

Abstract
Air pollution has been associated with significant adverse health effects leading to increased overall morbidity and mortality of worldwide significance. Epidemiological studies have shown that the largest portion of air pollution-related mortality is due to cardiovascular diseases, predominantly those of ischemic nature. Human studies suggest an association with atherosclerosis and increasing experimental animal data support that this association is likely to be causal. While both gasses and particles have been linked to detrimental health effects, more evidence implicates the particulate matter (PM) components as major responsible for a large portion of the proatherogenic effects. Multiple experimental approaches have revealed the ability of PM components to trigger and/or enhance free radical reactions in cells and tissues, both ex vivo as well as in vivo. It appears that exposure to PM leads to the development of systemic prooxidant and proinflammatory effects that may be of great importance in the development of atherosclerotic lesions. This article reviews the epidemiological studies, experimental animal, and cellular data that support the association of air pollutants, especially the particulate components, with systemic oxidative stress, inflammation, and atherosclerosis. It also reviews the use of transcriptomic studies to elucidate molecular pathways of importance in those systemic effects.
Keywords: Air pollution, Particulate matter, Ultrafine particles, Oxidative stress, Inflammation, Atherosclerosis, Dysfunctional HDL, Cardiovascular disease
Exposure to air pollution is associated with atherosclerosis
Extensive epidemiological evidence supports the association of air pollution with adverse health effects leading to increased morbidity and mortality of worldwide significance (Brook et al. 2004; Bhatnagar 2006; Brook et al. 2010) mostly due to cardiovascular diseases and predominantly those of ischemic character (Pope et al. 2004). Thus, exposure to air pollution is now recognized as a novel cardiovascular risk factor of great importance since it is modifiable and with the potential to affect large numbers of people around the globe. While air pollution is a complex mixture of compounds in gaseous (ozone, CO, and nitrogen oxides) and particle phases, the cardiovascular effects have been mostly ascribed to its particulate components (Bhatnagar 2006; Brook 2008; Araujo and Nel 2009). Ambient particles can be classified according to their aerodynamic diameter into size fractions such as particulate matter (PM)10 (“thoracic” particles, <10 μm), PM2.5–10 (“coarse” particles, 2.5–10 μm), PM2.5 (fine particles, <2.5 μm) and ultrafine particles (UFP, <0.1 μm; U.S.EPA 2004) that are derived from various sources and by a variety of processes characteristic of each size fraction. For instance, UFP are mostly generated through tailpipe emission from mobile sources (motor vehicles, aircrafts, etc.), while major sources of PM2.5 include power plants, oil refinery, wildfires, tailpipe, brake emissions from mobile sources and coarse particles are typically derived from soil, agricultural and road dust, construction debris, among others.
Various mechanisms have been proposed to explain how inhalation of ambient particulate could result in systemic cardiovascular effects such as: (1) activation of pulmonary receptors resulting in autonomic nervous system imbalance and the development of dysrhythmias; (2) induction of pulmonary and systemic inflammation; (3) access of particles or their chemical constituents to the systemic circulation (Fig. 1). The latter two mechanisms can lead to the induction of atherothrombotic effects responsible for acute coronary syndromes and ischemic heart disease. Indeed, several studies support the association between air pollution and atherosclerosis in humans (Table 1). Kunzli et al. reported in 2005 a cross-sectional study where the degree of carotid intima-medial thickness (CIMT) of 798 individuals correlated with an increase of 5.9% for every 10 μg/m3 rise in PM2.5 levels (Kunzli et al. 2005; Table 1). A recent study on a related population also showed that the annual rate of CIMT progression among individuals living within 100 m of a highway was accelerated and more than twice the population mean progression (Kunzli et al. 2010). Likewise, Hoffman et al. (2007) reported an association between long-term residential exposure to high traffic and coronary artery calcification (CAC) scores as a measure of coronary atherosclerosis. They found in a German cohort study including 4,494 participants, that as compared with subjects living >200 m away from a major road, subjects living within 101–200 m, 51–100 or less than 50 m showed 8%, 34%, and 63% increase in the probability of having a high CAC scores, respectively (Table 1). Data from the Multi-Ethnic Study of Atherosclerosis (Diez Roux et al. 2008; Allen et al. 2009) also support the association of PM with atherosclerosis. Diez Roux et al. (2008) reported that PM10 exposures assessed over long-term (20-year means and 2001 mean) and 20-year PM2.5 exposures did correlate with 1–3% increase in CIMT per 21 μg/m3 increase in PM10 or 12.5 μg/m3 increase in PM2.5, respectively. Likewise, Allen et al. (2009) reported that PM2.5 exposures correlated with increased risk for aortic calcification in a related study (Table 1).
Fig. 1.
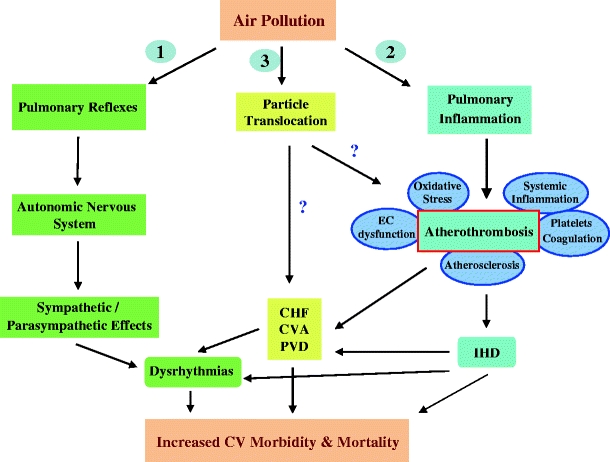
Potential mechanisms how exposure to PM leads to CV disease. Three main pathways could mediate PM-related cardiovascular effects: (1) induction of autonomic nervous system imbalance, (2) development of pulmonary oxidative stress and inflammation with systemic “spill-over” of inflammatory mediators (e.g., cytokines, activated cells), (3) translocation of particles and/or chemical constituents to the systemic circulation. CHF congestive heart failure, CV cardiovascular, CVA cerebrovascular accident, EC endothelial cells, IHD ischemic heart disease, PVD peripheral vascular disease. Pulmonary inflammation may range from activation of inflammatory molecular pathways without histological evidence of it to overt infiltration by inflammatory cells. Taken from Araujo and Book (2011)
Table 1.
Human studies linking exposure to air pollution with atherosclerosis
| Study | Air pollutant | Evaluation of atherosclerosis | Major findings | Reference |
|---|---|---|---|---|
| Kunzli et al. 2005 | PM2.5-Ozone | CIMT | 5.9% increased in CIMT per every 10 μg PM2.5/m3 | (Kunzli et al. 2005) |
| Hoffmann et al. 2007 | PM2.5-Distance to major road | CACS | Increased CAC scores with shorter distances to a major road | (Hoffmann et al. 2007) |
| Diez Roux et al. 2008 | PM10 -PM2.5 | CIMT CACS BAI | 1–3% increase in CIMT per every increase in 21 and 12.5 μg/m3 of PM10 and PM2.5 respectively | (Diez Roux et al. 2008) |
| Allen et al. 2009 | PM2.5-distance to major road | Aortic calcification | 6% increase in the risk of aortic calcification with a 10 μg/m3 increase in PM2.5 | (Allen et al. 2009) |
| Hoffman et al. 2009 | PM2.5-distance to major road | BAI | Decreased BAI in subjects living within 50 meters from a major road, especially women | (Hoffman et al. 2009) |
| Kunzli et al. 2010 | PM2.5-distance to highway or major road | CIMT | Greater annual progression of CIMT among individuals living <100 m from a highway | (Kunzli et al. 2010) |
Studies are listed in chronological order based on the year of publication. CIMT carotid intima-media thickness, CACS coronary artery calcium score, BAI brachial artery index. Modified from Araujo (2011)
While epidemiological studies with PM10 and PM2.5 data support that a smaller particle size correlates with larger cardiovascular effects, there are only few reports supporting the association of UFP with increased total or cardio-respiratory mortality (Wichmann et al. 2000; Yue et al. 2007) and there are no reports that support an association between UFP and atherosclerosis yet, partly due to the difficulty to perform reliable measurements of UFP particle number and mass concentrations since they are very dependent on the proximity to the source of generation. Despite the lack of direct evidence, it has been suggested that associations between PM and cardiovascular (CV) morbidity and mortality could be even stronger with the smaller particles. In a study conducted in four large retirement communities in the Los Angeles basin, Delfino et al. (2009) followed 60 elderly subjects with history of CAD over 7-month periods with a very detailed pollutant exposure characterization and blood collection for the determination of systemic inflammatory, antioxidant and coagulation markers. They found positive associations of particle number (dominated by UFPs) and outdoor quasi-ultrafine PM0.25 (<0.25 μm) with biomarkers of systemic inflammation such as interleukin (IL)-6, soluble tumor necrosis factor receptor II (sTNF-RII), and C-reactive protein (CRP; Delfino et al. 2008, 2009). This study is in agreement with a previous report where exposure to UFP correlated with increased plasma levels of soluble CD40 ligand (sCD40L), a marker for platelet activation that can cause increased coagulation and inflammation (Ruckerl et al. 2007) as well as other repeated-measure studies showing associations between ambient air pollution and biomarkers of systemic inflammation in healthy young adults (Chuang et al. 2007) and susceptible subjects with CAD (Dubowsky et al. 2006; Ying et al. 2009b).
Role of particle size and chemical composition
The cardiovascular toxicity of particles may depend on several physicochemical parameters that can determine their ability to travel through the tracheobronchial tree upon inhalation, interact with cellular components in the lungs and either penetrate into the systemic circulation or trigger responses in the lungs that can be transduced systemically. Particle cardiopulmonary toxicity appears to correlate with their prooxidant and proinflammatory potentials. In this article, we mostly refer to evidence accrued using ambient particulate matter and/or diesel exhaust particles (DEP) as a model pollutant. Multiple studies have demonstrated that DEP can promote the generation of reactive oxygen species (ROS) in macrophages, bronchial epithelial cells and lung microsomes incubated with the particles or their organic extracts (Hiura et al. 1999; Li et al. 2002). DEP can also induce oxidative stress and cellular effects in target vascular cells such as endothelial cells (Bai et al. 2001; Hirano et al. 2003; Gong et al. 2007; Li et al. 2009) as indicated in more details below.
It appears that DEP redox properties reside in their polar and aromatic fractions as they most actively induce the expression of heme oxygenase-1 (HO-1), glutathione S-transferase and other phase II enzymes in response to increased oxidative stress (Li et al. 2004). This antioxidant response is modulated by the p45-NFE2 related transcription factor 2 (Nrf2) (Li et al. 2004), which is able to escape proteosomal degradation in the cytoplasm and translocate to the nucleus under the influence of electrophiles (Li et al. 2004). Ambient PM can also induce tissue oxidative stress and trigger Nrf2-regulated antioxidant gene expression in response to its content of prooxidative and electrophilic chemicals that differ markedly among the various particle sizes. Different particle sizes are apparently married to different chemical compositions resulting in different prooxidant and proatherosclerotic effects as it is described below. Thus, UFP collected from various locations and seasons differ significantly in their ability to trigger Nrf2-regulated responses, in strong relationship with their chemical composition (e.g., content of polycyclic aromatic hydrocarbons; PAHs) and prooxidant potential (Li et al. 2003).
Of even larger importance to the data generated by in vitro approaches, it is the fact that several studies have demonstrated the ability of PM to promote systemic prooxidant and proinflammatory effects in vivo as well as enhanced atherosclerotic lesion development. This body of evidence not only confirms the relevance of the in vitro data but also brings support to the notion that the particle prooxidative and proinflammatory potentials may be important determinants of particle vascular toxicity. Table 2 summarizes several of those studies that support the causal association between particulate air pollution and atherosclerosis using various animal models. Exposures to air pollutants have been performed via intratracheal instillations (IT) of PM as well as inhalation of polluted air, concentrated ambient particles (CAPs), or diesel exhaust emissions. These various exposure modalities differ in their degree of difficulty and their ability to model “real life” scenarios. Thus, while IT inhalations can be manipulated experimentally at ease, they may be more limited to reflect physiologically relevant exposures. On the other hand, inhalation of CAPs can model physiological conditions better but are more difficult to be conducted. Importantly, the convergence of similar findings derived from different exposure modalities helps in data interpretation and brings robustness to the conclusions derived.
Table 2.
Animal studies evaluating the effect of particulate air pollution on atherosclerosis
| Study | PM fraction (mode of administration) | Animal model | Diet | Assessment of atherosclerosis (method) | Effect of PM on atherosclerosis | Ref |
|---|---|---|---|---|---|---|
| Suwa et al. 2002 | PM10 (IT) | Watanabe rabbits | Chow | Percent lesional volume in coronary arteries and aorta (histology) | Increase | (Suwa et al. 2002) |
| Chen and Nadziejko 2005 | PM2.5 (inhaled CAPs) | apoE−/−, LDL−/− mice and apoE−/− mice | Chow | Percent lesional area in whole aorta (histology) | No change (apoE−/−, LDL−/− mice) increase (apoE−/− mice) | (Chen and Nadziejko 2005) |
| Sun et al. 2005 | PM2.5 (inhaled CAPs) | apoE−/− mice | Chow or CED | Percent lesional area in cross-sections of aorta (histology) | N.S. increase (chow-fed mice). Increase (CED-fed mice) | (Sun et al. 2005) |
| Niwa et al. 2007 | Carbon black (IT) | LDL-R−/− mice | CED | Percent lesional area in whole aorta (histology) | Increase | (Niwa et al. 2007) |
| Sun et al. 2008 | PM2.5 (Inhaled CAPs) | apoE−/− mice | Chow or CED | Percent lesional area in aorta (ultrasound) | N.S. increase (chow-fed mice). Increase (CED-fed mice) | (Sun et al. 2008) |
| Yatera et al. 2008 | PM10 (IT) | Watanabe rabbits | Chow | Percent lesional volume and% lesional area in the aorta (histology) | Increase | (Yatera et al. 2008) |
| Araujo et al. 2008 | PM2.5 and UFP (Inhaled CAPs) | apoE−/− mice | Chow | Mean lesional area in aortic root (histology) | N.S. increase (PM2.5-exposed mice) Increase (UFP-exposed mice) | (Araujo et al. 2008) |
| Castro-Soares et al. 2009 | PM (inhaled polluted air) | LDLR−/− mice | CED | Percent lesional area in aortic root. Aortic wall thickness (histology) | No change in lesional area.Increase in wall thickness | (Soares et al. 2009) |
| Ying et al. 2009a | PM2.5 (inhaled CAPs) | apoE−/− mice | CED | Percent lesional area in cross-sections of aorta (histology) | Increase | (Ying et al. 2009a) |
| Campen et al. 2010 | PM + gasses (Inhaled DE) | apoE−/− mice | CED | Percent lesional area in the aortic root plaque composition | N.S. increase. Increase in macrophage content | (Campen et al. 2010) |
| Chen et al. 2010 | PM2.5 (inhaled CAPs) | apoE−/− mice | Chow | Percent lesional area in brachiocephalic and left common arteries (ultrasound) | Increase | (Chen et al. 2010) |
| Quan et al. 2010 | PM±/− gasses (Inhaled CAPs vs. Inhaled whole DE or DE gasses) | apoE−/− mice | Chow | Percent lesional area in brachiocephalic artery (ultrasound) | Increase (CAPs ≥ whole DE ≥ DE gasses = filtered air) | (Quan et al. 2010) |
Studies are shown in chronological order based on the year of publication. IT intratraqueal, CAPs concentrated ambient particles, DE diesel emissions, CED cholesterol-enriched diet, NS not significant. Modified from Araujo (2011)
Indeed, IT PM10 and IT carbon black have been reported to stimulate atherosclerotic lesion formation in Watanabe hyperlipidemic rabbits (Suwa et al. 2002; Yatera et al. 2008) and low-density lipoprotein-receptor (LDL-R)−/− mice (Niwa et al. 2007), respectively. In those studies, atherosclerosis was assessed both in the coronary arteries (Suwa et al. 2002) and in the aorta (Suwa et al. 2002; Niwa et al. 2007; Yatera et al. 2008). On the other hand, the effect of PM2.5 has been mostly evaluated by long-term inhalation exposures to concentrated PM2.5. Five studies have been reported where concentrated PM2.5 led to enhanced atherosclerosis in apoE null mice exposed to CAPs at Sterling Forest, New York (Chen et al. 2010; Chen and Nadziejko 2005; Sun et al. 2005; Sun et al. 2008) or in Manhattan, New York (Ying et al. 2009b). Chen and Nadziejko (2005) reported that 39–41 week-old apoE−/− mice fed a chow diet and exposed to 10X ambient concentrations of PM2.5 for 6 h per day, 5 days per week for 5 months led to a 57% increase in the percentage of atherosclerotic plaque area in the aortic root (Table 2). Sun et al. (2005) showed that younger 6-week-old apoE−/− mice fed a chow diet and exposed to similar conditions for 6 months displayed a 45% increase in the percentage of aortic atherosclerotic plaque area, which was not statistically significant however. The feeding of a high-fat diet resulted in a greater promotion of atherosclerosis that was statistically significant, as PM2.5-exposed mice displayed a 58% increase in aortic root plaque area (Sun et al. 2005), together with impaired vasomotor response (Table 2). In another study, Sun et al. (2008) reported that apoE−/− mice fed a high fat diet and exposed to concentrated PM2.5 also resulted in enhancement of aortic atherosclerosis as assessed by the percentage of the plaque area in the aortic arch by ultrasound bio-microscopy. PM2.5 exposures led to atherosclerotic plaques that were also richer in tissue factor (Sun et al. 2008), which could play a causative role or simply be an indicator of greater atherosclerotic plaque burden. When compared to inhaled side-stream tobacco smoke, concentrated PM2.5 appears to exert greater promotion of plaque formation as assessed by ultrasound bio-microscopy (Chen et al. 2010). Importantly, concentrated PM2.5 from a different location, such as an exposure laboratory in Manhattan, also resulted in similar proatherogenic effects (Ying et al. 2009b).
The different degrees how aortic atherosclerosis was enhanced by PM2.5 in the various CAPs studies can be due to several factors such as differences in the length of exposures, the type of experimental diet or the PM composition. In addition, while 4–6 month exposures to concentrated PM2.5 in New York (Chen et al. 2010; Chen and Nadziejko 2005; Sun et al. 2005; Sun et al. 2008; Ying et al. 2009a) have shown positive results, 5-week exposures to concentrated PM2.5 in Los Angeles only resulted in a non-statistically significant trend for increased atherosclerotic lesions (Araujo et al. 2008), which raises important questions about toxicological aspects such as effective dose, threshold or latency of effects. In addition, there have been discrepancies in whether the feeding of cholesterol-enriched diets and/or extreme hyperlipidemia exert or not an additive effect to the PM exposures. Thus, two studies have reported a significant enhancement of atherosclerosis when apoE-/- mice were fed a cholesterol-enriched diet (CED) but only suggestive trends when mice were fed a chow diet (Sun et al. 2005, 2008). On the other hand, one study showed that the PM2.5 proatherogenic effects were not seen under the condition of extreme hyperlipidemia exhibited by apoE−/−, LDL-R−/− mice (Chen and Nadziejko 2005).
Another important aspect relates to the different degree how PM promotes atherogenesis in different locations where particulates may originate from different sources, under different climatological conditions and resulting in different physicochemical characteristics. Thus, the study from Ying et al. (2009a) showed that 4-month inhalation exposures to concentrated PM2.5 in the Upper East side of Manhattan (6 h/day, 5 days/week) of CED-fed 6-week-old apoE−/− mice resulted in ~89% increase in aortic atherosclerotic plaques over controls. This is in contrast to the other CAPs PM2.5 studies where CED-fed mice were exposed for 5–6 months in Sterling Forest, New York resulting in an increase from 58% to 68% increase in atherosclerotic lesions over controls (Sun et al. 2005; 2008). Larger effects with the Manhattan PM2.5 could have been due to greater PM2.5 levels and/or greater relative content in organic carbon and elemental carbon as mentioned by their authors (Ying et al. 2009a).
In relation to the role of particle size, only one study has approached the question whether particle size has any role in PM-mediated proatherogenic effects. Thus, Araujo et al. tested the hypothesis that the smaller size and greater redox potential of UFP would result in greater proatherogenic effects than PM2.5 in an animal study at the University of California in Los Angeles. ApoE−/− mice were fed a chow diet and subjected to concentrated PM2.5, UFP or filtered air for 5 h/day, 3 days per week for 5 weeks. UFP-exposed mice developed 25% and 55% greater aortic atherosclerotic plaques, assessed by the mean atherosclerotic lesional area in the aortic root, as compared to PM2.5 or FA-exposed mice, respectively (Araujo et al. 2008), despite an exposure mass that was ~4 times less. Given that the concentrator technology employed (Versatile Aerosol Concentration Enrichment System) in this study generated overlapping CAPs aerosols, it is difficult to estimate the true relative proatherogenic strength of UFP vs. PM2.5. While it is possible that the UFP fraction could concentrate the PM proatherogenic effects, a clear demonstration of this will require the straight comparison of UFP to the accumulation mode particles in the 0.1–2.5 μm range (Araujo and Nel 2009). Interestingly, in this study, UFP exhibited a greater relative content of PAHs, a whole group of redox-active compounds that can facilitate free radical reactions (Li et al. 2003; Ntziachristos et al. 2007; Ayres et al. 2008). Additional studies are required to compare PM2.5 and UFP exposures in other locations in the world where the effects of different particle size and composition on atherosclerosis, can be assessed.
It has been debated whether PM at ambient levels could cause similar effects to those induced by CAPs over perhaps longer periods of time. Castro-Soares et al. addressed this issue by exposing LDL-R−/− mice to 1X ambient air (non-filtered) vs. filtered air at a location 20 m away from the roadside in downtown Sao Paulo, Brazil. Although mice exposed to non-filtered air developed atherosclerotic plaques that were comparable in lipid content to those developed by controls’, their aortic walls were thicker, suggesting a greater contribution of other components (either cellular or non-cellular; Soares et al. 2009). Another important point is whether the gasses could contribute to the vascular proatherogenic effects of particulates, perhaps by cooperative interactions with them. There is indeed experimental data that show that gaseous pollutants could also play a role in promoting atherosclerosis. Lewis et al. showed that exposures of CED-fed C57BL6 mice to carbon disulfide (CS2), a known volatile organic compound, at 500 and 800 ppm CS2 6 h/day, 5 days/week for up to 20 weeks, markedly enhanced aortic atherosclerosis (1999). In addition, Chuang et al. (2009) reported that apoE−/− mice exposed to 0.5 ppm O3 for 8 h/day, 5 days/week for 8 weeks exhibited more than double the degree of aortic atherosclerosis exhibited by filtered air-exposed controls (Table 2). Furthermore, Campen et al. examined the effect of whole vs. particle-filtered diesel exhausts emissions on atherosclerotic lesion formation of CED-fed 10-week-old male apoE−/− mice over 50 days. Although they did not find significant differences in the extent of aortic plaque formation between the various groups, mice exposed to whole diesel exhausts developed atherosclerotic lesions with greater macrophage content (Campen et al. 2010; Table 2). Interestingly, exposure to whole diesel exhausts led to upregulation of MMP-9 in the aorta, consistent with similar effects reported for gasoline exhausts (Lund et al. 2007). While particle-filtering tended to diminish the effects of the diesel emissions on plaque macrophage content, it did not alter the degree of aortic MMP-9 upregulation (Campen et al. 2010), suggesting that both gaseous and particulate components could be affecting different pathways. It is possible then that gaseous and particulate pollutants could exert different but cooperative effects, which will need to be explored in more details in the future, in spite of a recently published study that did not find any contribution of diesel exhaust gasses to the effects exerted by CAPs inhalations (Quan et al. 2010).
Altogether, the reported literature suggests that particle size and chemical composition are important determinants in the ability of particulates to enhance atherosclerotic lesion formation. In addition, while the epidemiological data mostly points towards the contribution of the particulate matter, the experimental animal data suggests that gaseous components may also play a role.
How does particulate matter promote atherosclerosis?
PM-enhanced atherosclerosis is most likely the result of systemic prooxidant and proinflammatory effects. These systemic effects could derive from increased ROS production and development of inflammation in the lungs that somehow may get transduced to the systemic tissues (e.g., release of inflammatory mediators into the blood; pathway 2 of Fig. 1) or they could be the result of translocation of particles or their chemical constituents into the systemic circulation with direct action at the target vascular sites (pathway 3 of Fig. 1). There are multiple reports that support the ability of PM to induce local prooxidant and proinflammatory effects in the lungs. The latter may include activation of inflammatory molecular pathways without histological evidence of overt pulmonary inflammation, local activation of cells leading to pan-systemic activation of the adaptive immune system or infiltration of the lungs by inflammatory cells with the development of histologically defined pulmonary inflammatory foci (Fig. 1). This article focuses however on the experimental evidence that supports the occurrence of those effects in systemic tissues, especially at the target vessels (summarized in Table 3) as follows:
Table 3.
Mechanisms mediating PM-induced proatherogenic effects. In vitro and experimental animal evidence
| Mechanisms | Indicators | References |
|---|---|---|
| Evidence in cellular and/or animal studies | ||
| Oxidative stress in systemic tissues | Increased liver peroxidation and upregulation of antioxidant genes in liver | (Gong et al. 2007; Araujo et al. 2008) |
| Alteration of circulating lipoproteins | Oxidative modification of LDL | (Soares et al. 2009) |
| Generation of dysfunctional HDL | (Araujo et al. 2008) | |
| Vascular prooxidant effects | Increased ROS production in ECs and macrophages | (Goldsmith et al. 1998; Hiura et al. 1999; Soukup et al. 2000; Bai et al. 2001; Lee and Kang 2002; Hirano et al. 2003; Li et al. 2004; Li et al. 2006; Chio et al. 2007; Li et al. 2010; Montiel-Davalos et al. 2010) |
| Upregulation of antioxidant genes in ECs and macrophages | (Hirano et al. 2003; Li et al. 2004; Gong et al. 2007; Li et al. 2010) | |
| Activation of NADPH oxidase in ECs and macrophages | (Beck-Speier et al. 2005; Li et al. 2006; Mo et al. 2009) | |
| Induction of electron leaks in mitochondrial electron transport complexes in ECs | (Li et al. 2006) | |
| Increased ROS in vessels with atherosclerotic plaques | (Sun et al. 2005; Ying et al. 2009a) | |
| Vascular proinflammatory effects | Greater proinflammatory cytokine production in ECs and macrophages | (Alfaro-Moreno et al. 2002; Fujii et al. 2002; Jimenez et al. 2002; Amakawa et al. 2003; Osornio-Vargas et al. 2003; Pozzi et al. 2003; Brown et al. 2004; Becker et al. 2005a; Ishii et al. 2005; Imrich et al. 2007; Li et al. 2010) |
| Increased expression of cell adhesion molecules in ECs | (Li et al. 2010) | |
| Greater monocyte adhesion to ECs | (Li et al. 2010) | |
| Monocyte migration and recruitment in atherosclerotic vessels | (Yatera et al. 2008) | |
| Activation of NF-kB in ECs and macrophages | (Mondal et al. 2000; Montiel-Davalos et al. 2010) | |
| Activation of MAPK in ECs and macropages | (Hiura et al. 1999; Sumanasekera et al. 2007; Mo et al. 2009) | |
| Activation of ERK1/2 pathways in ECs and macrophages | (Beck-Speier et al. 2005; Mo et al. 2009) | |
| Increased proinflammatory cytokines in circulating blood | (Mutlu et al. 2007; Tamagawa et al. 2008) | |
| Vascular toxic effects | Decrease in viability of ECs and macrophages | (Hiura et al. 1999; Bai et al. 2001; Lee and Kang 2002) |
| Increased in ECs and macrophages | (Hiura et al. 1999; Lee and Kang 2002; Montiel-Davalos et al. 2010) | |
| Increase in EC permeability and/or upregulation of genes involved in EC permeability | (Nadadur et al. 2009; Wang et al. 2010) | |
| Antiproliferative effects in ECs | (Yamawaki and Iwai 2006) | |
| Evidence in humans | Subclinical measures | |
| Systemic inflammation | Elevated circulating pro-inflammatory biomarkers/mediators (figrinogen, CRP, TNF-α, IL 1-ß, IL-6, IL-7, GM-CSF, ICAM-1) | (Ibald-Mulli et al. 2001; van Eeden et al. 2001; Pope et al. 2004; Riediker et al. 2004; Analitis et al. 2006; Peters et al. 2006; Zeka et al. 2006; Chuang et al. 2007; O’Neill et al. 2007; Ruckerl et al. 2007) |
| Systemic oxidative stress | Increased biomarkers of lipid, protein or DNA oxidation | (Sorensen et al. 2003; Vinzents et al. 2005; Brauner et al. 2007) |
| Elevated plasma homocysteine | (Baccarelli et al. 2007; Park et al. 2008) | |
| Inhibition of oxidative response by w-3 PUFA | (Romieu et al. 2008) | |
| Systemic hypertension | Elevated systemic diastolic and/or systolic blood pressure | (Ibald-Mulli et al. 2001; Lippmann et al. 2003; Choi et al. 2007; Chuang et al. 2007; Diez Roux et al. 2008; Dvonch et al. 2009; Zanobetti and Schwartz 2009) |
| Elevated endothelin-1 levels | (Peters et al. 2006) | |
CRP C-reactive protein, ECs endothelial cells, ET-1 endothelin 1, GM-CSF granulocyte macrophage colony-stimulating factor, ICAM-1 intercellular adhesion molecular 1; IL interleulin, PUFA polyunsaturated fatty acids, ROS reactive oxygen species, TNF-α tumor necrosis factor α
Systemic oxidative stress
Human studies have associated exposure to PM with increased systemic oxidative stress, which include the detection of biomarkers of oxidative alteration of proteins, lipids and/or DNA in the circulating blood or in products excreted in the urine, as recently reviewed (Moller and Loft 2010). Reports include controlled exposure studies, panel and cross-sectional studies, generally small in size. Measures of oxidation in circulating blood are highly relevant since they may imply the involvement of oxidatively modified plasma lipoproteins such as LDL and/or high-density lipoprotein (HDL), key players in the promotion or protection from atherosclerosis, respectively (Araujo and Nel 2009). Liu et al. (2009) reported the association of increases in exposure to black carbon and PM2.5 with elevation in plasma levels of thiobarbituric acid reactive substances (TBARS) of 28 nonsmoking seniors, in agreement with earlier reports of increased serum TBARS in association with PM2.5 (Sorensen et al. 2003) or after moving or living in a polluted urban location such as Mexico city (Medina-Navarro et al. 1997; Sanchez-Rodriguez et al. 2005).
Experimental animal work brings support to the causality notion. Indeed, we have reported that exposure to CAPs in the PM2.5 and UFP size ranges led to increased hepatic lipid peroxidation, accompanied by a higher upregulation of Nrf2-regulated antioxidant genes and unfolded protein response (UPR) genes of UFP-exposed mouse livers (Gong et al. 2007; Araujo et al. 2008). Castro-Soares et al. (2009) have shown that chronic exposure to urban air pollution led to enhanced susceptibility of LDL to oxidation in hyperlipemic LDL-R−/− mice fed a high fat diet as determined by a kinetics of LDL oxidation by CuSO4. Interestingly, animals exposed to polluted air also developed increased plasma levels of anti-oxidized LDL antibodies (Soares et al. 2009). Although LDL oxidation has not been demonstrated in any of the CAPs studies using apoE−/− mice, we have reported that 5-week exposures of apoE−/− mice to concentrated PM led to the development of dysfunctional HDL (Araujo et al. 2008) as it is discussed below. It is not known how PM inhalations can modify HDL function but it could likely be the result of oxidative modifications on either its protein and/or lipid components that may result in abrogation of its antioxidant enzymatic activities as well as anti-inflammatory and reverse cholesterol-transport properties. Thus, PM prooxidant effects could be enhancing the proatherogenic properties of LDLs and diminishing the antiatherogenic properties of protective HDLs.
Vascular prooxidative effects
PM has been shown to exert prooxidative effects in vitro in cell types that are key players in the development of atherosclerotic lesions such as endothelial cells (Bai et al. 2001; Hirano et al. 2003; Li et al. 2006, 2010; Montiel-Davalos et al. 2010), macrophages (Goldsmith et al. 1998; Hiura et al. 1999; Soukup et al. 2000; Lee and Kang 2002; Li et al. 2004; Chio et al. 2007; Ohyama et al. 2007) and possibly smooth muscle cells (Sun et al. 2008). Indeed, DEP or ambient PM have been reported to increase ROS production in human aortic endothelial cells (Li et al. 2010; Montiel-Davalos et al. 2010), likely by activation of the endothelial NADPH oxidase (Mo et al. 2009) and/or electron leaks in the mitochondrial electron transport complexes (Li et al. 2006). It seems that DEP in the ultrafine size range stimulates superoxide production in human aortic endothelial cells, partly mediated by JNK activation (Li et al. 2009). PM can also lead to increased nitric oxide production in pulmonary artery endothelial cells (Bai et al. 2001). Increased ROS can lead to the upregulation of antioxidant genes such as HO-1 in human aortic endothelial cells (Li et al. 2010), human microvascular endothelial cells (Gong et al. 2007) and rat heart microvessel endothelial cells (Hirano et al. 2003). PM can exert similar in vitro prooxidant effects in macrophages, which has been demonstrated in cultures of immortalized immune cells such as THP-1 (Hiura et al. 1999; Chio et al. 2007) and RAW264.7 cells (Hiura et al. 1999; Li et al. 2004) or in primary cell cultures of blood monocytes (Ohyama et al. 2007), thyglycolate-stimulated peritoneal macrophages (Lee and Kang 2002; Li et al. 2004) or alveolar macrophages (Goldsmith et al. 1998; Soukup et al. 2000; Beck-Speier et al. 2005). The use of the latter macrophages could give clues about the effects on systemic macrophages despite their different localization.
Exposure to PM also results in prooxidant vascular effects in vivo. Nurkiewicz et al. (2004, 2006)showed in rats that intratracheal instillation of residual oil fly ash (ROFA) led to greater vascular ROS generation, as assessed by the tetranitrobluetetrazolium reduction method (Nurkiewicz et al. 2006), resulting in a dose-dependent impairment of systemic endothelium-dependent arterial dilation as well as proinflammatory effects. Likewise, inhalation exposures to concentrated PM2.5 has led to enhanced ROS generation in the aortic plaques, increased formation of 3-nitrotyrosine residues (Sun et al. 2005; Ying et al. 2009b) and upregulation of the NADPH oxidase subunits p47phox and Rac1 (Ying et al. 2009b).
Systemic inflammation
PM exposure has been associated with increased proinflammatory mediators in the systemic circulation, both in animals (Mutlu et al. 2007; Tamagawa et al. 2008) and humans (van Eeden et al. 2001). IT administration of ROFA to rats led to increased leukocyte rolling and adhesion as well as deposition of myeloperoxidase in the spinotrapezius muscle microcirculation (Nurkiewicz et al. 2004, 2006) together with the prooxidant effects mentioned above. In New Zealand White Rabbits and mice, IT PM10 administration resulted in significantly increased serum IL-6 levels (Mutlu et al. 2007; Tamagawa et al. 2008) as well as TNF-α (Mutlu et al. 2007). Interestingly, lack of IL-6 ameliorated the PM10-mediated systemic proinflammatory response as well as a PM10 procoagulant state in mice (Mutlu et al. 2007), suggesting an important role for IL-6 in the mediation of PM systemic inflammatory effects. Likewise, CAPs PM2.5 inhalation exposures led to elevation in circulating IL-6 and TNF-alpha (Sun et al. 2009) together with increases in circulating adipokines, such as resistin and a change in the balance of macrophages in adipose tissue towards the proinflammatory M1 phenotype, suggestive of a systemic proinflammatory state (Sun et al. 2009). Furthermore, it has been shown that PM induces increased monocyte migration to atherosclerotic vessels in Watanabe rabbits (Yatera et al. 2008). In humans, PM exposures has been reported to associate with increased blood levels of granulocyte macrophage colony-stimulating factor (GM-CSF), IL-6, IL-1, sTNF-RII, CRP, and sCD40L (van Eeden et al. 2001; Dubowsky et al. 2006; Chuang et al. 2007; Ruckerl et al. 2007; Delfino et al. 2008).
We have shown that CAPs exposures led to the development of dysfunctional HDL in apoE−/− mice as mentioned above. Interestingly, the degree of HDL dysfunction was particle size-dependent since UFP exposures led to a greater degree of dysfunction than did PM2.5 (Araujo et al. 2008). Using a monocyte chemotactic assay in a co-culture of endothelial cells and smooth muscle cells that evaluated the HDL anti-inflammatory capacity by determining the degree of inhibition of LDL-induced chemotaxis, UFP exposures not only failed to inhibit LDL-mediated inflammatory effects as PM2.5 exposures did, but it promoted more monocyte migration. This greater degree of HDL dysfunction correlated with the greater degree of atherosclerosis and a systemic Nrf2-regulated antioxidant response, supporting the notion that dysfunctional HDL could either play a role in disease pathogenesis or serve as a marker for the PM-mediated systemic prooxidant and/or proinflammatory effects. Indeed, proinflammatory HDL has been reported to predict susceptibility to atherosclerosis in humans (Ansell et al. 2003) and in rabbits (Van Lenten et al. 2007) and although there is no firm evidence yet that greater levels of proinflammatory HDL do promote atherogenesis, better characterization of this phenotype would be helpful since it could contribute to a better understanding of the pathogenesis and/or lead to the identification of a useful biomarker for the exposures or PM systemic effects.
Vascular proinflammatory effects
In vitro work with endothelial cells in culture has shown that DEP and ambient PM can lead to the activation of NF-kB (Li et al. 2010; Montiel-Davalos et al. 2010), p38 MAPK (Sumanasekera et al. 2007; Mo et al. 2009), and ERK1/2 pathways (Mo et al. 2009) with subsequent upregulation of proinflammatory TNF-α (Li et al. 2010; Montiel-Davalos et al. 2010). Likewise, treatment of endothelial cells with PM results in increased expression of cell adhesion molecules such as VCAM (Li et al. 2010), E-selectin, P-selectin (Montiel-Davalos et al. 2007) and increased cytokine production such as IL-8 and monocyte chemotactic protein 1 (Li et al. 2010).
Similar proinflammatory effects occur in PM-treated macrophages of various sources (Table 3). Both DEP and ambient PM treatments of either human/rat/mouse alveolar macrophages, human blood monocytes, THP-1 monocytes, RAW 264.7 macrophages, and/or J774A macrophages have been reported to result in increased production of TNF-α, IL-6 (Alfaro-Moreno et al. 2002; Fujii et al. 2002; Amakawa et al. 2003; Osornio-Vargas et al. 2003; Pozzi et al. 2003; Becker et al. 2005a), IL-8 (Monn and Becker 1999; Becker et al. 2005b), IL1-α (Brown et al. 2004), IL1-β (Jimenez et al. 2002), GM-CSF (Fujii et al. 2002; Ishii et al. 2005), and macrophage-inflammatory protein-2 (Imrich et al. 2007). Furthermore, it has been reported that PM induces greater monocyte adhesion to human aortic endothelial cells (Li et al. 2010). Interestingly, the ability of DEP in the ultrafine range generated from a same engine, under an idling vs. driving mode led to particles with different chemical composition that diverged on their prooxidant vs proinflammatory potentials (Li et al. 2010) which indicate that PM could activate various pathways, not all ROS related.
Vascular cytotoxic effects
PM has been shown to exert toxic effects in endothelial cells (Li et al. 2010; Montiel-Davalos et al. 2010) and macrophages (Hiura et al. 1999; Lee and Kang 2002; Table 3). PM can lead to increased permeability of endothelial cells (Wang et al. 2010), which could facilitate systemic translocation of particles or their chemical constituents onto the systemic circulation, release of inflammatory mediators from the lungs or monocyte migration to vascular sites. PM toxic effects also lead to stimulation of apoptosis (Hiura et al. 1999; Lee and Kang 2002; Montiel-Davalos et al. 2010), inhibition of cellular proliferation (Yamawaki and Iwai 2006) and decreased cell viability in cultures (Hiura et al. 1999; Bai et al. 2001; Lee and Kang 2002). Overall, vascular cytotoxicity may have various effects on atherogenesis that can depend on the stage of plaque development. For instance, while apoptosis has been thought to limit early atherogenesis, it may actually do the opposite in more advanced lesions (Tabas 2007).
Vascular dysfunction
Several studies have reported the ability of both particulate and gaseous pollutants to cause vascular dysfunction, both on its constricting ability as well as its vasodilatory capacity, by mechanisms that could be either endothelial-dependent (O’Neill et al. 2005; Briet et al. 2007; Ruckerl et al. 2007; Rundell et al. 2007; Liu et al. 2009) or independent (O’Neill et al. 2005). Accumulating evidence suggests that endothelial dysfunction could be an early marker for atherosclerosis (Davignon and Ganz 2004) but less likely that it could independently increase the propensity for atherosclerosis per se. Therefore, the alteration in vascular reactivity function could be a consequence of the same PM-induced systemic prooxidant and proinflammatory effects described above, in addition to neurohormonal mechanisms (pathway 1 of Fig. 1).
Hypertension
Several studies report the association of air pollutants with hypertension (Ibald-Mulli et al. 2001; Lippmann et al. 2003; Choi et al. 2007; Chuang et al. 2007; Diez Roux et al. 2008; Dvonch et al. 2009; Zanobetti and Schwartz 2009; Table 3), a well-known risk factor for atherosclerosis. Pro-hypertensive and vasoconstrictive effects appear to occur both in the pulmonary and systemic circulation and seem to include a variety of mechanisms that range from increased ROS generation and endothelial cell dysfunction to neurohormonal activation. The latter may occur due to increased activity of the sympathetic nervous system as well as enhanced endothelin 1 and angiotensin II receptor pathways, all leading to decreased endothelial-dependent vasodilatation and enhanced vasoconstrictor activity in several studies. Thus, while some of the systemic prooxidant and proinflammatory effects mentioned above could be partially responsible, it is also likely that neurohormonal mechanisms could contribute in some degree to those effects (pathway 1 of Fig. 1; Brook 2008). Indeed, it is possible that PM-induced hypertension could play a role in the promotion of atherosclerosis; however, no hypertensive effects have been reported in any of the atherosclerosis studies conducted with experimental animals so far.
Altogether, cumulative evidence suggests that PM-induced systemic prooxidant and proinflammatory effects appear to be at the center of its ability to enhance atherosclerosis as shown by several studies using in vitro and in vivo approaches. While the data from some studies may carry higher significance than from others, it is the convergence of similar findings from different experimental designs and approaches what brings more robustness to this notion. These prooxidant and proinflammatory effects could be mediated by various pathways with various degrees of involvement of the lungs (Fig. 1). It appears that UFP may exhibit greater ability to enhance atherosclerosis than larger particulate. Various reasons that could explained their greater proatherogenic potential include larger particle number, greater lung retention, larger content of redox active compounds, and greater bioavailability as we have recently reviewed in details (Araujo and Nel 2009).
Effects of particulate matter on vascular gene expression
Several transcriptomic studies have been performed either in tissues harvested from animals exposed to PM or in cells treated in culture with PM or their chemical constituents. The large majority of studies have focused on the PM effects in the lungs which may help to determine the degree of pulmonary involvement in the development of systemic vascular effects. Fewer studies have been performed in vascular cells, either on endothelial cells (Yamawaki and Iwai 2006; Gong et al. 2007; Nadadur et al. 2009), macrophages (Verheyen et al. 2004; Huang et al. 2009) or atherosclerotic plaques from exposed mice (Floyd et al. 2009; Table 4), which have provided clues on some of the molecular pathways that may be responsible for the phenotypes observed. This article focuses on the latter.
Table 4.
Transcriptomic studies evaluating the effect of PM on vascular cells
| Study | PM source | Cells or tissue/platform | Design | Findings | Refs |
|---|---|---|---|---|---|
| Verheyen et al. 2004 | DEP | THP-1 differentiated macropahges/incyte cDNA clone set | PMA-differentiated cells were treated with DEP for 6 and 24 h vs. controls | 50 and 39 clones were up or downregulated at 6 h; 54 and 60 clones were up or downregulated at 24 h (>1.3-fold). Genes were enriched in immune response, cell adhesion, apoptosis, metabolism, signal transduction and transport pathways | (Verheyen et al. 2004) |
| Yamawaki and Iwai 2006 | Carbon black (CB) | HUVECs/Affimetrix | Cells treated with CB for 24 h vs. controls | Top 89 upregulated probesets included IL-8, CXCL2, CXCL3, VCAM1, HO-1, MMP10, BMP2. | (Yamawaki and Iwai 2006) |
| Gong et al. 2007 | DEP | HMECs/Illumina | Cells treated with DEP, oxPAPC and DEP + oxPAPC for 4 h vs. controls | 1,555 regulated genes > 1.5-folds. 664 genes of those exhibited synergistic effects between DEP and oxPAPC. Genes were enriched in antioxidant, inflammatory, unfolded protein response and apoptosis pathways | (Gong et al. 2007) |
| Nadadur et al. 2009 | ROFA and Vanadium | HUVECs/Clontech cDNA array | Cells treated with ROFA or Vanadium for 30 min vs. controls | 810 genes regulated by ROFA >2-folds (339 upregulated and 471 downregulated). Genes wereenriched in cytokines/groth factors, cell adhesion molecules, transporters, signal transduction families. Nine transcription factors were activated by ROFA (>2-fold by TranSignal Protein/DNA arrays) which included AP-1, Nrf2, and USF-1 | (Nadadur et al. 2009) |
| Huang et al. 2009 | PM2.5 | Alveolar macrophages/Agilent | Cells treated with PM2.5 for 4 h vs. control | 38 and 43 differentially up or downregulated genes, respectively. Upregulated genes were enriched in metal binding, antioxidant functions including Metallothioneins, NQO1, MMP9, ATOX1 | (Huang et al. 2009) |
| Floyd et al. 2009 | PM2.5 | Aortic atherosclerotic plaques/Affymetrix | Plaques harvested by LCM from apoE−/− exposed to PM2.5 vs. filtered air for 5 months | 611 differentially expressed genes (216 upregulated, 395 downregulated) enriched in inflammation, immunological disease, lipid metabolism, cell death ontological categories among others | (Floyd et al. 2009) |
DEP diesel exhaust particles, HMECs human microvascular endothelial cells, HUVECs human umbilical vein endothelial cells, LCM laser capture microdissection, ox-PAPC oxidized PAPC, oxidized PAPC (1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine), PMA phorbol myristate acetate, ROFA residual oil fly ash, AP-1 activating protein 1, ATOX1 Atx1 antioxidant protein 1 homolog, BMP2 Bone morphogenetic protein 2, CXCL2 chemokin (C-X-C motif) ligand 2, CXCL3 chemokin (C-X-C motif) ligand 3, HO-1 heme oxygenase 1, IL-8 interleukin 8, MMP10 matrix metalloproteinase 10, NQO1 NAD(P)H dehydrogenase, quinone 1, Nrf2 NFE-E2 related factor 2, USF1 upstream stimulating factor 1, VCAM1 vascular cell adhesion molecule 1
It is interesting how all these studies have shown the involvement of inflammatory pathway genes in vascular cells such as chemokin (C-X-C motif) ligand 2 (CXCL2), CXCL3, IL-8 which confirms both in vitro and experimental animal studies described in previous sections. Various antioxidant genes have also been found upregulated in endothelial cells (Yamawaki and Iwai 2006), macrophages (Huang et al. 2009; Table 4) and atherosclerotic plaques (Floyd et al. 2009) such as HO-1, superoxide dismutase 2, NAD(P)H:quinone oxidoreductase 1, and various metallothioneins, likely in response to the PM-mediated enhancement of ROS in the vasculature. ROS and inflammatory stressors result in the activation of UPR genes (e.g., activating factor 3, activating factor 4, and X box-binding protein 1; XBP1) as we have shown in endothelial cells treated with DEP (Gong et al. 2007), also found to be upregulated after treatment with carbon black (Yamawaki and Iwai 2006). Importantly, the identification of this pathway by an in vitro transcriptomic approach (Gong et al. 2007) led to the identification of the involvement of that same pathway in vivo (Araujo et al. 2008), which underscores the importance of these approaches in identifying molecular footprints of relevance.
While the individual contribution of air pollutants to atherogenesis might be small, we have thought that their effects could be exacerbated in synergy with other known proatherogenic factors. We tested this hypothesis by evaluating the antioxidant response of microvascular endothelial cells to DEP in the absence and presence of oxidized PAPC (1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine), one of the key prooxidative components generated in LDL particles. As both DEP and ox-PAPC led to prooxidative effects that were expected (Bai et al. 2001; Hirano et al. 2003), the combination of both treatments synergized in increasing antioxidant gene expression, including HO-1 (Gong et al. 2007). In addition, network analysis of the genomic profiles of cells subjected to the various treatments revealed groups of genes enriched in synergistic interactions between DEP and ox-PAPC leading to activation of several pathways (e.g., inflammation, apoptosis, unfolded protein response) of relevance in atherogenesis. Some representative examples included proinflammatory genes such as IL-8 and CXCL1 production, immune response genes such as IL-11 and UPR genes such as ATF 4, heat shock 70 kDa protein 8 and XBP1. Here again, the use of a transcriptomic approach helped us to identify cooperative interactions between PM and oxidized lipids that could also be of importance in the development of atherosclerotic lesions in vivo.
Conclusions and perspectives
Exposure to air pollution leads to increased cardiovascular morbidity and mortality, predominantly of ischemic nature. Epidemiological studies show there is a positive association between exposure to PM and measures of atherosclerosis, that is likely to be causal as supported by animal studies. PM-mediated enhancement of atherosclerosis is likely to be the result of its systemic prooxidant and proinflammatory effects.
It will be important to dissect the mechanisms for the development of vascular systemic effects and to determine whether the lungs play an active role. Effects on circulating lipoproteins need to be characterized in details and confirmed in humans, since they may yield a better understanding of the PM-mediated pathogenesis and could lead to the identification of a biomarker of exposures and/or development of PM systemic effects. Likewise, there is need of epidemiological and exposure studies to evaluate UFP toxicity in humans. Transcriptomic and proteomic studies in cells and/or tissues harvested from animals or subjects exposed to air pollutants are highly promising tools to give clues on the molecular pathways involved. Genomic studies are also required to unravel gene–environment interactions of importance and to identify profiles that could result in increased genetic susceptibility to air pollution-mediated CV effects.
Acknowledgments
Writing of this article was supported by the National Institute of Environmental Health Sciences, National Institutes of Health (“Outstanding New Investigator in the Environmental Health Sciences” RO1 Award ES016959 to Jesus A. Araujo).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Alfaro-Moreno E, Martinez L, Garcia-Cuellar C, Bonner JC, Murray JC, et al. Biologic effects induced in vitro by PM10 from three different zones of Mexico City. Environ Health Perspect. 2002;110(7):715–720. doi: 10.1289/ehp.02110715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RW, Criqui MH, Diez Roux AV, Allison M, Shea S, et al. Fine particulate matter air pollution, proximity to traffic, and aortic atherosclerosis. Epidemiology. 2009;20(2):254–264. doi: 10.1097/EDE.0b013e31819644cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amakawa K, Terashima T, Matsuzaki T, Matsumaru A, Sagai M, et al. Suppressive effects of diesel exhaust particles on cytokine release from human and murine alveolar macrophages. Exp Lung Res. 2003;29(3):149–164. doi: 10.1080/01902140303770. [DOI] [PubMed] [Google Scholar]
- Analitis A, Katsouyanni K, Dimakopoulou K, Samoli E, Nikoloulopoulos AK et al (2006) Short-term effects of ambient particles on cardiovascular and respiratory mortality. Epidemiology 17(2):230–233 [DOI] [PubMed]
- Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108(22):2751–2756. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- Araujo JA (2011) Ultrafine particles and Atherosclerosis. In: Bhatnagar A, editor. Environmental Cardiology: Pollution and Heart Disease. Cambridge, U.K: Royal Society of Chemistry. Issues in Toxicology No.8, pp. 198–219
- Araujo JA, Nel AE. Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress. Part Fibre Toxicol. 2009;6:24. doi: 10.1186/1743-8977-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo JA, Brook RD. Cardiovascular effects of particulate matter air pollution: an overview and perspectives. In: Bhatnagar A, editor. Environmental Cardiology. Cambridge: Royal Society of Chemistry; 2010. [Google Scholar]
- Araujo JA, Barajas B, Kleinman M, Wang X, Bennett BJ, et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102(5):589–596. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JG, Borm P, Cassee FR, Castranova V, Donaldson K, et al. Evaluating the toxicity of airborne particulate matter and nanoparticles by measuring oxidative stress Potentialâ—a workshop report and consensus statement. Inhal Toxicol. 2008;20(1):75–99. doi: 10.1080/08958370701665517. [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Zanobetti A, Martinelli I, Grillo P, Hou L et al (2007) Effects of exposure to air pollution on blood coagulation. J Thromb Haemost 5(2):252–260 [DOI] [PubMed]
- Bai Y, Suzuki AK, Sagai M. The cytotoxic effects of diesel exhaust particles on human pulmonary artery endothelial cells in vitro: role of active oxygen species. Free Radic Biol Med. 2001;30(5):555–562. doi: 10.1016/S0891-5849(00)00499-8. [DOI] [PubMed] [Google Scholar]
- Beck-Speier I, Dayal N, Karg E, Maier KL, Schumann G, et al. Oxidative stress and lipid mediators induced in alveolar macrophages by ultrafine particles. Free Radic Biol Med. 2005;38(8):1080–1092. doi: 10.1016/j.freeradbiomed.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Becker S, Mundandhara S, Devlin RB, Madden M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol Appl Pharmacol. 2005;207(2 Suppl):269–275. doi: 10.1016/j.taap.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Becker S, Dailey L, Soukup JM, Silbajoris R, Devlin RB. TLR-2 is involved in airway epithelial cell response to air pollution particles. Toxicol Appl Pharmacol. 2005;203(1):45–52. doi: 10.1016/j.taap.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ Res. 2006;99(7):692–705. doi: 10.1161/01.RES.0000243586.99701.cf. [DOI] [PubMed] [Google Scholar]
- Brauner EV, Forchhammer L, Moller P, Simonsen J, Glasius M et al. (2007) Exposure to ultrafine particles from ambient air and oxidative stress-induced DNA damage. Environ Health Perspect 115(8):1177–1182 [DOI] [PMC free article] [PubMed]
- Briet M, Collin C, Laurent S, Tan A, Azizi M, et al. Endothelial function and chronic exposure to air pollution in normal male subjects. Hypertension. 2007;50(5):970–976. doi: 10.1161/HYPERTENSIONAHA.107.095844. [DOI] [PubMed] [Google Scholar]
- Brook RD. Cardiovascular effects of air pollution. Clin Sci. 2008;115(6):175–187. doi: 10.1042/CS20070444. [DOI] [PubMed] [Google Scholar]
- Brook RD, Franklin B, Cascio W, Hong Y, Howard G, et al. Air pollution and cardiovascular disease: a statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation. 2004;109(21):2655–2671. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- Brook RD, Rajagopalan S, Pope CA, III, Brook JR, Bhatnagar A et al. (2010) Particulate matter air pollution and cardiovascular disease. An update to the scientific statement from the American Heart Association. Circulation: CIR.0b013e3181dbece3181. [DOI] [PubMed]
- Brown DM, Donaldson K, Stone V. Effects of PM10 in human peripheral blood monocytes and J774 macrophages. Respir Res. 2004;5:29. doi: 10.1186/1465-9921-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campen MJ, Lund AK, Knuckles TL, Conklin DJ, Bishop B, et al. Inhaled diesel emissions alter atherosclerotic plaque composition in ApoE(−/−) mice. Toxicol Appl Pharmacol. 2010;242(3):310–317. doi: 10.1016/j.taap.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LC, Nadziejko C. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal Toxicol. 2005;17(4–5):217–224. doi: 10.1080/08958370590912815. [DOI] [PubMed] [Google Scholar]
- Chen LC, Quan C, Hwang JS, Jin X, Li Q, et al. Atherosclerosis lesion progression during inhalation exposure to environmental tobacco smoke: a comparison to concentrated ambient air fine particles exposure. Inhal Toxicol. 2010;22(6):449–459. doi: 10.3109/08958370903373845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio C-P, Chen S-C, Chiang K-C, Chou W-C, Liao C-M. Oxidative stress risk analysis for exposure to diesel exhaust particle-induced reactive oxygen species. Sci Total Environ. 2007;387(1–3):113–127. doi: 10.1016/j.scitotenv.2007.07.045. [DOI] [PubMed] [Google Scholar]
- Choi JH, Xu QS, Park SY, Kim JH, Hwang SS, et al. Seasonal variation of effect of air pollution on blood pressure. J Epidemiol Community Health. 2007;61(4):314–318. doi: 10.1136/jech.2006.049205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang GC, Yang Z, Westbrook DG, Pompilius M, Ballinger CA, et al. Pulmonary ozone exposure induces vascular dysfunction, mitochondrial damage, and atherogenesis. Am J Physiol Lung Cell Mol Physiol. 2009;297(2):L209–216. doi: 10.1152/ajplung.00102.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang KJ, Chan CC, Su TC, Lee CT, Tang CS. The effect of urban air pollution on inflammation, oxidative stress, coagulation, and autonomic dysfunction in young adults. Am J Respir Crit Care Med. 2007;176(4):370–376. doi: 10.1164/rccm.200611-1627OC. [DOI] [PubMed] [Google Scholar]
- Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109(23):27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- Delfino RJ, Staimer N, Tjoa T, Gillen DL, Polidori A, et al. Air pollution exposures and circulating biomarkers of effect in a susceptible population: clues to potential causal component mixtures and mechanisms. Environ Health Perspect. 2009;117(8):1232–1238. doi: 10.1289/journla.ehp.0800194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino RJ, Staimer N, Tjoa T, Polidori A, Arhami M, et al. Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ Health Perspect. 2008;116(7):898–906. doi: 10.1289/ehp.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez Roux AV, Auchincloss AH, Franklin TG, Raghunathan T, Barr RG, et al. Long-term exposure to ambient particulate matter and prevalence of subclinical atherosclerosis in the multi-ethnic study of atherosclerosis. Am J Epidemiol. 2008;167(6):667–675. doi: 10.1093/aje/kwm359. [DOI] [PubMed] [Google Scholar]
- Dubowsky SD, Suh H, Schwartz J, Coull BA, Gold DR. Diabetes, obesity, and hypertension may enhance associations between air pollution and markers of systemic inflammation. Environ Health Perspect. 2006;114(7):992–998. doi: 10.1289/ehp.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvonch JT, Kannan S, Schulz AJ, Keeler GJ, Mentz G, et al. Acute effects of ambient particulate matter on blood pressure: differential effects across urban communities. Hypertension. 2009;53(5):853–859. doi: 10.1161/HYPERTENSIONAHA.108.123877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd HS, Chen LC, Vallanat B, Dreher K. Fine ambient air particulate matter exposure induces molecular alterations associated with vascular disease progression within plaques of atherosclerotic susceptible mice. Inhal Toxicol. 2009;21(5):394–403. doi: 10.1080/08958370802317745. [DOI] [PubMed] [Google Scholar]
- Fujii T, Hayashi S, Hogg JC, Mukae H, Suwa T, et al. Interaction of alveolar macrophages and airway epithelial cells following exposure to particulate matter produces mediators that stimulate the bone marrow. Am J Respir Cell Mol Biol. 2002;27(1):34–41. doi: 10.1165/ajrcmb.27.1.4787. [DOI] [PubMed] [Google Scholar]
- Goldsmith CA, Imrich A, Danaee H, Ning YY, Kobzik L. Analysis of air pollution particulate-mediated oxidant stress in alveolar macrophages. J Toxicol Environ Health A. 1998;54(7):529–545. doi: 10.1080/009841098158683. [DOI] [PubMed] [Google Scholar]
- Gong KW, Zhao W, Li N, Barajas B, Kleinman M, et al. Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol. 2007;8(7):R149. doi: 10.1186/gb-2007-8-7-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Furuyama A, Koike E, Kobayashi T. Oxidative-stress potency of organic extracts of diesel exhaust and urban fine particles in rat heart microvessel endothelial cells. Toxicology. 2003;187(2–3):161–170. doi: 10.1016/S0300-483X(03)00053-2. [DOI] [PubMed] [Google Scholar]
- Hiura TS, Kaszubowski MP, Li N, Nel AE. Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. J Immunol. 1999;163(10):5582–5591. [PubMed] [Google Scholar]
- Hoffmann B, Moebus S, Mohlenkamp S, Stang A, Lehmann N, et al. Residential exposure to traffic is associated with coronary atherosclerosis. Circulation. 2007;116(5):489–496. doi: 10.1161/CIRCULATIONAHA.107.693622. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Moebus S, Kroger K, Stang A, Mohlenkamp S et al (2009) Residential exposure to urban air pollution, ankle-brachial index, and peripheral arterial disease. Epidemiology 20(2):280–288 [DOI] [PubMed]
- Huang YC, Li Z, Carter JD, Soukup JM, Schwartz DA, et al. Fine ambient particles induce oxidative stress and metal binding genes in human alveolar macrophages. Am J Respir Cell Mol Biol. 2009;41(5):544–552. doi: 10.1165/rcmb.2008-0064OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibald-Mulli A, Stieber J, Wichmann HE, Koenig W, Peters A. Effects of air pollution on blood pressure: a population-based approach. Am J Public Health. 2001;91(4):571–577. doi: 10.2105/AJPH.91.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imrich A, Ning Y, Lawrence J, Coull B, Gitin E, et al. Alveolar macrophage cytokine response to air pollution particles: oxidant mechanisms. Toxicol Appl Pharmacol. 2007;218(3):256–264. doi: 10.1016/j.taap.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Hayashi S, Hogg JC, Fujii T, Goto Y, et al. Alveolar macrophage-epithelial cell interaction following exposure to atmospheric particles induces the release of mediators involved in monocyte mobilization and recruitment. Respir Res. 2005;6:87. doi: 10.1186/1465-9921-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez LA, Drost EM, Gilmour PS, Rahman I, Antonicelli F, et al. PM(10)-exposed macrophages stimulate a proinflammatory response in lung epithelial cells via TNF-alpha. Am J Physiol Lung Cell Mol Physiol. 2002;282(2):L237–248. doi: 10.1152/ajplung.00024.2001. [DOI] [PubMed] [Google Scholar]
- Kunzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, et al. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect. 2005;113(2):201–206. doi: 10.1289/ehp.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzli N, Jerrett M, Garcia-Esteban R, Basagana X, Beckermann B, et al. Ambient air pollution and the progression of atherosclerosis in adults. PLoS ONE. 2010;5(2):e9096. doi: 10.1371/journal.pone.0009096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Kang JJ. Extract of motorcycle exhaust particles induced macrophages apoptosis by calcium-dependent manner. Chem Res Toxicol. 2002;15(12):1534–1542. doi: 10.1021/tx0255727. [DOI] [PubMed] [Google Scholar]
- Lewis JG, Graham DG, Valentine WM, Morris RW, Morgan DL, et al. Exposure of C57BL/6 mice to carbon disulfide induces early lesions of atherosclerosis and enhances arterial fatty deposits induced by a high fat diet. Toxicol Sci. 1999;49(1):124–132. doi: 10.1093/toxsci/49.1.124. [DOI] [PubMed] [Google Scholar]
- Li N, Wang M, Oberley TD, Sempf JM, Nel AE. Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. J Immunol. 2002;169(8):4531–4541. doi: 10.4049/jimmunol.169.8.4531. [DOI] [PubMed] [Google Scholar]
- Li N, Sioutas C, Cho A, Schmitz D, Misra C, et al. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. 2003;111(4):455–460. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173(5):3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- Li R, Ning Z, Majumdar R, Cui J, Takabe W, et al. Ultrafine particles from diesel vehicle emissions at different driving cycles induce differential vascular pro-inflammatory responses: implication of chemical components and NF-kappaB signaling. Part Fibre Toxicol. 2010;7:6. doi: 10.1186/1743-8977-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Ning Z, Cui J, Khalsa B, Ai L, et al. Ultrafine particles from diesel engines induce vascular oxidative stress via JNK activation. Free Radic Biol Med. 2009;46(6):775–782. doi: 10.1016/j.freeradbiomed.2008.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hyseni X, Carter JD, Soukup JM, Dailey LA, et al. Pollutant particles enhanced H2O2 production from NAD(P)H oxidase and mitochondria in human pulmonary artery endothelial cells. Am J Physiol Cell Physiol. 2006;291(2):C357–365. doi: 10.1152/ajpcell.00365.2005. [DOI] [PubMed] [Google Scholar]
- Lippmann M, Frampton M, Schwartz J, Dockery D, Schlesinger R, et al. The US Environmental Protection Agency particulate matter health effects research centers program: a midcourse report of status, progress, and plans. Environ Health Perspect. 2003;111:1074–1092. doi: 10.1289/ehp.5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Ruddy T, Dalipaj M, Poon R, Szyszkowicz M, et al. Effects of indoor, outdoor, and personal exposure to particulate air pollution on cardiovascular physiology and systemic mediators in seniors. J Occup Environ Med. 2009;51(9):1088–1098. doi: 10.1097/JOM.0b013e3181b35144. [DOI] [PubMed] [Google Scholar]
- Lund AK, Knuckles TL, Obot Akata C, Shohet R, McDonald JD, et al. Gasoline exhaust emissions induce vascular remodeling pathways involved in atherosclerosis. Toxicol Sci. 2007;95(2):485–494. doi: 10.1093/toxsci/kfl145. [DOI] [PubMed] [Google Scholar]
- Medina-Navarro R, Lifshitz A, Wacher N, Hicks JJ. Changes in human serum antioxidant capacity and peroxidation after four months of exposure to air pollutants. Arch Med Res. 1997;28(2):205–208. [PubMed] [Google Scholar]
- Mo Y, Wan R, Chien S, Tollerud DJ, Zhang Q. Activation of endothelial cells after exposure to ambient ultrafine particles: the role of NADPH oxidase. Toxicol Appl Pharmacol. 2009;236(2):183–193. doi: 10.1016/j.taap.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Moller P, Loft S (2010) Oxidative Damage to DNA and Lipids as Biomarkers of Exposure to Air Pollution. Environ Health Perspect 118(8): doi:10.1289/ehp.0901725. [DOI] [PMC free article] [PubMed]
- Mondal K, Stephen Haskill J, Becker S. Adhesion and pollution particleinduced oxidant generation is neither necessary nor sufficient for cytokine induction in human alveolar macrophages. Am J Respir Cell Mol Biol. 2000;22(2):200–208. doi: 10.1165/ajrcmb.22.2.3661. [DOI] [PubMed] [Google Scholar]
- Monn C, Becker S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol Appl Pharmacol. 1999;155(3):245–252. doi: 10.1006/taap.1998.8591. [DOI] [PubMed] [Google Scholar]
- Montiel-Davalos A, Alfaro-Moreno E, Lopez-Marure R. PM2.5 and PM10 induce the expression of adhesion molecules and the adhesion of monocytic cells to human umbilical vein endothelial cells. Inhal Toxicol. 2007;19(1):91–98. doi: 10.1080/08958370701495212. [DOI] [PubMed] [Google Scholar]
- Montiel-Davalos A, Ibarra-Sanchez Mde J, Ventura-Gallegos JL, Alfaro-Moreno E, Lopez-Marure R. Oxidative stress and apoptosis are induced in human endothelial cells exposed to urban particulate matter. Toxicol In Vitro. 2010;24(1):135–141. doi: 10.1016/j.tiv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, et al. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J Clin Invest. 2007;117(10):2952–2961. doi: 10.1172/JCI30639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadadur SS, Haykal-Coates N, Mudipalli A, Costa DL. Endothelial effects of emission source particles: acute toxic response gene expression profiles. Toxicol In Vitro. 2009;23(1):67–77. doi: 10.1016/j.tiv.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa Y, Hiura Y, Murayama T, Yokode M, Iwai N. Nano-sized carbon black exposure exacerbates atherosclerosis in LDL-receptor knockout mice. Circ J. 2007;71(7):1157–1161. doi: 10.1253/circj.71.1157. [DOI] [PubMed] [Google Scholar]
- Ntziachristos L, Froines J, Cho A, Sioutas C. Relationship between redox activity and chemical speciation of size-fractionated particulate matter. Particle Fib Toxic. 2007;4(1):5. doi: 10.1186/1743-8977-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurkiewicz TR, Porter DW, Barger M, Castranova V, Boegehold MA. Particulate matter exposure impairs systemic microvascular endothelium-dependent dilation. Environ Health Perspect. 2004;112(13):1299–1306. doi: 10.1289/ehp.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurkiewicz TR, Porter DW, Barger M, Millecchia L, Rao KM, et al. Systemic microvascular dysfunction and inflammation after pulmonary particulate matter exposure. Environ Health Perspect. 2006;114(3):412–419. doi: 10.1289/ehp.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill MS, Veves A, Zanobetti A, Sarnat JA, Gold DR, et al. Diabetes enhances vulnerability to particulate air pollution-associated impairment in vascular reactivity and endothelial function. Circulation. 2005;111(22):2913–2920. doi: 10.1161/CIRCULATIONAHA.104.517110. [DOI] [PubMed] [Google Scholar]
- O'Neill MS, Veves A, Sarnat JA, Zanobetti A, Gold DR, et al. Air pollution and inflammation in type 2 diabetes: a mechanism for susceptibility. Occup Environ Med. 2007;64(6):373–379. doi: 10.1136/oem.2006.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama M, Otake T, Adachi S, Kobayashi T, Morinaga K. A comparison of the production of reactive oxygen species by suspended particulate matter and diesel exhaust particles with macrophages. Inhal Toxicol. 2007;19(Suppl 1):157–160. doi: 10.1080/08958370701496103. [DOI] [PubMed] [Google Scholar]
- Osornio-Vargas AR, Bonner JC, Alfaro-Moreno E, Martinez L, Garcia-Cuellar C, et al. Proinflammatory and cytotoxic effects of Mexico City air pollution particulate matter in vitro are dependent on particle size and composition. Environ Health Perspect. 2003;111(10):1289–1293. doi: 10.1289/ehp.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, O'Neill MS, Vokonas PS, Sparrow D, Spiro A, 3rd et al (2008) Traffic-related particles are associated with elevated homocysteine: the VA normative aging study. Am J Respir Crit Care Med 178(3):283–289 [DOI] [PMC free article] [PubMed]
- Peters A, Veronesi B, Calderon-Garciduenas L, Gehr P, Chen L et al (2006) Translocation and potential neurological effects of fine and ultrafine particles a critical update. Particle and Fibre Toxicology 3(1):13 [DOI] [PMC free article] [PubMed]
- Pope CA, 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, et al. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109(1):71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- Pozzi R, De Berardis B, Paoletti L, Guastadisegni C. Inflammatory mediators induced by coarse (PM2.5–10) and fine (PM2.5) urban air particles in RAW 264.7 cells. Toxicology. 2003;183(1–3):243–254. doi: 10.1016/S0300-483X(02)00545-0. [DOI] [PubMed] [Google Scholar]
- Quan C, Sun Q, Lippmann M, Chen LC. Comparative effects of inhaled diesel exhaust and ambient fine particles on inflammation, atherosclerosis, and vascular dysfunction. Inhal Toxicol. 2010;22(9):738–753. doi: 10.3109/08958371003728057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riediker M, Cascio WE, Griggs TR, Herbst MC, Bromberg PA. Particulate matter exposure in cars is associated with cardiovascular effects in healthy young men. Am J Respir Crit Care Med. 2004;169(8):934–940. doi: 10.1164/rccm.200310-1463OC. [DOI] [PubMed] [Google Scholar]
- Romieu I, Garcia-Esteban R, Sunyer J, Rios C, Alcaraz-Zubeldia M et al (2008) The effect of supplementation with omega-3 polyunsaturated fatty acids on markers of oxidative stress in elderly exposed to PM(2.5). Environ Health Perspect 116(9): 1237–1242 [DOI] [PMC free article] [PubMed]
- Ruckerl R, Phipps RP, Schneider A, Frampton M, Cyrys J, et al. Ultrafine particles and platelet activation in patients with coronary heart disease—results from a prospective panel study. Part Fibre Toxicol. 2007;4:1. doi: 10.1186/1743-8977-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundell KW, Hoffman JR, Caviston R, Bulbulian R, Hollenbach AM. Inhalation of ultrafine and fine particulate matter disrupts systemic vascular function. Inhal Toxicol. 2007;19(2):133–140. doi: 10.1080/08958370601051727. [DOI] [PubMed] [Google Scholar]
- Sanchez-Rodriguez MA, Retana-Ugalde R, Ruiz-Ramos M, Munoz-Sanchez JL, Vargas-Guadarrama LA, et al. Efficient antioxidant capacity against lipid peroxide levels in healthy elderly of Mexico City. Environ Res. 2005;97(3):322–329. doi: 10.1016/j.envres.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Soares SR, Carvalho-Oliveira R, Ramos-Sanchez E, Catanozi S, da Silva LF, et al. Air pollution and antibodies against modified lipoproteins are associated with atherosclerosis and vascular remodeling in hyperlipemic mice. Atherosclerosis. 2009;207(2):368–373. doi: 10.1016/j.atherosclerosis.2009.04.041. [DOI] [PubMed] [Google Scholar]
- Sorensen M, Daneshvar B, Hansen M, Dragsted LO, Hertel O, et al. Personal PM2.5 exposure and markers of oxidative stress in blood. Environ Health Perspect. 2003;111(2):161–166. doi: 10.1289/ehp.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup JM, Ghio AJ, Becker S. Soluble components of Utah Valley particulate pollution alter alveolar macrophage function in vivo and in vitro. Inhal Toxicol. 2000;12(5):401–414. doi: 10.1080/089583700196112. [DOI] [PubMed] [Google Scholar]
- Sumanasekera WK, Ivanova MM, Johnston BJ, Dougherty SM, Sumanasekera GU, et al. Rapid effects of diesel exhaust particulate extracts on intracellular signaling in human endothelial cells. Toxicol Lett. 2007;174(1–3):61–73. doi: 10.1016/j.toxlet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294(23):3003–3010. doi: 10.1001/jama.294.23.3003. [DOI] [PubMed] [Google Scholar]
- Sun Q, Yue P, Kirk RI, Wang A, Moatti D, et al. Ambient air particulate matter exposure and tissue factor expression in atherosclerosis. Inhal Toxicol. 2008;20(2):127–137. doi: 10.1080/08958370701821482. [DOI] [PubMed] [Google Scholar]
- Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation. 2009;119(4):538–546. doi: 10.1161/CIRCULATIONAHA.108.799015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwa T, Hogg JC, Quinlan KB, Ohgami A, Vincent R, et al. Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol. 2002;39(6):935–942. doi: 10.1016/S0735-1097(02)01715-1. [DOI] [PubMed] [Google Scholar]
- Tabas I. Apoptosis and efferocytosis in mouse models of atherosclerosis. Curr Drug Targets. 2007;8(12):1288–1296. doi: 10.2174/138945007783220623. [DOI] [PubMed] [Google Scholar]
- Tamagawa E, Bai N, Morimoto K, Gray C, Mui T, et al. Particulate matter exposure induces persistent lung inflammation and endothelial dysfunction. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L79–85. doi: 10.1152/ajplung.00048.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S.EPA . Air quality criteria for particulate matter (final report, Oct 2004) Washington: US Environmental Protection Agency; 2004. [Google Scholar]
- van Eeden SF, Tan WC, Suwa T, Mukae H, Terashima T, et al. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)) Am J Respir Crit Care Med. 2001;164(5):826–830. doi: 10.1164/ajrccm.164.5.2010160. [DOI] [PubMed] [Google Scholar]
- Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hama S, et al. Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J Lipid Res. 2007;48(11):2344–2353. doi: 10.1194/jlr.M700138-JLR200. [DOI] [PubMed] [Google Scholar]
- Verheyen GR, Nuijten JM, Van Hummelen P, Schoeters GR. Microarray analysis of the effect of diesel exhaust particles on in vitro cultured macrophages. Toxicol In Vitro. 2004;18(3):377–391. doi: 10.1016/j.tiv.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Vinzents PS, Moller P, Sorensen M, Knudsen LE, Hertel O et al (2005) Personal exposure to ultrafine particles and oxidative DNA damage. Environ Health Perspect 113(11):1485–1490 [DOI] [PMC free article] [PubMed]
- Wang T, Chiang ET, Moreno-Vinasco L, Lang GD, Pendyala S, et al. Particulate matter disrupts human lung endothelial barrier integrity via ROS- and p38 MAPK-dependent pathways. Am J Respir Cell Mol Biol. 2010;42(4):442–449. doi: 10.1165/rcmb.2008-0402OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann HE, Spix C, Tuch T, Wolke G, Peters A, et al. Daily mortality and fine and ultrafine particles in Erfurt, Germany part I: role of particle number and particle mass. Res Rep Health Eff Inst. 2000;98:5–86. [PubMed] [Google Scholar]
- Yamawaki H, Iwai N. Mechanisms underlying nano-sized air-pollution-mediated progression of atherosclerosis: carbon black causes cytotoxic injury/inflammation and inhibits cell growth in vascular endothelial cells. Circ J. 2006;70(1):129–140. doi: 10.1253/circj.70.129. [DOI] [PubMed] [Google Scholar]
- Yatera K, Hsieh J, Hogg JC, Tranfield E, Suzuki H, et al. Particulate matter air pollution exposure promotes recruitment of monocytes into atherosclerotic plaques. Am J Physiol Heart Circ Physiol. 2008;294(2):H944–953. doi: 10.1152/ajpheart.00406.2007. [DOI] [PubMed] [Google Scholar]
- Ying Z, Kampfrath T, Thurston G, Farrar B, Lippmann M, et al. Ambient particulates alter vascular function through induction of reactive oxygen and nitrogen species. Toxicol Sci. 2009;111(1):80–88. doi: 10.1093/toxsci/kfp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Yue P, Xu X, Zhong M, Sun Q, et al. Air pollution and cardiac remodeling: a role for RhoA/Rho-kinase. Am J Physiol Heart Circ Physiol. 2009;296(5):H1540–1550. doi: 10.1152/ajpheart.01270.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue W, Schneider A, Stolzel M, Ruckerl R, Cyrys J, et al. Ambient source-specific particles are associated with prolonged repolarization and increased levels of inflammation in male coronary artery disease patients. Mutat Res. 2007;621(1–2):50–60. doi: 10.1016/j.mrfmmm.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Zanobetti A, Schwartz J. The effect of fine and coarse particulate air pollution on mortality: a national analysis. Environ Health Perspect. 2009;117(6):898–903. doi: 10.1289/ehp.0800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeka A, Sullivan JR, Vokonas PS, Sparrow D, Schwartz J (2006) Inflammatory markers and particulate air pollution: characterizing the pathway to disease. Int J Epidemiol 35(5):1347–1354 [DOI] [PubMed]