


Abstract
Guide RNAs (gRNAs), key components of the RNA editing reaction in Trypanosoma brucei, direct the insertion and deletion of uridylate (U) residues. Analyses of gRNAs reveal three functional elements. The 5′-end of the gRNA contains the anchor, which is responsible for selection and binding to the pre-edited mRNA. The second element (the guiding region) provides the information required for editing. At the 3′-end of the gRNA is a non-encoded U-tail, whose function remains unclear. However, the cleavage–ligation model for editing proposes that the U-tail binds to purine-rich regions upstream of editing sites, thereby strengthening the interaction and holding onto the 5′ cleavage product. Our previous studies demonstrated that the U-tail interacts with upstream sequences and may play roles in both stabilization and tethering. These studies also indicated that the U-tail interactions involved mRNA regions that were to be subsequently edited. This raised the question of what happens to the mRNA–U-tail interaction as editing proceeds in the 3′→5′ direction. We examined gCYb-558 and its U-tail interaction with 5′CYbUT and two partially edited 5′CYb substrates. Our results indicate that the 3′-end of the U-tail interacts with the same sequence in all three mRNAs. Predicted secondary structures using crosslinking data suggest that a similar structure is maintained as editing proceeds. These results indicate that the role of the U-tail may also involve maintenance of important secondary structure motifs.
INTRODUCTION
In kinetoplastid protozoa, several mitochondrial mRNAs undergo RNA editing (1–4). In this essential phenomenon, uridylate (U) residues are precisely inserted into and deleted from mitochondrial pre-mRNAs to produce translatable mature mRNAs. This post-transcriptional process is directed by guide RNAs (gRNAs) which are small RNA molecules (55–70 nt) (5). The specificity for the gRNA–pre-mRNA interaction is provided by a 5′ anchor sequence in the gRNA. This anchor is complementary to the mRNA sequence immediately downstream of the region to be edited. At the 3′-end of the gRNA is a post-transcriptionally added poly U tail (U-tail) that has been shown to interact with purine-rich mRNA sequences (6,7). Between the anchor and the U-tail is the information sequence, which determines the sequence of the mature mRNA.
The U-tail is particularly interesting, as its functions have not been clearly defined. Several roles have been previously proposed by various models of editing. In the cleavage–ligation model for editing, it is hypothesized that the U-tail helps to stabilize the interaction of the gRNA and mRNA by binding to purine-rich regions upstream of editing sites (ESs) (5,6). In vitro studies by Seiwert et al. (8) also demonstrated that removal of the U-tail did not reduce gRNA-directed cleavage of the mRNA. However, formation of the edited product was strongly suppressed. This led to the hypothesis that in addition to stability, the U-tail was involved in tethering the 5′ cleavage product during the editing reaction. Our previous crosslinking study of three different gRNA–mRNA pairs provided direct evidence that the U-tail was binding to the upstream purine-rich sequences (7). The crosslinking data indicated that the U-tail interacted with upstream purines, just 5–28 nt upstream of the anchor duplex. Although the crosslinking data identified a favored crosslinking site, the data did suggest that the U-tail was able to interact with a range of upstream sequences. Incorporation of this data into computer-predicted secondary structure models of the different gRNA–mRNA pairs generated structures that were very similar. In all cases, the anchor duplex region was correctly paired and the secondary structure in the mRNA editing domain eliminated. In addition, the gRNA guiding region formed a stem–loop positioned across from the first few ESs. These results suggest that the U-tail may act not only to increase the stability of the RNA interactions, but may also work to ‘iron out’ any secondary structure in the mRNA in the immediate editing domain, possibly increasing the accessibility of the editing complex to the proper ESs. In addition, the formation of common structures suggests that the U-tail may aid in the formation of common core architecture important for the assembly of an editing complex.
Crosslinking of the 3′-end of the U-tail with sequences near the first ES indicated that the U-tail was interacting with mRNA regions that were to be subsequently edited. This raised the interesting question as to what happens to the mRNA–U-tail interaction as editing proceeds in the 3′→5′ direction. To examine this question, we looked at gCYb-558 and analyzed its interaction with three apocytochrome b (CYb) mRNA substrates: 5′CYbUT, which is unedited, and two partially edited substrates (PES), 5′CYbPES1T and 5′CYbPES3T, which have ES1–ES3 fully edited, respectively. By placing an azidophenacyl (APA) group at the 3′-end of gCYb-558, we were able to use photoaffinity crosslinking techniques to examine how the changes in sequence caused by the editing reaction affect the positioning of the U-tail. Surprisingly, reverse transcriptase (RT) analyses of the major crosslinked species for all three different CYb substrates consistently revealed strong termination products at the same five bases. This region is the same region previously identified as being involved in U-tail binding and is located only 4–8 nt upstream of the growing anchor in the most edited substrate. This is striking because editing through ES3 requires the addition of six U residues and essentially doubles the length of the gRNA–mRNA duplex. Using this crosslink data, secondary structure models suggest that the gRNA stem–loop is maintained as editing proceeds through ES3. This is made possible by incorporating part of the U-tail into the stem–loop. Therefore, as editing proceeds, the U-tail may have the additional role of maintaining important secondary structure motifs.
We have also shown that 3′-crosslinked 5′CYbUT and gCYb-558 molecules are biologically relevant as they are recognized and specifically cleaved by the gRNA-directed endonuclease at the correct ES. This demonstrates the usefulness of these crosslinked molecules in the development of a model for gRNA and mRNA interactions in editing.
MATERIALS AND METHODS
Oligodeoxynucleotides
Big SK, 5′-GGCCGCTCTAGAACTAGTGG-3′ (20 nt); T7, 5′-AATTAATACGACTCACTATAG-3′ (22 nt); CYbCS BamHI, 5′-CCGGATCCATATATTCTATATAAACAACC-3′ (29 nt); CYbN, 5′-GGAGGTACCGTTAAGAATAATGGTTATAAATTTTATATAA-3′ (40 nt); CYbH-1, 5′-CAACCTGACATT-3′ (12 nt); CYbH-2, 5′-ACCATTATTCT-3′ (11 nt); CYbPES1, 5′-CTATATAAACAACCTGACATTAAAAGACAACCTTTCTTTTTTC-3′ (43 nt); CYbPES3, 5′-CTATATAAACAACCTGACATTAAAAGACAACACAAATTTCTT-TTTTC-3′ (47 nt); NgCYb-558(sU), 5′-TTATTCCCTTTATCACCTAGAAATTCACATTGTCTTTTAATCCCTATAGTGAGTCGTATTAAATT-3′ (65 nt); NgCYb-558B, 5′-AAAAAAAAAAAAAAATTATTCCCTTTATCA-3′ (30 nt).
DNA templates and RNA synthesis
5′CYbUT has been described previously (9). Partially edited 5′CYb substrates were created using PCR. To synthesize 5′CYbPES1T and 5′CYbPES3T, the 5′CYbUT DNA template was amplified using T7 and CYbPES1 or CYbPES3 oligodeoxyribonucleotides, respectively. The PCR products obtained from this step were re-amplified using 5′CYbN and CYbCS oligonucleotides. These PCR products were subjected to BamHI and KpnI digestion and cloned into pBluescript SK– (Stratagene). Templates for transcription were obtained from the appropriate plasmids using T7 and Big SK oligonucleotides for PCR. 5′CYbUT and the partially edited RNAs were synthesized by T7 RNA polymerase using a Ribomax kit (Promega) according to manufacturer’s instructions. mRNAs were gel purified on 6% denaturing polyacrylamide gels. The RNAs were passively eluted in 10 mM Tris pH 7.8, 0.1% SDS, 2 mM EDTA and 0.3 M NaOAc pH 7.0. gCYb-558 RNA was synthesized using NgCYb-558sU and T7 oligodeoxyribonucleotides via the Uhlenbeck single-stranded T7 transcription method (10). The sequence for the oligodeoxyribonucleotide template for gCYb-558 was redesigned to more closely match the native gRNA sequence without a U-tail (11). To improve the homogeneity at the 3′-end of the transcribed gRNA, transcription was carried out under low Mg2+ conditions (10 mM) (30 mM HEPES pH 8.0, 3 mM Spermidine, 10 mM DTT and 5 mM KCl). A U15-tail was then added to the gRNA by ligation of a U15 RNA oligonucleotide (Dharmacon) using a bridging oligodeoxyribonucleotide (NgCYb-558B) and T4 DNA ligase (12). Before ligation, the U15 RNA oligonucleotide was 5′-end-labeled with [32P]ATP. This involved drying down 1.8 nmol of U15 RNA oligonucleotide, 250 µCi of [γ-32P]ATP and 5.25 nmol of cold ATP. The pellet was resuspended in 20 µl of 1× T4 DNA kinase buffer and 75 U of T4 DNA kinase (NEB). Kinase reactions were incubated at 37°C for 1–1.5 h. To inactivate the kinase, the reaction was incubated at 65°C for 20 min. Equimolar amounts of NgCYb-558B (bridging oligonucleotide) and NgCYb-558sU (no U-tail) RNA were added and heated to 70°C for 2 min. The molecules were annealed by cooling to 37°C at a rate of 2°C/min. The reaction conditions for the ligase reaction were as follows: 1× T4 DNA ligase buffer, 35 U of T4 DNA ligase (Boehringer Mannheim), 15% PEG 8000 and 120 U of RNasin (Promega). The ligation was incubated overnight at room temperature. The full-length product was then gel purified on an 8% denaturing polyacrylamide gel and recovered.
RNA modifications
Photoagent attachment, crosslinking of gRNAs and mRNAs and primer extension mapping were described previously (7). The mRNA to gRNA ratio was 10:1, using a gRNA concentration of 2.5–3.0 µM. Efficiency of crosslinking was quantitated using a Storm PhosphorImager (Molecular Dynamics).
RNase H analysis
Each RNase H reaction contained a total of 10 pmol of RNA. If necessary, crosslinks (gCYb-558 was already radioactively labeled as above) and uniformly labeled mRNAs were supplemented with the appropriate cold mRNA to make a total of 10 pmol of RNA per reaction. 5′-end-labeled crosslinks and uniformly labeled mRNAs were incubated with 20–30 pmol of each oligodeoxyribonucleotide in 120 mM HEPES pH 8.0, 210 mM NH4Cl, 7 mM MgCl2, 0.75 mM DTT and 5 U of RNase H (Takara). Reactions were incubated at 55°C for 30 min. Products were run out on a 6% denaturing gel and subjected to autoradiography for analysis.
Cleavage reactions
The mRNA of crosslinks (0.25–0.5 pmol) were 3′-end-labeled with 10 µCi of [5′-32P]pCp using T4 RNA ligase as described by Wahle and Keller (13). Only the mRNA was end-labeled in this process as the 3′-end of the gRNA was crosslinked to the mRNA. Glycerol gradients were obtained as described previously (8,14). RNAs were heated to 60°C and cooled slowly to 27°C at 2°C/min before the addition of 10 µl of an active glycerol fraction. Reaction conditions were as follows: 20 mM HEPES pH 7.9, 50 mM KCl, 10 mM MgOAc, 0.05 mM DTT, 1 mM EDTA and 5 mM CaCl2. Aliquots of 0.5–2 fmol of crosslink or 3.5–20 fmol of 5′CYbUT were used in cleavage reactions. Excess (10–20-fold) gCYb-558 was used where specified. Control lane conditions were identical except no mitochondrial proteins were added. A T1 ladder was created by incubating 3′-end-labeled 5′CYbUT (60 000 c.p.m., 1 fmol) in 12.8 M urea, 40 mM sodium citrate pH 8.3 and 2 mM EDTA pH 8.0 with 2 U of T1 ribonuclease (Boehringer Mannheim) for 2 min at 55°C. Reaction products were run out on an 8% denaturing polyacrylamide gel and exposed to film.
Secondary structure predictions
The program RNAstructure v. 3.5 (15) was used to predict secondary structures. Crosslink data were used to force the last U to basepair with the appropriate base in the mRNA. RNAdraw was then used to graphically display the connect files (16). Lower case a’s were used to create a single molecule for the RNAstructure program as described previously (7).
RESULTS
CYb substrates and gCYb-558
The editing of apocytochrome b pre-mRNA is limited to a small region near its 5′-end and is a developmentally regulated process. Editing inserts 34 U’s over 13 sites only during the procyclic (insect) and stumpy bloodstream stages of the trypanosome life cycle (17). Maturation of the first seven ESs is guided by gCYb-558, which directs the insertion of 21 U’s. gCYb-558 is 59 nt long (including a U15-tail) and is able to interact with unedited CYb via an anchor of 13 nt with one mismatch. The unedited mRNA used in this study, 5′CYbUT, contains 88 nt of the 5′-end of CYb and has been described previously (7,9). The partially edited substrates, CYbPES1T and CYbPES3T, are identical in sequence to CYbUT except for the editing events at sites 1–3. CYbPES1T is edited at site 1 by the addition of two U’s, extending the anchor duplex region by 3 bp. CYbPES3T is fully edited at sites 1–3, with a total of six U insertions. These editing events extend the anchor duplex by 13 bp (Fig. 1A–C).
Figure 1.

Sequence of the 5′CYb mRNA substrates in the editing region (A–C) and full-length gCYb-558 (D). Bold U’s, those added to create partially edited substrates. The gRNA–mRNA anchor is shown for each 5′CYb substrate with the gRNA aligned below the mRNA. The base pairing between the gRNA anchor and the mRNA is shown by Watson–Crick (vertical line) and non-Watson–Crick (colon) base pairs. Changes to the sequence of gCYb-558 are underlined (see Materials and Methods).
In our previous crosslinking study, we used gRNAs with a U10-tail. The average length of a gRNA U-tail is approximately 15 residues (6). However, we found that the T7 RNA polymerase used for transcription of the gRNAs stuttered extensively with a U15 template, generating considerable heterogeneity at the 3′-ends of the gRNAs. In pursuing the question of how the U-tail interacts as editing proceeds, we made two improvements to our assay. (i) The sequence of gCYb-558 was redesigned to more closely match that found in vivo (an additional three guiding nucleotides; Fig. 1D) (11). (ii) gCYb-558 is synthesized with T7 RNA polymerase using a template with no U-tail encoded. A U15 RNA oligonucleotide is then ligated to the 3′-end of the gRNA using a deoxyoligonucleotide as a bridge and T4 DNA ligase (12). This method produces a much more homogeneous population of gRNAs for use in our crosslinking studies.
gRNA U-tail interactions
We have previously analyzed gCYb-558–5′CYbUT crosslinks generated using gRNAs with two different U-tail lengths, U10 and U5. Analysis of the most abundant crosslinked species generated with U10 gCYb produced a ladder of termination products beginning just 5′ of the anchor duplex and extending ∼17 nt upstream (7). The strongest stops observed were 14–16 nt upstream of the anchor duplex. Analyses of the crosslinks generated with the U5 gCYb showed a series of termination products that spanned the same nucleotides as those observed with U10. However, the dominant termination products now correlated to crosslinks with the nucleotides that flank the first ES. Crosslinking this close to ES1 indicated that the U-tail was interacting with mRNA regions that were to be subsequently edited by the interacting gRNA. This led us to investigate what happens to the mRNA–U-tail interaction as editing proceeds through this region.
Crosslinks were produced by annealing gCYb-558 (modified with a 3′ APA group on the terminal U) to each of the different CYb substrates and exposing them to 312 nm UV light. Crosslinked RNAs were then separated using denaturing PAGE. As previously observed for the gCYb-558 U10–5′CYbUT crosslinks, a dominant band (B1, the species of slowest mobility) and two minor, faster bands (B2 and B3) were obtained for each mRNA–gCYb-558 combination used. 3′-crosslinking efficiencies were measured for the major B1 bands on a phosphorimager. The efficiencies were very similar for the CYbUT and CYbPES1T substrates (∼1%, average of four different crosslinking experiments). However, the efficiency of crosslinking for the CYbPES3T substrate was much lower (0.34%, data not shown).
The position of the minor B3 crosslink was mapped using RT and confirmed our previous results that this crosslink occurs in the 5′ vector sequence of the mRNA. B2 did not produce any strong terminations. Surprisingly, RT analyses of the dominant B1 species for all three different CYb substrates consistently revealed strong termination products (highlighted by closed circles, see Fig. 2) at the same five bases (C51 G52 G53 A54 G55). These invariable termination products indicate that crosslinking occurs within G52–A56, the RT stops one nucleotide 3′ of a crosslink (18). Termination products just upstream were observed inconsistently (Fig. 2A). A strong stop was also often observed in the middle of the anchor duplex region of 5′CYbPES3T (Fig. 2C). However, RNase H digestion (see below) did not indicate the presence of a crosslink. This termination product is more easily explained by the RT having difficulty reading through the long (26 nt) anchor duplex region. The above crosslinks fall within the same sequence that was previously identified using the slightly different gCYb-558 sequence and the shorter U10-tail. What is remarkable is the fact that the U-tail is interacting only 4–8 nt upstream of the growing anchor in 5′CYbPES3T.
Figure 2.
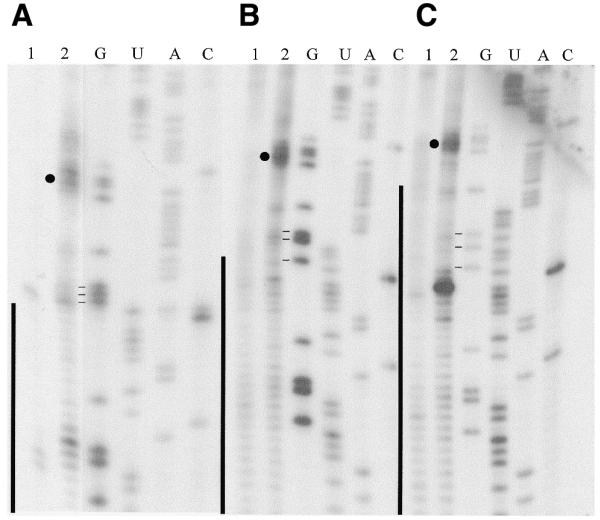
Primer extension analyses of 3′ APA modified gCYb-558 crosslinked to three CYb mRNA substrates. (A) 5′CYbUT, (B) 5′CYbPES1T and (C) 5′CYbPES3T. Lane 1, RT of mRNA alone; lane 2, RT of crosslink. G, U, A and C are sequencing lanes. Vertical lines, anchor duplex. Closed circle, the major crosslink. Horizontal lines, three G’s flanking the first three editing events.
Additional evidence for crosslinks in this region was provided by RNase H digestion. Using several oligonucleotides complementary to the mRNA, two oligonucleotides (CYbH-1 and -2) were found to flank the crosslink (Fig. 3). RNase H digestion with both oligonucleotides removed a total of ∼60 nt from the CYb substrates. Treatment of the crosslink with CYbH-1, -2 or both resulted in an RNA species with a faster mobility. However, the digestion products still ran slower than full-length mRNA (Fig. 3, lanes 5–8). This indicated that gCYb-558 was crosslinked to the mRNA, creating a branched molecule that runs with a slower mobility. This analysis narrowed the location of the crosslink to a region of 43 nt of 5′CYbUT (45 and 49 nt for 5′CYbPES1T and 5′CYbPES3T, respectively) spanning the location of the strong upstream RT termination sites. Complete digestion of the crosslinks was not obtained; hence the lighter, full-length crosslink bands (Fig. 3, lanes 6–8). In lanes 2 and 4, the short products (less than ∼70 nt) are not shown. Similar analyses done with crosslinked 5′CYbUT and 5′CYbPES3T showed identical results (data not shown). RNase H digestion with both CYbH-1 and -2 also indicated that the strong RT termination product observed in the anchor of 5′CYbPES3T was not due to a crosslink. Only a single RNA species was observed after RNase treatment. One would expect that if both termination products were the result of crosslinks, a second RNA species of different mobility would be observed. Furthermore, RNase H digestion of 5′CYbPES3T generated the same pattern of bands with mobilities identical for those observed for 5′CYbUT and 5′CYbPES1T crosslinks, where no RT termination product was observed in the anchor.
Figure 3.
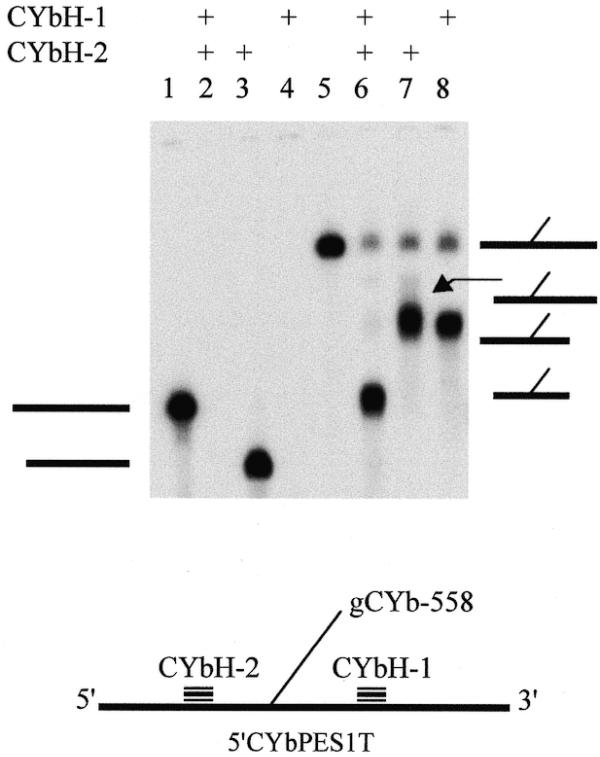
RNase H mapping of gCYb-558 and 5′CYbPES1T crosslink. Lanes 1–4, 5′CYbPES1T mRNA only; lanes 5–8, 3′-crosslinked gCYb-558 and 5′CYbPES1T. All lanes were treated with RNase H. Thick horizontal lines on the left, mRNA alone; thick and thin lines on the right, crosslinked mRNA and gRNA, respectively.
Cleavage of crosslinked substrates
In order to demonstrate that these crosslinks represented biologically relevant molecules, we wanted to determine whether they were substrates for the RNA editing machinery. Direct visualization of editing was not possible due to the branched structure of the crosslinked RNA (8). In addition, detection of editing using the poison primer assay was hindered by the presence of the crosslinked gRNA and its ability to bind to the mRNA via its anchor sequence (19). Therefore, we examined whether these molecules could be accurately cleaved by the gRNA-directed endonuclease previously identified in mitochondrial fractions (20,21).
For these assays, 3′ crosslinked molecules were generated using 32P-trace-labeled gRNAs and unlabeled mRNAs. The 3′-end of purified crosslinked mRNAs were then end-labeled to a high specific activity using T4 RNA ligase and [5′-32P]pCp and again gel-purified. 3′-end-labeled crosslinks (30 000 c.p.m., 0.5–2 fmol) were then incubated in an editing reaction (8,22). UTP and ATP were not included in the reaction mix in order to inhibit ligation and enhance the production of the cleavage product. The 3′-crosslinked molecules were accurately cleaved at ES1 in the presence of active mitochondrial fractions (Fig. 4). Crosslinked 5′CYbUT yielded a 3′ cleavage product that is 59 nt in length as expected for cleavage at ES1 (where two U’s are inserted). This cleavage is consistent with cleavage by the editing endonuclease and not the two other mitochondrial endonucleases identified previously (21). A crosslinked species that ran just below the full-length crosslink was observed very weakly in the control lane, and enhanced in the presence of mitochondrial proteins. This might indicate the presence of a hypersensitive cleavage site in the crosslinked RNA that is susceptible to cleavage by mitochondrial RNases. The location of this cleavage site was not determined. However, if cleavage was in the mRNA, its mobility shows that the cleavage site has to be upstream of the crosslink. It is also possible that cleavage of the gRNA could be responsible for this RNA species as well. An additional product with a mobility of ∼91 nt is also observed in the presence of active lysate. It has not been determined whether this product results from cleavage of free mRNA from broken crosslinks or crosslinked RNA. Cleavage of free mRNA would indicate that the cleavage site is upstream of the CYb editing domain within the 5′-UTR. If the product is a crosslinked molecule, the cleavage site cannot be determined without additional experiments.
Figure 4.
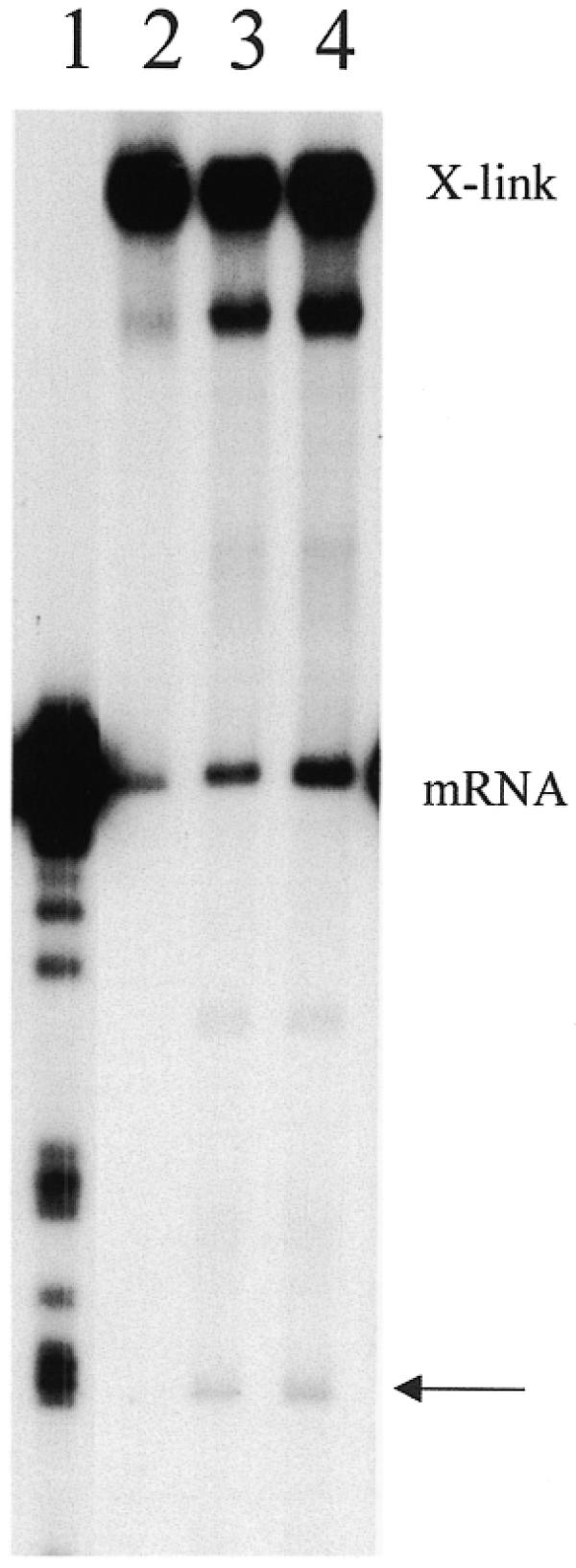
Accurate cleavage of 3′-crosslinked 5′CYbUT and gCYb-558. Crosslinks (mRNA 3′-end-labeled) were assayed for gRNA-directed cleavage using standard cleavage conditions. Lane 1, T1 digest of 5′CYbUT; lanes 2–4, 3′-crosslinked RNAs: lane 2, without mitochondrial fraction; lane 3, with mitochondrial fraction; lane 4, with mitochondrial fraction and a 10-fold excess of free gCYb-558. X-link, 3′-crosslinks; mRNA, free 5′CYbUT from broken crosslinks; arrow, cleavage products.
Isolation and subsequent handling of the crosslinked substrates always results in crosslink breakage and subsequent release of free mRNA (Fig. 4, lanes 2–4). Therefore, we considered whether the released free mRNA could generate the cleavage products. Breakage of the mRNA–gRNA crosslinks would release the two RNAs in equimolar amounts. In the in vitro cleavage assays described to date, generation of cleavage product requires the addition of excess gRNA in order to drive the reaction (20,21,23). No gRNA-directed cleavage of free mRNA is detected when utilizing a 1:1 mRNA:gRNA ratio (data not shown). Efficient cleavage is only observed when the gRNA is supplied in excess. In contrast, cleavage of the crosslinked substrates was relatively efficient in the absence of any added free gRNA (Fig. 4, lane 3) and the addition of free gRNA to the reaction did not increase the efficiency of cleavage (Fig. 4, lane 4). These data indicate that the 3′-crosslinked substrates support accurate gRNA-directed cleavage, suggesting that these crosslinked substrates have been captured in a biologically active state.
Predicted secondary structures
To understand how the U-tail could interact with the same sequence in all three cases, we incorporated the crosslink data into computer-predicted secondary structures using the computer program, RNAstructure v. 3.5 (Fig. 5) (15). This was done by instructing the program to pair the 3′-terminal U of gCYb-558 with G53 of the mRNA, one of the dominant crosslinked nucleotides. The structures predicted were very similar to one another (Fig. 5). The anchor duplex regions are correctly paired and the previously described stem–loop structure formed within the guiding region of the gRNAs (6,7,24) is maintained in all three folds. The predicted structure for gCYb-558 and 5′CYbPES3T (Fig. 5C) is particularly interesting as part of the U-tail is involved in maintaining the stem–loop structure. This shortens the length of the predicted U-tail–mRNA interaction from 13–14 bp (in CYbPES1T and CYbUT) to only 7 bp for 5′CYbPES3T. One would predict that this would weaken the interaction of the U-tail with 5′CYbPES3T and may explain the decrease in U-tail crosslinking efficiency observed with the CYbPES3T substrate.
Figure 5.

Secondary structure predictions incorporating 3′-crosslink data. (A and B) In all three gRNA–mRNA pairs, the 5′- and 3′-ends of the mRNA produced identical folds. (C) The predicted folds for the different gRNA–mRNA pairs highlighting the interaction with the gRNA. The gRNA sequence is in bold and the gRNA–mRNA anchors are underlined. The gRNA and mRNA were linked together via a linker of 10 non-pairing bases (a).
DISCUSSION
Understanding the mechanism and regulation of the RNA editing process involves not only the identification of the RNA and protein components of the system, but also the elucidation of how the individual components interact to function as a complex unit. Both the specificity and the catalytic activity of editing are likely to be dependent on the three-dimensional structure of the ribonucleoprotein complex. The pre-mRNAs to be edited are defined by the proteins they encode and have very different primary sequences. Likewise, the gRNAs have very little in common (with the exception of the 3′ U-tail), as their primary sequence is also defined by the protein sequence they must create. For the common components of the editing machinery to assemble, they must be able to recognize common secondary or tertiary structures formed by the mRNA, the gRNA or the interacting molecules. We have begun to analyze the interactions between gRNAs and mRNAs using comparative photoaffinity crosslinking techniques. In our initial studies utilizing three different gRNA–mRNA pairs, we characterized the initiating gRNA’s U-tail interaction with unedited mRNA sequences. In these studies, we found that the U-tail did interact with upstream purine-rich sequences, preferring purines located near the first few ESs. Interaction of the U-tail in this region prevented the formation of mRNA stem–loop structures in the immediate editing domain possibly increasing the accessibility of this region to the editing machinery. In addition, the data argue for the formation of two intermolecular helices (the 5′ gRNA anchor–mRNA duplex and the 3′ U-tail–mRNA duplex) flanking the first few ESs. A third intramolecular helix formed by the guiding region of the gRNA is also predicted. These computer predictions indicated that the different gRNA–mRNA pairs, despite having very different primary sequences, could form very similar secondary structures and argue for the formation of a common core architecture that may be important in the assembly of a functional editing complex.
In this current study, we have investigated the interaction of gCYb-558’s U-tail with partially edited 5′CYb substrates in order to determine how the change in sequence associated with the editing reaction might affect the U-tail–mRNA interaction. Our previous studies indicated that the U-tail was interacting with mRNA regions that were to be subsequently edited. We wanted to determine how the increase in the anchor duplex region might affect this interaction. One possibility was that the interaction with the mRNA is flexible, with the U-tail ‘sliding’ up the mRNA as editing progressed. Alternatively, the U-tail may move 5′ along the mRNA in a stepwise fashion. Once editing proceeds beyond a specific threshold, the U-tail would interact at a new position, further upstream. This threshold might be defined by the maintenance of a predicted gRNA stem–loop across from the current ES. Editing could break this threshold by creating a large enough anchor duplex, employing nucleotides previously involved in the gRNA stem–loop, thus destabilizing the stem–loop and the interaction of the U-tail.
Here we report our observation that, despite the progression of editing up to ES3 (an increase of 13 bp within the anchor duplex region), the U-tail continues to interact with the same purine-rich sequence (G52–A56) observed with the unedited mRNA. This crosslink data indicates that the U-tail does not slide up the mRNA as editing proceeds. The computer-predicted structures generated using this crosslink data, indicate that as the anchor duplex extends, the stem–loop structure in the gRNA can be maintained by alternate base pairs that include the U-tail. This suggests that the role of the U-tail may change as editing proceeds. During the initial stages, its role may be to provide stability during the initial gRNA–mRNA interaction and to help tether the 5′ cleavage product. However, as editing proceeds, increasing the size of the gRNA–anchor duplex, the U-tail’s contribution to the stability is less important. Furthermore, with the 3′-end of the U-tail continuing to interact with the same sequence, the number of U’s interacting with the 5′ cleavage product decreases, thereby reducing the U-tail’s ability to hold on to the 5′ cleavage product. This function may be taken over by protein–RNA interactions as suggested by Burgess et al. (25) and Kapushoc and Simpson (26). Instead of these initial roles, it may be that the U-tail functions to maintain important secondary structure motifs (such as the gRNA stem–loop) by feeding into the structure as editing progresses. This stem–loop could potentially function as a protein binding site (6,7,24,27). Alternatively, as base stacking is a major factor in the stabilization of RNA structures, it may be that the presence of multiple helices that can stack may be important for editing complex stability.
These functions may help explain why the gRNA has a U-tail. Uridines are able to interact with the upstream purine-rich sequences found in pre-mRNA, allowing the U-tail to help stabilize the gRNA–mRNA interaction and bind to the 5′ cleavage product. Progression of editing diminishes this requirement and instead the U-tail can interact with its own guiding region to maintain the gRNA stem–loop. We hypothesize that because uridines are able to bind both A and G, a series of uridines may be the best universal sequence able to carry out the above functions.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by NIH grant AI34155 to D.K.
References
- 1.Alfonzo J.D., Thiemann,O. and Simpson,L. (1997) The mechanism of U insertion/deletion RNA editing in kinetoplastid mitochondria. Nucleic Acids Res., 25, 3751––3759.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stuart K., Allen,T.E., Heidmann,S. and Seiwert,S.D. (1997) RNA editing in kinetoplastid protozoa. Microbiol. Mol. Biol. Rev., 61, 105––120.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hajduk S.L. and Sabitini,R.S. (1998) Mitochondrial mRNA editing in kinetoplastid protozoa. In Grosjean,H. and Benne,R. (eds), Modification and Editing of RNA. ASM Press, Washington, DC, pp. 377––393..
- 4.Estevez A.M. and Simpson,L. (1999) Uridine insertion/deletion RNA editing in trypanosome mitochondria–a review. Gene, 240, 247––260.. [DOI] [PubMed] [Google Scholar]
- 5.Blum B., Bakalara,N. and Simpson,L. (1990) A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell, 60, 189––198.. [DOI] [PubMed] [Google Scholar]
- 6.Blum B. and Simpson,L. (1990) Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ oligo(U) tail involved in recognition of the preedited region. Cell, 62, 391––397.. [DOI] [PubMed] [Google Scholar]
- 7.Leung S.S. and Koslowsky,D.J. (1999) Mapping contacts between gRNA and mRNA in trypanosome RNA editing. Nucleic Acids Res., 27, 778––787.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seiwert S.D., Heidmann,S. and Stuart,K. (1996) Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell, 84, 831––841.. [DOI] [PubMed] [Google Scholar]
- 9.Koslowsky D.J., Kutas,S.M. and Stuart,K. (1996) Distinct differences in the requirements for ribonucleoprotein complex formation on differentially regulated pre-edited mRNAs in Trypanosoma brucei. Mol. Biochem. Parasitol., 80, 1––14.. [DOI] [PubMed] [Google Scholar]
- 10.Milligan J.F., Groebe,D.R., Witherell,G.W. and Uhlenbeck,O.C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res., 15, 8783––8798.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riley G.R., Corell,R.A. and Stuart,K. (1994) Multiple guide RNAs for identical editing of Trypanosoma brucei apocytochrome b mRNA have an unusual minicircle location and are developmentally regulated. J. Biol. Chem., 269, 6101––6108.. [PubMed] [Google Scholar]
- 12.Moore M.J. and Sharp,P.A. (1992) Site-specific modification of pre-mRNA: the 2′-hydroxyl groups at the splice sites. Science, 256, 992––997.. [DOI] [PubMed] [Google Scholar]
- 13.Wahle E. and Keller,W. (1994) 3′ end processing of mRNA. In Higgins,S.J. and Hames,B.D. (eds), RNA Processing. A Practical Approach. IRL Press, New York, NY, Vol. 2, pp. 1––33..
- 14.Pollard V.W., Harris,M.E. and Hajduk,S.L. (1992) Native mRNA editing complexes from Trypanosoma brucei mitochondria. EMBO J., 11, 4429––4438.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911––940.. [DOI] [PubMed] [Google Scholar]
- 16.Matzura O. and Wennborg,A. (1996) RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. Comput. Appl. Biosci., 12, 247––249.. [DOI] [PubMed] [Google Scholar]
- 17.Feagin J.E., Jasmer,D.P. and Stuart,K. (1987) Developmentally regulated addition of nucleotides within apocytochrome b transcripts in Trypanosoma brucei. Cell, 49, 337––345.. [DOI] [PubMed] [Google Scholar]
- 18.Burgin A.B. and Pace,N.R. (1990) Mapping the reactive site of ribonuclease P RNA using a substrate containing a photoaffinity agent. EMBO J., 9, 4111––4118.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seiwert S.D. and Stuart,K. (1994) RNA editing: transfer of genetic information from gRNA to precursor mRNA in vitro. Science, 266, 114––117.. [DOI] [PubMed] [Google Scholar]
- 20.Adler B.K. and Hajduk,S.L. (1997) Guide RNA requirement for editing-site-specific endonucleolytic cleavage of preedited mRNA by mitochondrial ribonucleoprotein particles in Trypanosoma brucei. Mol. Cell. Biol., 17, 5377––5385.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piller K.J., Rusché,L.N., Cruz-Reyes,J. and Sollner-Webb,B. (1997) Resolution of the RNA editing gRNA-directed endonuclease from two other endonucleases of Trypanosoma brucei mitochondria. RNA, 3, 279––290.. [PMC free article] [PubMed] [Google Scholar]
- 22.Cruz-Reyes J., Rusché,L.N., Piller,K.J. and Sollner-Webb,B. (1998) T. brucei RNA editing: adenosine nucleotides inversely affect U-deletion and U-insertion reactions at mRNA cleavage. Mol. Cell, 1, 401––409.. [DOI] [PubMed] [Google Scholar]
- 23.Cruz-Reyes J., Rusché,L.N. and Sollner-Webb,B. (1998) Trypanosoma brucei U insertion and U deletion activities co-purify with an enzymatic editing complex but are differentially optimized. Nucleic Acids Res., 26, 3634––3639.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmid B., Riley,G.R., Stuart,K. and Göringer,H.U. (1995) The secondary structure of guide RNA molecules from Trypanosoma brucei. Nucleic Acids Res., 23, 3093––3102.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgess M.L., Heidmann,S. and Stuart,K. (1999) Kinetoplastid RNA editing does not require the terminal 3′ hydroxyl of guide RNA, but modifications to the guide RNA terminus can inhibit in vitro U insertion. RNA, 5, 883––892.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapushoc S.T. and Simpson,L. (1999) In vitro uridine insertion RNA editing mediated by cis-acting guide RNAs. RNA, 5, 656––669.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hermann T., Schmid,B., Heumann,H. and Göringer,H.U. (1997) A three-dimensional working model for a guide RNA from Trypanosoma brucei. Nucleic Acids Res., 25, 2311––2318.. [DOI] [PMC free article] [PubMed] [Google Scholar]