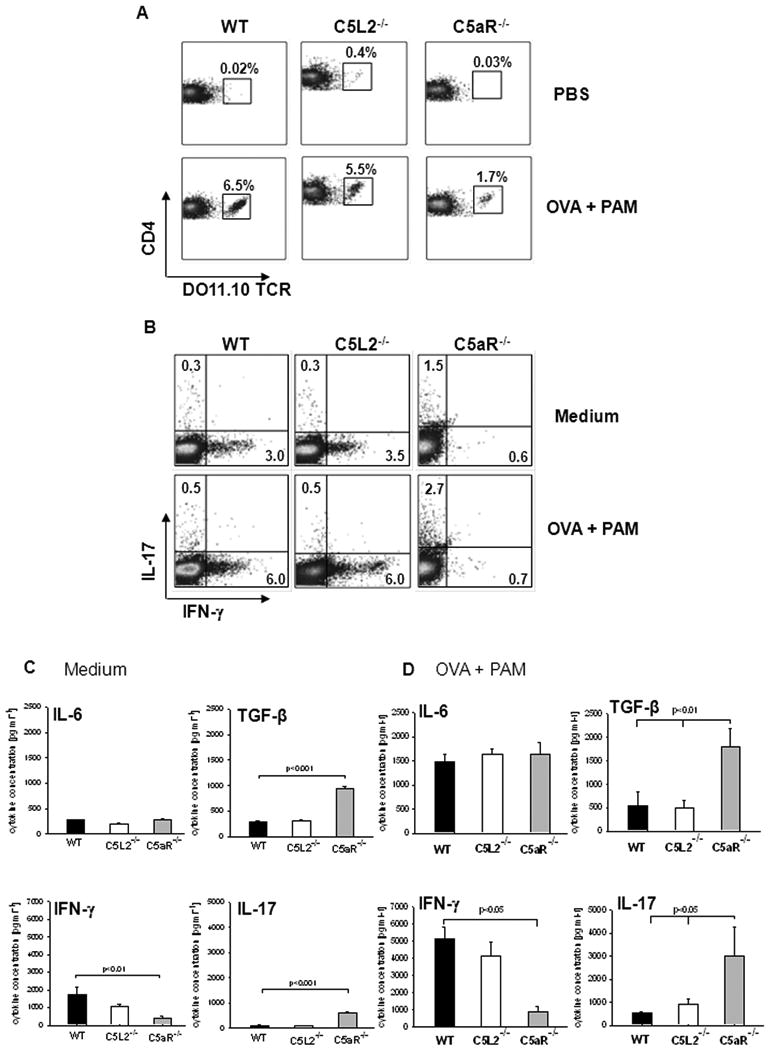
(A) CD4+ T cells from DO11.10/RAG2-/- mice mice were adoptively transferred into WT, C5L2-/-, and C5aR-/- mice. These mice were immunized i. p. with PBS or OVA + PAM. On day 4, splenocytes were isolated and the frequency of TCR transgenic CD4+ cells was assessed by flow cytometry. (B) Splenocytes from WT, C5L2-/-, and C5aR-/- mice from (A) were re-stimulated in vitro with medium or OVA + PAM and the frequency of IL-17A+ and IFN-γ+ cells among CD4+ T cells was determined by intracellular staining. (C, D) Cytokine levels in splenocyte culture supernatants re-stimulated with either medium (C) or OVA+PAM (D) as determined by ELISA. Data show mean ± SD (n=3). Data are representative of 3 independent experiments.
ELISA results support these findings. We found higher concentrations of IL-17A and TGF-β in C5aR-/- than in WT and C5L2-/- splenocyte cultures (Figs. 4C, D). IFN-γ concentrations were higher in WT and C5L2-/- than in C5aR-/- cultures (Figs. 4C, D). IL-6 secretion was similar in the different groups (Figs. 4C, D). We found no differences in the frequency of Foxp3+ Treg populations between the different treatment groups at day 4 (data not shown).
C5aR but not C5L2 ligation promotes MAPK activation in spleen-derived DC
To define potential mechanisms underlying the distinct impact of C5aR and C5L2 on Th cell differentiation and proliferation, we determined the expression and the function of both receptors on sDC. We found C5aR and C5L2 surface expression on CD11c+/MHC-II+ spleen cells as well as on CD11c+-sorted sDC (Supporting Information Fig. 4A and B). An important signaling pathway underlying C5a-mediated effector functions is the activation of ERK1/2. Consistent with previous findings, we noted p-ERK1/2 as early as 0.5 minutes in WT sDC, which was absent in C5aR-/- sDC (Supporting Information Fig. 4C). In contrast to neutrophils and macrophages [7], we found that C5a-induced p-ERK1/2 in C5L2-/- sDC was identical to WT sDC, suggesting that at least in sDC, C5L2 is not functional in terms of driving ERK1/2 phosphorylation.
Reduced steady state MHC-II expression in sDC from C5aR-deficent mice
In order to further assess functional changes on C5aR-/- sDC that impact their ability to promote Th cell differentiation, we determined the expression of MHC-II. We found moderate surface expression of MHC-II in CD11c+ WT sDC (Fig. 5A) under steady state conditions. Surprisingly, surface expression of MHC-II in C5aR-/- sDC was substantially lower than in sDC from WT or C5L2-/- mice (Fig. 5A). sDC numbers in the spleen of WT, C5aR-/- and C5L2-/- were indistinguishable (data not shown). Stimulation of sDC with PAM had no impact on sDC MHC-II expression of either strain (Fig. 5A).
Figure 5. C5aR signaling has a direct regulatory impact on MHC-II expression, costimulatory molecule expression and cytokine production from sDC.
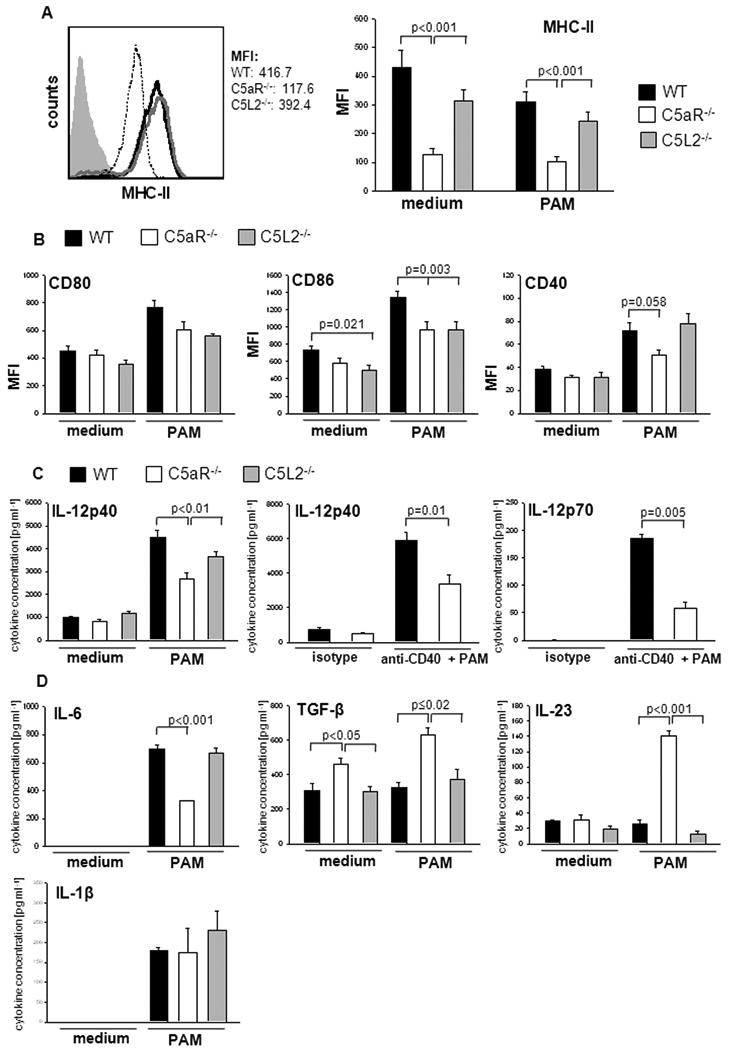
(A) Left panel: Surface expression of MHC-II molecules as determined by flow cytometry. WT- black line, C5aR-/-- dotted line, C5L2-/-- gray line. Gray filled histogram depicts isotype control. Right panel: sDC from WT, C5aR-/-, and C5L2-/- mice were incubated in the absence and presence of PAM for 24h. Cells were gated on CD11chi cells; MFI: median fluorescence intensity. (B) Surface expression of CD80, CD86 and CD40 on sDC. (C and D) Cytokine production from PAM- or PAM + anti-CD40-challenged sDC. Cells were cultured in the absence or presence of PAM or PAM + anti-CD40 for 24 hours. Concentrations of IL-12, IL-6, IL-23, TGF-β and IL-1β were measured by ELISA in culture supernatants. Data show mean ± SD. Data are representative of 5-8 independent experiments.
This finding prompted us to assess whether this regulatory impact on MHC-II expression is of a broader nature. In addition to sDC, we found reduced expression levels of MHC-II in spleen B cells from C5aR-/- mice (Supporting Information Fig. 5A). Further, MHC-II expression was also reduced in sDC and B cells from C5aR-/- mice on the C57BL/6 background, suggesting a strain-independent effect of C5a on MHC-II expression (Supporting Information Fig. 5B).
C5aR signaling regulates costimulatory molecule surface expression on sDC
Next, we determined the expression levels of costimulatory molecules. Under steady state conditions, we found moderate CD80 and CD86 surface expression whereas CD40 expression was low (Fig. 5B). CD86 expression in C5L2-/- sDC was reduced as compared with WT sDC (Fig. 5B). TLR2 stimulation upregulated CD80, CD86 and CD40 expression in sDC from all strains (Fig. 5B). However, compared with WT sDC, TLR2-induced upregulation of CD86 and CD40 expression was impaired in C5aR-deficient DC (Fig. 5B). PAM-induced CD86 expression was also impaired in C5L2-/- sDC. These data suggest that the reduced expression of MHC-II in C5aR-/- sDC and their impaired ability to promote TLR-2-induced upregulation of CD86/CD40 may account for the reduced efficiency of C5aR-/- sDC to induce Th cell proliferation. The isolated impact on CD86 expression observed with C5L2-/- APC is insufficient to translate into reduced Th cell proliferation.
C5aR-deficient sDC produce Th17 promoting cytokines after TLR2 challenge
Finally, we determined the direct impact of C5aR and C5L2 deficiency on TLR2-driven cytokine production from sDC independent of CD4+ T cells. Although TLR2 challenge significantly increased IL-12p40 production from WT, C5aR- and C5L2-deficient cells, IL-12p40 levels were consistently lower in C5aR-/- sDC cultures (Fig. 5C).
Activation of CD40 during DC/Tcell interactions is an important positive regulator of IL-12 production. As expected, ligation of CD40 using an agonist anti-CD40 Ab increased IL-12p40 and p70 production in WT cells (Fig. 5C). IL-12 production was reduced in C5aR-/- sDC, suggesting that C5a not only enhances TLR2- induced IL-12 production but also CD40- mediated amplification of IL-12 production.
No IL-6 or IL-1β and low levels of IL-23 were detected in the supernatants from cells of either strain under steady state conditions (Fig. 5D). TGF-β levels were higher in C5aR-/- as compared with WT or C5L2-/- sDC cultures (Fig. 5D). PAM administration had no impact on IL-23 and TGF-β production from WT and C5L2-/- cells but markedly upregulated IL-23 and TGF-β production from C5aR-/- sDC (Fig. 5D).
TLR2 challenge upregulated IL-1β and IL-6 production in all strains. Of note, the increase in IL-6 production was less intense in the C5aR-/- sDC cultures (Fig. 5D). The opposing effect of C5aR deficiency on IL-6 production in sDC as compared with sDC/Tcell co-cultures (Fig. 1A and 5D) suggests a T cell-derived IL-17 feedback on C5aR-/- DC [24].
In summary, our data suggest that C5aR signaling in sDC contributes to TLR2-induced IL-12 production. In the absence of C5aR, DC are reprogrammed in a way that TLR2 ligation induces the production of high levels of IL-23 and TGF-β, which instructs the development of Th17 instead of Th1 cells.
Discussion
We demonstrate that C5aR activation in sDC provides a critical innate immune signal that regulates differentiation of naïve T cells into Th1, Th17 and Treg. We have previously shown that in contrast to DC, C5aR activation on peritoneal macrophages negatively regulates TLR-driven IL-12 production [8], suggesting cell-specific differences. Recently, C3aR and C5aR expression on both APC and CD4+ T cells has been found to control B7-CD28 and CD40-CD40L costimulation during cognate APC-CD4+ T cell interaction, regulating Th1 responses [12,25]. In agreement with these data, we found decreased MHC-II expression in C5aR-deficient sDC and an impaired ability of such DC to upregulate co-stimulatory molecules and produce IL-12 in response to TLR2 stimulation. Further, we show that this effect is strictly C5aR-related with C5L2 being at best marginally involved.
As expected, the negative regulatory impact of C5aR-deficiency on sDC reduced the potency of such cells to drive T cell proliferation and promote TLR2-driven Th1 cell differentiation. Unexpectedly, sDC lacking functional C5aR promoted de novo Treg differentiation. This is the first report showing a role for C5aR in Treg differentiation. Up to now, only the complement regulatory protein CD46 has been implicated in Treg development. Interestingly, CD46 ligation promotes the induction of another population of Treg, i.e. Tr1 cells [26].
The reciprocal relationship between the ability of DC to drive proliferation and the conversion rate of naive T cells into Treg has been shown before [27]. Our data add to this observation by showing that C5aR signaling positively correlates with the expression of MHC-II and DC-induced T cell proliferation in vitro and in vivo.
The higher TGF-β concentrations in C5aR-/- as compared with WT DC/T cell co-cultures and the fact the TGF-β is key to Treg formation suggest that C5aR signaling regulates Treg differentiation through an impact on DC-derived TGF-β production. The requirement of IL-2 and TGF-β for Th17 development and our data that Treg already appear after 12h of DC/T cell co-culture may indicate an association between Treg and Th17 differentiation. Indeed, it has been shown that Treg can function as inducers of Th17 cells and that Treg may serve as a source for the increased TGF-β levels [28] and/or may regulate DC-driven TGF-β production [29]. In vivo, we found no differences in Treg frequencies after 4 days of adoptive transfer of DO11.10 CD4+ T cells into the different mouse strains. Whether transient Treg differentiation is induced in the C5aR-/- environment in vivo and contributes to C5aR-regulated Th17 differentiation awaits further studies. We are currently generating mice which will allow for specific depletion of OVA-specific regulatory T cells in a DO11.10RAG2-/- environment to address this question.
Bacteria as well as fungi trigger Th17 responses [13]. More specifically, activation of NOD-like receptors and membrane bound C-type lectins have been shown to induce the production of proinflammatory cytokines such as IL-6, IL-1ß and IL-23 which instruct Th17 differentiation in concert with TGF-β [30,31]. In contrast, TLR ligands preferentially promote IL-12 production leading to IFN-γ producing Th1 cells [32]. As shown by the Stockinger lab, TLR ligation on DC can stimulate Th17 differentiation in the presence of exogenously added Treg [15]. However, the pathways leading to Treg formation in the presence of TLR-driven DC activation remain elusive. Our data suggest that in the absence of C5aR signaling, naïve Th cells can develop into CD25+Foxp3+ T cells.
Further, we show that in the absence of C5aR signaling, TLR2-activated sDC are reprogrammed to produce high concentrations of IL-6 and IL-23 instead of IL-12, which in concert with TGF-β induce Th17 cell differentiation. At this point, we do not know whether such Treg suppress the proliferation of OVA Tg effector T cells or whether the low IL-12p70 prevents the development of Th1 cells. Whatever the dominant mechanism may be, our findings suggest that cross-talk between TLR2 and C5aR defines the quality of the cytokine milieu emerging during infection which is important for Th cell lineage commitment. Importantly, the impact of C5a on Th17 differentiation is complex. C5aR and TLR4 signaling synergize in macrophages to drive IL-6 production and to promote Th17 differentiation of CD3/CD28-stimulated CD4+ T cells [33]. Thus, depending of the nature of the APC, i.e. macrophage or DC, C5aR signaling can either promote or suppress Th17 differentiation in the context of TLR activation.
Importantly, pathogens have evolved several strategies to interfere with such cross-talk. Many pathogens express ligands for TLRs and are recognized by soluble C-type lectins such as mannan-binding lectin or ficolins that activate the complement cascade. Based on our findings, such simultaneous activation of TLR2 and complement would result in TLR-driven Th1 adaptive immune responses. Strikingly, many strains of S. aureus secrete proteins to evade complement-mediated immunity such as the potent C5a receptor antagonist CHIPS [34] or complement convertase inhibitors [35]. Further, group A streptococci secrete a C5a peptidase to inactivate C5a [36]. Thus, by interfering with C5a generation, C5a degradation and C5a/C5aR interaction, microorganisms can affect DC function and instruct distinct adaptive immune responses. Further, many bacteria and fungi bind the potent complement regulator factor H which efficiently prevents activation of the amplification loop of the alternative pathway on the pathogen surface, thus preventing generation of AT [37].
In summary we demonstrate a novel function for C5aR signaling in DC that shapes adaptive immune responses at the DC/T cell interface. C5aR signaling in DC balances the sophisticated network of DC-derived cytokines, MHC-II and costimulatory molecule expression that defines the fate of naïve T cells. As DC are of critical importance in immunotherapy, C5aR may become an interesting target for DC-based therapeutic approaches. Importantly, several potent C5aR antagonists are available that could be helpful to modulate DC function [38].
Materials and Methods
Mice
BALB/c mice were purchased from Jackson Laboratories (Bar Harbor, ME) and TCR transgenic DO11.10/RAG2-/- mice from Taconic (Germantown, PA). C5aR-/- and C5L2-/- mice were backcrossed onto the BALB/c background for 9 generations. Experimental groups were sex-matched and 8-12 weeks of age. All animals were held under specific pathogen-free conditions and the experiments were approved by the institutional animal care and use committee of Cincinnati Children's Hospital.
Splenic cell isolation
Spleens were treated with 0.5 mg/ml Liberase Cl (Boehringer) and 0.5 mg/ml DNase (Sigma) in RPMI for 45 min at 37°C, followed by red blood cell lysis (155 mM NH4Cl, 10 mM NaHCO3, 0.1 mM EDTA). DC enrichment was achieved after positive selection using CD11c MicroBeads (Miltenyi Biotec). CD4+ naïve T cells were enriched from DO11.10/RAG2-/- spleens by negative selection using CD4+ T cell isolation Kit (Miltenyi Biotec). The purity was ≥ 95%.
Stimulation of DC
Splenic DC (1 × 106) were incubated in 500μl of RPMI and stimulated with the synthetic triacyl-lipopeptide (S)-(2,3-bis(palmitoyloxy)-(2RS)-propyl)-N-palmitoyl-(R)-Cys-(S)-Ser(S)-Lys4-OH, trihydrochloride [PAM3CSK4 (PAM)- 100 ng/ml, Invivogen] for 24 h. In some experiments cells were stimulated simultaneously with Pam3Cys and an agonist anti-CD40 antibody (FGK45- 10 μg/ml) or the appropriate isotype control.
Stimulation of OVA-specific CD4+ Th cells for intracellular cytokine staining
DC (2 × 104) incubated with and without PAM3CSK4 were cultured with CD4+ T cells (1 × 105) in a total volume of 200 μl. Cultures were performed in the presence of 1 μM OVA (Sigma) as indicated. On d4, cells were re-stimulated with 50 ng/ml PMA (Sigma) and 500 ng/ml ionomycin (EMD Biosciences) for 4 h in the presence of brefeldin A (10 μg/ml- Sigma), followed by fixation, permeabilization and intracellular staining. C5aR was blocked using 10 μM of the C5aR antagonist A8Δ71-73 [39]. The endotoxin concentration was < 0.1 EU/ml. Anti-CD25 (PC61.5, eBiosciences) or the appropriate isotype was used at 20 μg/ml.
Flow cytometry
Surface expression of C5aR (CD88), C5L2, MHC class II, CD40, CD80, CD86, CD4, CD25 and the transgenic DO11.10 TCR was determined using the following mAbs: CD88-PE (MCA2457, AbD serotec), C5L2-PE (kind gift of Dr. P. Ward, University of Michigan), IAd-FITC (39-10-8), CD80-PE (16-10A1), CD86-PE (GL1) (all from BD Pharmingen), CD40-APC (IC10), CD4-FITC or APC (L3T4), CD25-APC (PC61.5) and KJ1-26-FITC (all from eBioscience). Alternatively, cells were stained with the respective isotypes. Cells were analyzed using FACSCalibur flow cytometer (BD Biosciences) and FCS Express software. Intracellular cytokine detection in CD4+ T cells was determined using anti-IL-17A-PE (17B7), and anti-IFN-γ-APC (XMG1.2) (e-Bioscience). For detection of Foxp3 expression, the FJK-16S anti-mouse/rat Foxp3 staining set was used per manufacturer's instructions (eBioscience).
Before gating on CD4+ T cells, we excluded all small cells with low granularity (by FSC/SSC) that we found to comprise >95% of dead cells as determined by 7-amino-actinomycin D (7AAD) staining as described [11].
Cytokine ELISA
IL-12p40, IL-12p70, IFN-γ, IL-6, TGF-β (total), IL-21 and IL-17A concentrations in supernatants of DC or DC-T cell cultures were determined using DuoSet ELISA kits (R&D Systems) according to the manufacture's protocol. Detection limit was 15.6 pg/ml. Quantikine ELISA kit (R&D Systems) was used for IL-23 determinations as per manufacturer's instructions. Detection limit was 4.17 pg/ml.
In vivo experiments
WT, C5aR-/- and C5L2-/- mice were injected i.v. with CD4+ DO11.10 RAG2-/- cells (2.5 × 106) and i.p. with PBS or OVA + PAM (50 μg). After 4d, spleens were removed and 1×106 cells were used for CD4/DO11.10 TCR staining. Remaining spleen cells (1 × 106) were re-stimulated with medium or OVA (1μM) + PAM (100 ng/ml) for another 4d and then used for intracellular cytokine staining. Supernatants were used in ELISA experiments.
Statistical Analysis
Statistical analysis was performed with the SigmaStat version 10.0 statistical package (Jandel, Germany). Normal distribution was assumed. Unpaired t test was used to analyze differences between two groups. Comparisons of the means of more than two groups were performed using one way analysis of variance (ANOVA). When the mean values of the groups showed a significant difference, pairwise comparison was done using the Tukey test. P<0.05 was considered a significant difference. Data were taken from three to five individual experiments and expressed as mean values ± standard deviation (s.d.).
Supplementary Material
Acknowledgments
This work was funded by National Institute of Health (AI057839 to J.K. and HL051366 to C.G.). We thank D. Hildeman (CCHMC) for kindly providing the anti-CD40 antibody. E.S Reis was supported by a research fellowship of the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil.
Abbreviations
- AT
anaphylatoxins
- C5aRA
C5aR antagonist
- CHIPS
chemotaxis inhibitory protein of staphylococci
- MHC-II
MHC class II
- PAM
Pam3CysSerLys4
- sDC
spleen-derived DC
Footnotes
Conflict-of-interest disclosure: The authors declare financial or commercial conflict of interest.
References
- 1.Köhl J. The role of complement in danger sensing and transmission. Immunol Res. 2006;34:157–176. doi: 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- 2.Gerard C, Gerard NP. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol. 1994;12:775–808. doi: 10.1146/annurev.iy.12.040194.004015. [DOI] [PubMed] [Google Scholar]
- 3.Lee H, Whitfeld PL, Mackay CR. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol. 2008;86:153–160. doi: 10.1038/sj.icb.7100166. [DOI] [PubMed] [Google Scholar]
- 4.Cain SA, Monk PN. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg(74) J Biol Chem. 2002;277:7165–7169. doi: 10.1074/jbc.C100714200. [DOI] [PubMed] [Google Scholar]
- 5.Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, Brodbeck RM, et al. C5L2, a Nonsignaling C5A Binding Protein. Biochemistry. 2003;42:9406–9415. doi: 10.1021/bi034489v. [DOI] [PubMed] [Google Scholar]
- 6.Gerard NP, Lu B, Liu P, Craig S, Fujiwara Y, Okinaga S, Gerard C. An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. J Biol Chem. 2005;280:39677–39680. doi: 10.1074/jbc.C500287200. [DOI] [PubMed] [Google Scholar]
- 7.Chen NJ, Mirtsos C, Suh D, Lu YC, Lin WJ, McKerlie C, Lee T, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature. 2007;446:203–207. doi: 10.1038/nature05559. [DOI] [PubMed] [Google Scholar]
- 8.Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Köhl J. C5a negatively regulates Toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–426. doi: 10.1016/j.immuni.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, Wetsel RA, et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–236. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karp CL, Grupe A, Schadt E, Ewart SL, Keane-Moore M, Cuomo PJ, Köhl J, et al. Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat Immunol. 2000;1:221–226. doi: 10.1038/79759. [DOI] [PubMed] [Google Scholar]
- 11.Köhl J, Baelder R, Lewkowich IP, Pandey MK, Hawlisch H, Wang L, Best J, et al. A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J Clin Invest. 2006;116:783–796. doi: 10.1172/JCI26582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–453. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 17.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 19.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 21.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 22.Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, Sakaguchi S. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol. 1999;162:5317–5326. [PubMed] [Google Scholar]
- 23.Letourneau S, Krieg C, Pantaleo G, Boyman O. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J Allergy Clin Immunol. 2009;123:758–762. doi: 10.1016/j.jaci.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 25.Lalli PN, Strainic MG, Lin F, Medof ME, Heeger PS. Decay accelerating factor can control T cell differentiation into IFN-gamma-producing effector cells via regulating local C5a-induced IL-12 production. J Immunol. 2007;179:5793–5802. doi: 10.4049/jimmunol.179.9.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–392. doi: 10.1038/nature01315. [DOI] [PubMed] [Google Scholar]
- 27.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von BH. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 28.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 30.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 31.Fritz JH, Le BL, Sellge G, Magalhaes JG, Fsihi H, Kufer TA, Collins C, et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26:445–459. doi: 10.1016/j.immuni.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 33.Fang C, Zhang X, Miwa T, Song WC. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with Toll-like receptor signaling and interleukin-6 production. Blood. 2009;114:1005–1015. doi: 10.1182/blood-2009-01-198283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, Poppelier MJ, et al. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med. 2004;199:687–695. doi: 10.1084/jem.20031636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jongerius I, Köhl J, Pandey MK, Ruyken M, van Kessel KP, van Strijp JA, Rooijakkers SH. Staphylococcal complement evasion by various convertase-blocking molecules. J Exp Med. 2007;204:2461–2471. doi: 10.1084/jem.20070818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wexler DE, Chenoweth DE, Cleary PP. Mechanism of action of the group A streptococcal C5a inactivator. Proc Natl Acad Sci U S A. 1985;82:8144–8148. doi: 10.1073/pnas.82.23.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology. 2000;49:149–157. doi: 10.1016/s0162-3109(00)80300-8. [DOI] [PubMed] [Google Scholar]
- 38.Köhl J. Drug evaluation: The C5a receptor antagoist PMX-53. Curr Opin Mol Ther. 2006;8:529–538. [PubMed] [Google Scholar]
- 39.Otto M, Hawlisch H, Monk PN, Muller M, Klos A, Karp CL, Köhl J. C5a mutants are potent antagonists of the C5a receptor (CD88) and of C5L2: position 69 is the locus that determines agonism or antagonism. J Biol Chem. 2004;279:142–151. doi: 10.1074/jbc.M310078200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.