

Abstract
Cyclic phosphatidic acid (CPA) is a naturally occurring analog of lysophosphatidic acid (LPA) in which the sn-2 hydroxy group forms a 5-membered ring with the sn-3 phosphate. Here we describe the synthesis of R-3-CCPA and S-3-CCPA along with their pharmacological properties as inhibitors of lysophospholipase D/autotaxin, agonists of the LPA5 GPCR, and blockers of lung metastasis of B16-F10 melanoma cells in a C57BL/6 mouse model. S-3CCPA was significantly more efficacious in the activation of LPA5 compared to the R stereoisomer. In contrast, no stereoselective differences were found between the two isomers toward the inhibition of autotaxin or lung metastasis of B16-F10 melanoma cells in vivo. These results extend the potential utility of these compounds as potential lead compounds warranting evaluation as cancer therapeutics.
Keywords: lysophosphatidic acid, NPP2, autotaxin, GPR92, lysophospholipase D
Lysophosphatidic acid (LPA) is a pleiotrophic phospholipid growth factor with multiple roles in cancer metastasis and progression1.LPA elicits numerous biological effects including the promotion of cellular survival, mitogenesis, angiogenesis, migration, and cancer invasion that are mediated, at least in part, by specific cell surface G protein-coupled receptors (GPCR) and intracellular targets that include the nuclear hormone receptor peroxisome proliferator-activated receptor (PPARγ).2 Cylic-phosphatidic acid (1-acyl-2,3-glycerophosphate, CPA) is a naturally occurring analog of LPA in which the sn-2 hydroxy group forms a 5-membered ring with the sn-3 phosphate.3 CPA affects numerous cellular functions, including inhibition of cell cycle progression, induction of stress fiber formation, inhibition of tumor cell invasion and metastasis, and regulation of differentiation and survival of neuronal cells.4 CPA is a weak agonist of the LPA1 and LPA2 GPCR.5 Substitution of the sn-2 or sn-3 oxygen with a methylene in CPA yields carba-CPA (CCPA), a stabilized analog of CPA.6 Previous work has shown that 3-CCPA does not activate the LPA1-4 GPCR5 but is a weak agonist of LPA5.7
Autotaxin (ATX) was initially identified as an autocrine tumor cell motility factor from melanoma cell conditioned medium.8ATX has lysophospholipase D enzyme activity and is responsible for the hydrolysis of lysophophatidylcholine leading to the generation of LPA9;10 and CPA.11 While ATX can also produce sphingosine 1 phosphate (S1P) in vitro, it does not appear to contribute in a major way to S1P production in vivo. High levels of autotaxin are generated by a wide variety of metastatic human tumor cell lines including human teratocarcinoma,12 hepatocellularcarcinoma,13 metastatic breast cancer,1 ovariancancer,14 thyroid carcinoma,15 prostate cancer,16 follicular lymphoma17 and glioblastomamultiforme.18 ATX also plays an important role in the chemotherapeutic resistance of breast19 and ovarian cancer cells14 to chemotherapeutic agents. ATX is under feedback inhibition by its hydrolytic products LPA, CPA, and sphingosine-1-phosphate (S1P).20 Racemic 2-CCPA and 3-CCPA are potent inhibitors of ATX activity and 3-CCPA has been shown to reduce lung metastasis of B16-F10 melanoma cells injected intravenously into C57BL/6 mice.5 To further explore the therapeutic utility of 3-CCPA, stereochemically pure isomers are needed. For this reason we describe the synthesis and characterization of both the R-3-CCPA and S-3-CCPA.
The approach used for the synthesis of the two stereoisomers of 3-CCPA is shown in scheme 1. Dimethylphosphonate derivatives 2R and 2S were generated from compound 1 using n-butyllithium, BF3 etherate and either the R- or S-isomer of benzylglycidyl ether. The corresponding 3-carbacyclic analogs 3R and 3S resulted from treatment with pyridinium p-toluenesulfonate (PPTS).Following benzyl group deprotection by hydrogenation,the resulting alcohols 4R and 4S were converted to oleoyl esters 5R and 5S using N,N’-diisopropylcarbodiimide (DIC) and dimethylaminopyridine (DMAP). Final products R-3-CCPA and S-3-CCPA were prepared by methyl group deprotection and conversion to the corresponding sodium salts using TMSBr and dilute NaOH, respectively. Optical rotations were determined by dissolving the compounds in methanol, to be +7.3° for R-3-CCPA and for S-3-CCPA, the optical rotation was −7.9°. Each compound was purified by silica gel column chromatography and verified by mass spectrometry, NMR and HRMS.
Scheme 1.

(a) i ) THF, n-BuLi (2.5M in hexane), −78°C, 0.5hr; ii) R-Benzyl glycidyl ether (R) / S-Benzyl glycidyl ether (S); iii) THF,BF3OEt2,−78°C, 2hr; iv) −20°C, 2hr, 68%; (b) PPTS, Toluene, Reflux, 5hr, 65% ; (c) H2, Pd(OH)2 / C,MeoH, 82.5%; (d) C17H33COOH, DMAP, DIC, DCM, 18hr, 78%; (e)TMSBr,CH2Cl2, 1hr, 53%; (f) 0.05M NaOH
Compounds R-3-CCPA and S-3-CCPA were examined for their ability to block ATX-mediated hydrolysis of FS-3(Echelon Biosciences, Inc. Salt Lake City, UT) using a fluorescence resonance energy transfer-based assay.21 Recombinant ATX (25 nmol) in the presence of various concentrations of R-3-CCPA, S-3-CCPA, or LPA 18:1 (positive control) in assay buffer consisting of 1 mM MgCl2, 1 mM CaCl2, 3 mM KCl, 140 mM aCl, 50 mM Tris-HCl, pH 8.0 and 15 μM fatty acid free BSA was added to FS-3 (final concentration 1 μM). Assays were carried out in white wall 96-well plates (Corning Inc., Corning, NY) and the fluorescence (excitation 485 nm, emission 538 nm) was measured at the beginning and after 2 hours of incubation at 37 °C using a FLEX stationII plate reader (Molecular Devices, Sunnyvale, CA). Data were normalized to the corresponding vehicle control, and the mean ±standard deviation of triplicate wells was used to calculate ATX activity as per cent of vehicle control. The dose response-relationship of ATX inhibition showed little difference between the R-3-CCPA, S-3-CCPA or the racemate. However, R-3-CCPA was approximately 2 fold more potent in this assay than S-3-CCPA. The kinetic mechanism by which R-3-CCPA and S-3-CCPA inhibited recombinant ATX-mediated hydrolysis of FS-3 were determined by varying the concentration of the substrate (FS-3, ranging from 0.3 to 20 μM) in the presence of three concentrations of each inhibitor (0, 0.5 and 2 times the IC50). Simultaneous non-linear regression using WinNonLin ® 6.1 (Pharsight, Mountain View, CA) was used to fit experimental data and calculate Ki and Ki’ using the Michaelis-Menten equations for competitive, uncompetitive, mixed-mode, and non-competitive inhibition as we have described in recent work. 22-24 Mechanism of inhibition was assigned based on the lowest averaged percent residuals for each mechanism derived from curve fitting. Using this procedure R-3-CCPA and S-3-CCPA were determined to be mixed mode ATX inhibitors with Ki values of 0.8 and 1.6 μM, respectively.
The lack of ligand stereospecificity of the LPA1, LPA2, and LPA3 receptors has been published previously25 but no information of stereoselective ligand acitvitaion for LPA5 is currently available at the present time. Racemic-3-CCPA has previously been shown to bean agonist of the LPA5 GPCR.7 Here we compared the dose-response curves of LPA5 activation for R-3-CCPA and S-3-CCPA with that of the racemate. These experiments were performed in B103 cells stably expressing LPA5. Wild type B103 cells do not produce Ca2+ transients in response to LPA and are widely used as a host cell for LPA receptor expression studies. B103-LPA5 cells were loaded with Fura-2AM for 30 min in modified Krebs buffer containing 2% (v/v) pluronic acid, rinsed with Krebs buffer, and changes in the intracellular Ca2+ concentration were monitored by determining the ratio of emitted light intensities at 520 nm in response to excitation at 340 and 380 nm using a FLEX station II plate reader (Molecular Devices, Sunnyvale, CA).26 Compound S-3-CCPA showed significantly higher (p < 0.05) efficacy than did R-3-CCPA for LPA5-mediated calcium mobilization at concentrations above 1 μM (Figure 2). Thus, the LPA5 receptor shows a slight stereoselectivity for the S- over the R-stereoisomer which contrasts the weak preference (~2-fold) shown by ATX for the R-isomer.
Figure 2.
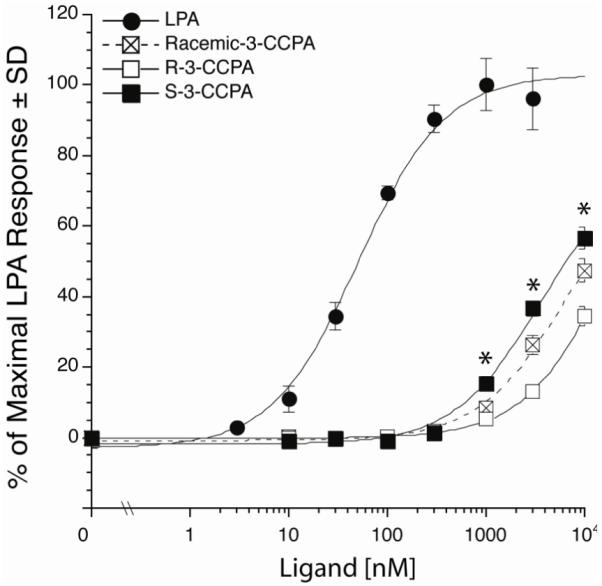
Dose-response relationship of LPA5 mediated
We have previously shown that racemic 3-CCPA inhibited lung metastasis of B16-F10 melanoma in a mice model. To extend this observation, the stereoisomers were characterized in this model.4;5 Eight-week-old female C57Bl/6 mice were inoculated with 5 × 104melanoma cells via the tail vein and divided randomly into 4 groups. The groups then received either saline vehicle, R-3-CCPA, S-3-CCPA, or racemate (at 0.5 mg/kg intraperitoneally) 30 min after the B16-F10 inoculation and daily for an additional 10 days. Animals in all groups were monitored for an additional 10 days without further treatments. On day 21, all mice were sacrificed and lungs were dissected, fixed with formalin and the numbers of black melanoma nodules on the lung surface were counted in each sample (Figure 3). All 3-CCPA treated groups (R-3-CCPA, S-3-CCPA and Racemic-3-CCPA) significantly reduced the number of lung metastases compared to the vehicle treated group. However, no statistically significant differences were found between the 3-CCPA treated groups using ANOVA followed by Newman-Keuls multiple comparison test.
Figure 3. Lack of stereoselectivityin lung metastasis of B16-F10 melanoma cells by R-3-CCPA, S-3-CCPA and Racemic -3-CCPA analogs in a mouse model.
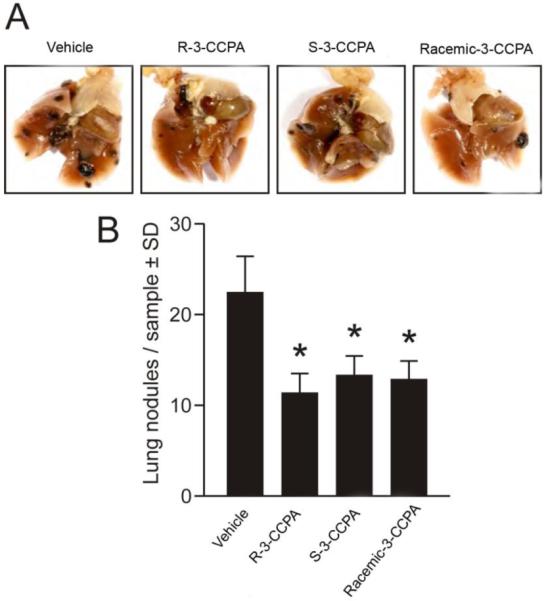
A) Representative images show fixed intrathoracic organs including lung lobules with visiblenodules on the surfaces in black. Total numbers of nodules werereduced insamples treated with 7R, 7S or racemate compared to vehicle. Scale bar is 0.5 cm.
B) Lung nodules of B16-F10 melanoma cells were quantified. The number of lung nodules was significantly decreased in groups treated with R-3-CCPA, S-3-CCPA, and the Racemic-3-CCPA compared to vehicle. However, no statistically significant differences were found either between the stereoisomers or the racemate. Data represent the mean ± SEM, n = 6-8 mice. *p< 0.05 compared to vehicle analyzed by one-way ANOVA followed by Newman-Keuls multiple comparison test.
In conclusion, we have synthesized pure stereoisomers of 3-CCPA and found that they inhibited ATX in vitro and B16-F10 melanoma metastasis in vivo without significant stereochemical preference. The lack of stereoselectivity is underlined by the equal efficacy of the racemic mixture. Interestingly, at the LPA5 GPCR the S-stereoisomer (S-3-CCPA) showed significantly higher efficacy. This is the first indication that the LPA5 receptor, unlike the LPA1,2,3 receptors shows stereo-selective activation by CCPA ligands.
Figure 1.
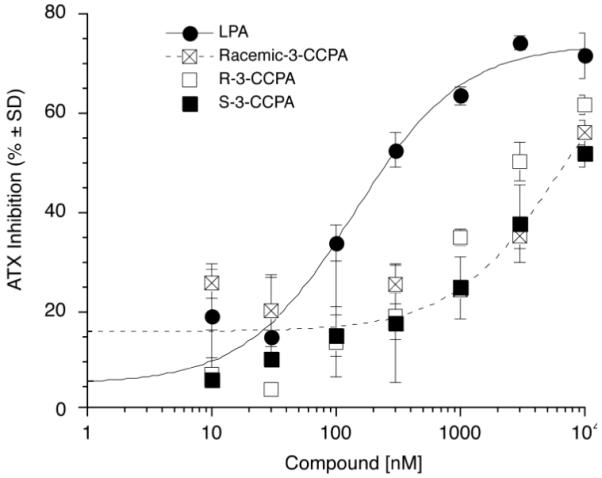
Dose response relationship of ATX inhibition by LPA, R-3-CCPA, S-3-CCPA and Racemic-3-CCPA analogs.
Acknowledgements
This research was supported by NIH grant CA92160 (G.T.), Van Vleet Professorship (D.M.), Breast Cancer Research Foundation (N.P.) and Lpath Inc. (G.M.).
Abbreviations
- ATX
Autotaxin
- BSA
Bovine serum albumin
- CCPA
Carbacyclic phosphatidic acid
- CPA
Cyclic phosphatidic acid
- DIC
Di-isopropyl carbodiimide
- DMAP
Dimethyl amino pyridine
- GPCR
G-protein coupled receptors
- HRMS
High resolution mass spectrometry
- LPA
Lysophosphatidic acid
- NMR
Nuclear magnetic resonance
- PPTS
Pyridinium -p-toluene sulfonate
- TMSBr
Trimethyl silyl bromide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, Yu S, Stephens LC, Cui X, Murrow G, Coombes K, Muller W, Hung MC, Perou CM, Lee AV, Fang X, Mills GB. Cancer Cell. 2009;15:539. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parrill AL. Biochim.Biophys.Acta. 2008;1781:540. doi: 10.1016/j.bbalip.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murakami-Murofushi K, Uchiyama A, Fujiwara Y, Kobayashi T, Kobayashi S, Mukai M, Murofushi H, Tigyi G. Biochim.Biophys.Acta. 2002;1582:1. doi: 10.1016/s1388-1981(02)00131-2. [DOI] [PubMed] [Google Scholar]
- 4.Fujiwara Y. Biochim.Biophys.Acta. 2008;1781:519. doi: 10.1016/j.bbalip.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker DL, Fujiwara Y, Pigg KR, Tsukahara R, Kobayashi S, Murofushi H, Uchiyama A, Murakami-Murofushi K, Koh E, Bandle RW, Byun HS, Bittman R, Fan D, Murph M, Mills GB, Tigyi G. J.Biol.Chem. 2006;281:22786. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uchiyama A, Mukai M, Fujiwara Y, Kobayashi S, Kawai N, Murofushi H, Inoue M, Enoki S, Tanaka Y, Niki T, Kobayashi T, Tigyi G, Murakami-Murofushi K. Biochim.Biophys.Acta. 2007;1771:103. doi: 10.1016/j.bbalip.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams JR, Khandoga AL, Goyal P, Fells JI, Perygin DH, Siess W, Parrill AL, Tigyi G, Fujiwara Y. J.Biol.Chem. 2009;284:17304. doi: 10.1074/jbc.M109.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stracke ML, Arestad A, Levine M, Krutzsch HC, Liotta LA. Melanoma Res. 1995;5:203. doi: 10.1097/00008390-199508000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. J.Cell Biol. 2002;158:227. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. J.Biol.Chem. 2002;277:39436. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 11.Tsuda S, Okudaira S, Moriya-Ito K, Shimamoto C, Tanaka M, Aoki J, Arai H, Murakami-Murofushi K, Kobayashi T. J.Biol.Chem. 2006;281:26081. doi: 10.1074/jbc.M602925200. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Mou L, Liu N, Tsao MS. Am.J.Respir.Cell Mol.Biol. 1999;21:216. doi: 10.1165/ajrcmb.21.2.3667. [DOI] [PubMed] [Google Scholar]
- 13.Wu JM, Xu Y, Skill NJ, Sheng H, Zhao Z, Yu M, Saxena R, Maluccio MA. Mol.Cancer. 2010;9:71. doi: 10.1186/1476-4598-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vidot S, Witham J, Agarwal R, Greenhough S, Bamrah HS, Tigyi GJ, Kaye SB, Richardson A. Cell Signal. 2010;22:926. doi: 10.1016/j.cellsig.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 15.Kehlen A, Englert N, Seifert A, Klonisch T, Dralle H, Langner J, Hoang-Vu C. Int.J.Cancer. 2004;109:833. doi: 10.1002/ijc.20022. [DOI] [PubMed] [Google Scholar]
- 16.Nouh MA, Wu XX, Okazoe H, Tsunemori H, Haba R, Abou-Zeid AM, Saleem MD, Inui M, Sugimoto M, Aoki J, Kakehi Y. Cancer Sci. 2009;100:1631. doi: 10.1111/j.1349-7006.2009.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masuda A, Nakamura K, Izutsu K, Igarashi K, Ohkawa R, Jona M, Higashi K, Yokota H, Okudaira S, Kishimoto T, Watanabe T, Koike Y, Ikeda H, Kozai Y, Kurokawa M, Aoki J, Yatomi Y. Br.J.Haematol. 2008;143:60. doi: 10.1111/j.1365-2141.2008.07325.x. [DOI] [PubMed] [Google Scholar]
- 18.Kishi Y, Okudaira S, Tanaka M, Hama K, Shida D, Kitayama J, Yamori T, Aoki J, Fujimaki T, Arai H. J.Biol.Chem. 2006;281:17492. doi: 10.1074/jbc.M601803200. [DOI] [PubMed] [Google Scholar]
- 19.Samadi N, Gaetano C, Goping IS, Brindley DN. Oncogene. 2009;28:1028. doi: 10.1038/onc.2008.442. [DOI] [PubMed] [Google Scholar]
- 20.van Meeteren LA, Ruurs P, Christodoulou E, Goding JW, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH. J.Biol.Chem. 2005;280:21155. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson CG, Bigman CS, Richardson RD, van Meeteren LA, Moolenaar WH, Prestwich GD. Org.Lett. 2006;8:2023. doi: 10.1021/ol060414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoeglund AB, Bostic HE, Howard AL, Wanjala IW, Best MD, Baker DL, Parrill AL. J.Med.Chem. 2010;53:1056. doi: 10.1021/jm9012328. [DOI] [PubMed] [Google Scholar]
- 23.North EJ, Osborne DA, Bridson PK, Baker DL, Parrill AL. Bioorg.Med.Chem. 2009;17:3433. doi: 10.1016/j.bmc.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.North EJ, Howard AL, Wanjala IW, Pham TC, Baker DL, Parrill AL. J.Med.Chem. 2010;53:3095. doi: 10.1021/jm901718z. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama K, Baker DL, Virag T, Liliom K, Byun HS, Tigyi G, Bittman R. Biochim.Biophys.Acta. 2002;1582:295. doi: 10.1016/s1388-1981(02)00184-1. [DOI] [PubMed] [Google Scholar]
- 26.Durgam GG, Virag T, Walker MD, Tsukahara R, Yasuda S, Liliom K, van Meeteren LA, Moolenaar WH, Wilke N, Siess W, Tigyi G, Miller DD. J.Med.Chem. 2005;48:4919. doi: 10.1021/jm049609r. [DOI] [PubMed] [Google Scholar]