

Abstract
The underlying defects in Angelman syndrome (AS) and autism spectrum disorder (ASD) may be in part due to basic defects in synaptic plasticity and function. In some individuals serotonin reuptake inhibitors, which decrease pre-synaptic re-uptake of serotonin, can ameliorate symptoms, as can resperidone, which blocks both dopamine and serotonin receptors. Loss of maternal UBE3A expression causes AS, while maternal duplications of chromosome 15q11.2-q13 that include the UBE3A gene cause ASD, implicating the maternally expressed UBE3A gene in the ASD phenotype. In a Drosophila screen for proteins regulated by UBE3A, we identified a key regulator of monoamine synthesis, the gene Punch, or GCH1, encoding the enzyme GTP cyclohydrolase I. Here we show that Dube3a, the fly UBE3A ortholog, regulates Punch/GCH1 in the fly brain. Over-expression of Dube3a elevates tetrahydrobiopterin (THB), the rate-limiting cofactor in monoamine synthesis while loss of Dube3a has the opposite effect. The fluctuations in dopamine levels were associated with hyper- and hypoactivity, respectively, in flies. We show that changes in Punch/GCH1 and dopamine levels do not depend on the ubiquitin ligase catalytic domain of Dube3a. In addition, both wild type Dube3a and a ubiquitination-defective Dube3a-C/A form were found at high levels in nuclear fractions and appear to be poly-ubiquitinated in vivo by endogenous Dube3a. We propose that the transcriptional co-activation function of Dube3a may regulate GCH1 activity in the brain. These results provide a connection between monoamine synthesis (dopamine/serotonin) and Dube3a expression that may explain why some individuals with ASD or AS respond better to selective serotonin reuptake inhibitors than others.
Introduction
The regulation of neurotransmitters like dopamine and serotonin in the brain is critical for proper synaptic function as well as plasticity. These neurotransmitters may be inappropriately regulated in the brains of autistic individuals accounting for at least some of the synaptic stability defects observed in autism (Makkonen et al., 2008). Although the underlying genetic heterogeneity of idiopathic autism has complicated the analysis of neurotransmitter levels, it has become apparent that at least some individuals with autism have altered tetrahydrobiopterin (THB) levels in cerebrospinal fluid (Tani et al., 1994). THB is a regulatory cofactor for the synthesis of monoamines including both dopamine and serotonin, neurotransmitters that have been linked to many of the behavioral traits manifested in ASD. Resperidone, an atypical neuroleptic that blocks dopamine and serotonin receptors has been widely employed to ameliorate monoamine-related behaviors is autistic individuals (c.f., (Canitano and Scandurra, 2008)). Treatment of the anxiety and hyperactivity associated with autism often involves the use of selective serotonin reuptake inhibitors (SSRIs), which inhibit the presynaptic re-uptake of serotonin thereby increasing postsynaptic activation (Kolevzon et al., 2006). Consistent with mounting evidence of monoamine variation in ASD are reports of haplotype associations in monoamine-related genes. For example, Hettinger et al. (2008) reported over-transmission in ASD families of two D1 dopamine receptor haplotypes (Hettinger et al., 2008). Thus, the improper regulation of synaptic monoamine levels may be a key aspect of autism pathogenesis.
The most common cytogenetic abnormalities detected in autistic individuals are maternally derived DNA duplications of the 15q11.2-q13 region, also known as the Prader-Willi/Angelman syndrome critical region (reviewed in (Hogart et al., 2010). The primary candidate gene in this region, UBE3A, encodes an E3 ubiquitin ligase protein that targets other proteins for degradation by the ubiquitin proteasome system (Kishino et al., 1997; Schroer et al., 1998). In the majority of cases maternally derived deletions of UBE3A result in the devastating neurogenetic disorder Angelman syndrome (AS), but other molecular defects that can turn off maternal UBE3A also result in an AS phenotype (Jiang et al., 1999). Mechanisms of UBE3A regulation may not be so simple, however, since UBE3A can also act as a transcriptional co-activator of steroid hormone receptors (Ramamoorthy and Nawaz, 2008) and mono-ubiquitination by UBE3A may be involved in the trafficking of synaptic proteins (Haas and Broadie, 2008; Hicke, 2001). The observation that UBE3A displays maternal allele-specific expression in the mammalian brain clearly implicates it in the pathology of both AS and the autism phenotype found in 15q11.2-q13 duplications (Albrecht et al., 1997; Lalande and Calciano, 2007; Rougeulle et al., 1997). In addition, individuals with Prader-Willi syndrome (PWS) who have two maternal copies of the 15q11.2-q13 region due to uniparental disomy for chromosome 15 also present with autistic features not typically found in PWS patients who have inherited paternal deletions of 15q11.2-q13 (Descheemaeker et al., 2006; Veltman et al., 2005). Finally, UBE3A levels are indeed elevated in lymphocytes from individuals with larger isodicentric 15q duplications, again implicating elevations of UBE3A in the pathogenesis of autism (Baron et al., 2006).
Using the powerful genetic tools available in Drosophila melanogaster we have been investigating the molecular etiology of both duplication 15q autism and AS. Our approach is to elevate fly and human UBE3A levels in the brains of Drosophila and then use proteomic profiling to identify potential UBE3A target proteins. Using this method we successfully identified a UBE3A-interacting protein involved in actin cytoskeletal rearrangements and neurogenesis, the Pbl/ECT2 protein (Reiter et al., 2006). Here we describe the identification of another Dube3a-regulated protein that is a key regulator of monoamine synthesis in flies as well as humans. In this report we show that expression of both wild type and ubiquitination defective Dube3a is able to regulate the activity of the Punch protein, an enzyme that produces tetrahydrobiopterin (THB), the rate-limiting co-factor in monoamine synthesis. This is the first report of a UBE3A target involved in neurotransmitter regulation and has broad implications for how the function of this target may contribute to the pathogenesis of AS, duplication 15q autism as well as idiopathic autism linked to UBE3A regulated pathways. We propose that the regulation of dopamine/serotonin levels by UBE3A may be responsible for some of the anxiety and hyperactivity observed as part of the clinical phenotype in AS and duplication 15q autism providing a mechanistic basis for therapeutic interventions that target the monoamine synthesis pathway.
Materials and Methods
Drosophila stocks and crosses
All fly crosses were performed at 25°C on standard corn meal based media. The following stocks were obtained from the Bloomington Stock Center (Bloomington, IN): HS-GAL4, gmr-GAL4, elav-GAL4, Pur1, PuEY4414 and PuEY2616. The UAS-RNAi-Dube3a (6190-R3) line was obtained from the National Institute of Genetics RNAi stock center (Mishima, Shizuoka, Japan). The UAS-FLAG-Dube3a lines were constructed by PCR cloning the insert from Gold Collection cDNA LD21888 into the Gateway (Invitrogen) acceptor vector pTWF available from the Drosophila Genomics Resource Center (DGRC). This vector was injected into embryos and transformants identified by standard methods. The UAS-hUBE3A lines have been described previously (Reiter et al., 2006). All transgenes were maintained in either w1118 or a y1 w* backgrounds. The Drosophila Dube3a mutant lines, w1118; Dube3a15b/TM6b, Tb and w1118; Dube3a80/TM6b, were a gift from Janice Fischer (Wu et al., 2008). A homozygous Dube3a15b mutant line as well as the w; UAS-Dube3a-C/A-FLAG; Dube3a15b and w, elav-GAL4; Dube3a15b lines were subsequently derived from the Dube3a15b heterozygous stock.
Proteomic analysis
Details of the 2D-gel analysis from fly head extracts were described previously (Reiter et al., 2006) with the following exceptions: 1) 2-D gels were run in triplicate in our laboratory using the Bio-Rad DODECA II system and then stained with SYPRO Ruby fluorescent protein stain to identify spots that changed in intensity, as opposed to relying on a commercial 2-D gel separation service, 2) Protein spots were excised from the acrylamide gels and mass spectra were recorded on a Bruker Ultraflex MALDI-ToFToF reflecting time-of-flight mass spectrometer (Bruker Daltonics, Billerica, MA). The mass spectrometry methods and protein identifications have been described elsewhere (Cummings et al., 2007).
Quantitative Western Blot Analysis
Transgenic and control flies, 3-5 days old, were subjected to a 37°C heatshock for 1hr, allowed to recover for 10min and then frozen in liquid nitrogen. Cytoplasmic and nuclear extracts were prepared from ∼30-50μl of fly heads using the ReadyPrep Protein Extraction Kit (Biorad, Hercules, CA). Samples were resolved on a NuPage 4-12% Bis-Tris 1.5mm Gel using MOPS Buffer (Invitrogen, Carlsbad, CA) and transferred to Immobilon-FL PVDF membrane (Millipore). The membrane was blocked with 5% milk, 3% BSA, and 0.1% Tween-20 in PBS. The following primary antibodies were used at 1:5000: α-Drosophila Dube3a, a gift of Janice Fischer (Wu et al., 2008), α-Punch (Krishnakumar et al., 2000) or α-alpha tubulin (from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA). IR labeled secondary antibodies (α-Guinea pig 680, α-rat 680 and α-mouse 800) were purchased from Li-Cor (Lincoln, NE) and used at a dilution of 1:10,000. The blot was imaged and analyzed using the Odyssey Infrared Imaging System (LiCor, Lincoln, NE). After adjustment for background fluorescence, lanes were normalized using the signal from α-alpha tubulin as the reference channel and signal intensity was then adjusted based on the calculated normalization factor assigned to each lane in the channel. Fold change (Fc) was calculated by dividing the normalized intensity values of each sample by the values for either w-; HS-GAL4 or w- control lane intensity.
qRT-PCR Analysis
Total RNA was extracted from ∼30-50μl of fly heads using 200μl of RNABee solution (AMS biotechnology, United Kingdom). RNA was quantified by spectrophotometry (NanoDrop Technologies, Wilmington, DE) and the integrity of the 18S and 26S rRNA verified using an Agilent 2100 Bioanlyzer (Agilent Technologies, Santa Clara, CA). Two μg of total RNA were input for each cDNA reaction synthesized using random hexamer primers via the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA). Primers and product-specific probe sets for each gene were selected using the Universal ProbeLibrary Assay Design Center software (Roche Applied Science, Indianapolis, IN). For a complete list of primers and probes used, refer to Supplemental Table 1. All assays for each cDNA sample were performed in triplicate at 40ng/reaction using the default cycling parameters of the Roche Light Cycler 480 system. The crossing point (Cp) value for each sample was calculated using Roche absolute quantitation algorithm. The average Cp value among three technical replicates was calculated and the average sample Cp values were normalized for loading by comparison to the average Cp value of TATA-binding protein TBP. The fold change (Fc) in gene expression in fly heads was then calculated by comparing the difference of the normalized mean Cp values between w- and the Dube3a15b mutant or between elav-GAL4 and each over-expression construct using the equation Fc = (2)Δcp. The ΔΔCt Method suggested by ABI was used for the statistical analysis of qRT-PCR (http://tinyurl.com/2cyjby8) to propagate the standard error illustrated in Table 1.
Table 1. qRT-PCR Fold Change.
Gray shaded boxes changed by a less than 2-fold average. Fold change in Dube3a15b mutant is compared to w- and elav-GAL4 expressing lines are compared to elav-GAL4 alone. Error is standard deviation for three technical replicate samples.
| Dube3a15b vs w- | elav>Dube3a vs elav | elav>Dube3a-C/A vs elav | |
|---|---|---|---|
| Pu-RB | -4.7±1.4 | +3.6±1.6 | +15.7±7.6 |
| Pu-RA/C | +1.3±0.2 | +1.6±0.2 | +1.5±0.4 |
| Tbh-RB | -1.7±1.0 | -1.5±0.2 | +1.7±0.6 |
| Pale | -1.6±0.3 | +1.5±0.1 | -2.0±1.0 |
| Ddc | -1.2±0.1 | +1.5±0.2 | +1.3±0.2 |
HPLC Analysis
Monoamines and pteridines were separated by HPLC analysis using a CoulArray HPLC system (model 5600A; ESA, Chelmsford, MA) and a Synergi 4μm Hydro-RP column (4.6 × 150 mm; Phenomenex, Torrance, CA), according to previously published methods (McClung and Hirsh, 1999), as modified by Chaudhuri et al. (Chaudhuri et al., 2007). Heads from 75 to 200 adults were extracted in 100–200 of 0.1M perchloric acid; extracts were filtered through 0.2μm filters. Ten microliters of each extract were injected for each sample. The mobile phase contained 75mM sodium phosphate, pH 3.0, 1.4mM octanesulfonic acid, 25μM ethylenediamine tetraacetic acid, 100μl/L triethylamine, and 7% acetonitrile. Separations were performed with isocratic flow at 1 ml/min. Amines were detected with an ESA electrochemical analytical cell (model 5011; channel 1 at 50 mV, channel 2 at 300 mV). Pteridines were detected with a linear model LC305 fluorescence detector at excitation wavelength 360nm and emission wavelength 465nm. Pool sizes were determined relative to freshly prepared standards (Sigma-Aldrich, St. Louis, MO). Analysis was performed using ESA CoulArray software.
Locomotion Assay
Locomotion was assayed as described previously (Carbone et al., 2006) with modifications. Adult males and females (15-20 individuals of each genotype), 2-3 days post-eclosion, were collected and transferred to individual vials containing normal media for 1hr prior to assays. The locomotor assay was started by subjecting the fly to gentle mechanical disturbance. The mobility of the fly was measured by counting the number of seconds the fly was in motion during the 45sec period immediately after the disturbance. All the assays were conducted during the same period of the day.
Results
The Punch protein is elevated in fly neurons expressing human or fly UBE3A
Our approach to the identification of UBE3A protein targets using proteomic profiling in Drosophila head extracts has been described previously (Reiter et al., 2006). In short, using the Drosophila GAL4/UAS system (Brand and Perrimon, 1993; Duffy, 2002) we expressed high levels of hUBE3A protein using the Heatshock-GAL4 driver (HS-GAL4). We then made cytoplasmic protein extracts from HS-GAL4 alone and HS-GAL>UAS-hUBE3A fly heads. Using this method, we identified a protein spot at approximately pI 6 and molecular weight ∼30-40kDa which dramatically increased in intensity upon UAS-hUBE3A transgene induction (Fig. 1). This protein spot was cut from three independent polyacrylamide gels for LC-MS-MS identification and found to be an enzyme involved in monoamine synthesis known as Punch (CG9441-PB), also known as GTP cyclohydrolase I in mammals and flies (Mackay and O'Donnell, 1983; Weisberg and O'Donnell, 1986). It is important to note that we expected our screen to identify proteins that would be down-regulated by hUBE3A transgene expression because mono-ubiquitination by hUBE3A should in most cases lead to poly-ubiquitination and subsequent degradation by the ubiquitin proteasome system (UPS). That is clearly not the case for this form of the Punch protein, which shows elevated protein levels on 2D analysis using both human UAS-hUBE3A and fly UAS-Dube3a over-expression constructs (data not shown). Based on the estimated size of this protein on both 2D and 1D polyacrylamide gels, we propose that the Punch isoform we identified is the 32kDa Pu-RB form (NP_726037). In contrast, GTP cyclohydrolase isoforms Pu-RA (NP_726038) and Pu-RC (NP_523801), which like Pu-RB are catalytically active (Funderburk et al., 2006), but whose transcripts initiate from an alternative promoter in the same locus (McLean et al., 1993), are not responsive to changes in Dube3a levels (see below).
Figure 1. Expression of hUBE3A up-regulates Punch expression in head extracts.
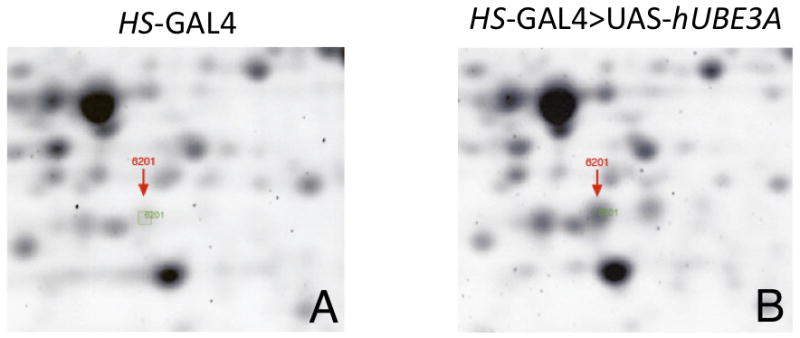
Head extracts from HS-GAL4 alone (A) or HS-GAL4> UAS-hUBE3A were subjected to 2D-gel analysis. Sypro Ruby protein staining of the gels revealed spot #6201, indicated by the red arrow, was elevated in response to increased hUBE3A (A vs B). This protein spot corresponds to an ∼30-40 kDa protein with an isoelectric point of ∼ 6.0. The spot was cut from 3 different gels and identified by mass spectrometry to be the Drosophila GTPCH/Punch protein.
Punch mutations are able to enhance Dube3a over-expression phenotypes in the eye
We had previously determined that over-expression of Dube3a using the eye specific GAL4 driver gmr-GAL4 results in a rough eye phenotype suitable for genetic screening (Peterson and Reiter, 2006). In order to determine if Punch and Dube3a are involved in overlapping genetic pathways, we used a transgenic Dube3a line that due to transgene position-effect shows only a slightly rough appearance at 25°C when crossed to gmr-GAL4 (Fig. 2B vs 2A). Three different heterozygous mutations in the Punch gene enhanced the gmr>Dube3a rough eye phenotype causing a glazed appearance, loss of inter-ommatidial bristles and often displaying yellowish discoloration indicative of underlying neurodegeneration (Fig. 2C-E). In the heterozygous state these mutations in Punch showed no eye phenotype when crossed to the gmr-GAL4 driver alone and the individual UAS-Dube3a stocks without gmr-GAL4 do not have rough eyes (data not shown). These results suggest that mutations in Punch can act as genetic enhancers of Dube3a over-expression phenotypes.
Figure 2. Mutations in Punch enhance a Dube3a over-expression rough eye phenotype in the eye.
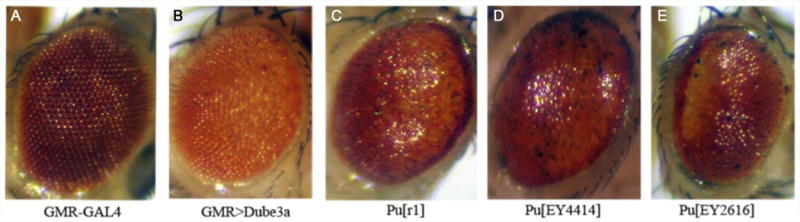
Drosophila eye phenotypes were photographed using a dissecting microscope at 6.3× magnification. (A) Control flies expressing one copy of gmr-GAL4 alone. (B) Expression of Dube3a using gmr-GAL4 causes a mild rough eye phenotype at 25°C with some disorganization of inter-ommatidial bristles, but no glazing or loss of individual ommatidia boundaries. (C-E) This phenotype is enhanced when any of three different alleles of the Punch mutant alleles are also included in the background (Pur1, PuEY4414 and PuEY2616). In all three cases there is loss of and/or fusion of interommatidial bristles and an overall glazed appearance to the eye. Heterozygous mutations in Punch alone do not result in a rough eye phenotype when crossed to gmr-GAL4 and look identical to the eye in panel A (data not shown).
Punch protein isoform B levels change as a result of Dube3a over-expression or loss of function in the fly brain
We performed quantitative infrared (IR) Western blot analysis on fly head extracts from HS-GAL4 alone as well as HS>UAS-Dube3a, human UAS-hUBE3A, fly ubiquitination defective UAS-Dube3a-C/A and a Dube3a loss of function mutant (Dube3a15b) (Fig. 3A). The UAS-Dube3a-C/A construct is the fly orthologue of the human UBE3A-C833A mutation that has been shown in vitro to bind substrates but is unable to transfer a ubiquitin molecule to these substrates (Huibregtse et al., 1993). The α-Punch antibody detects 3 bands at 32kDa, 36kDa, and 38kDa, proteins previously shown to be catalytically active GCH1 enzymes (Funderburk et al., 2006). We observed a >1.5 fold increase in the level of expression of the lowest, ∼32kDa, Punch protein band when we expressed either hUBE3A or the Dube3a-C/A mutant (Fig. 3A). We also observed a slight decrease in the ∼32kDa form in the Dube3a15b homozygous mutant animals, but no apparent change in Punch protein when the wild type Dube3a was expressed. The 36kDa and 38kDa bands also were elevated by either hUBE3A or Dube3a-C/A over-expression (Fig. 3).
Figure 3. Quantitative Western blot analysis of Punch and Dube3a in head extracts from Dube3a over and under-expressing flies.
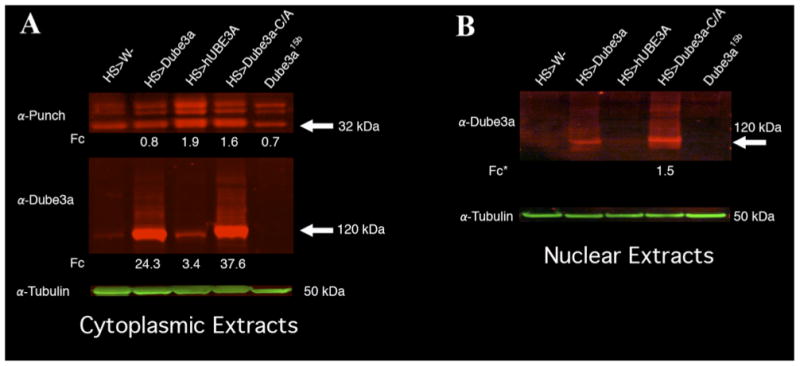
(A) Three distinct Punch bands could be detected in cytoplasmic protein extracts from fly heads expressing HS-GAL4 (1), HS>Dube3a (2), HS>Dube3a-C/A or Dube3a15b mutants. The ∼32kDa band changed intensity in HS>Dube3a-C/A and HS>hUBE3A extracts as compared to HS alone. We could also clearly detect an increase in Dube3a protein levels upon Dube3a transgene expression for both wild type and the C/A mutant (lower panel). A polyubiquitin-like smear could be seen in both the Dube3a and Dube3a-C/A over-expressing lanes. Although the expression of hUBE3A did not appear to polyubiquitinate endogenous Dube3a, it did result in a >3 fold increase in signal. The green band at the bottom is the α-Tubulin loading control for both panels. Quantification is indicated as fold change (Fc) in expression of normalized levels relative to control. (B) Nuclear extracts from fly heads expressing HS-GAL4, HS>Dube3a, HS>Dube3a-C/A or Dube3a15b mutants. A band of ∼120kDa could be detected using an α-Dube3a antibody in all samples except for the homozygous Dube3a15b mutants (last lane). There was also a pronounced polyubiquitin-like smear in the Dube3a and Dube3a-C/A expressing lanes indicating that polyubiquitinated Dube3a is present in the nucleus. The fold change (Fc*) is relative to wild type Dube3a expression on this blot since there were almost undetectable levels of Dube3a in the HS>w- fraction alone (first lane).
Since the ubiquitination-defective Dube3a-C/A form could significantly increase the observed protein levels of all Punch bands, we investigated the possibility that this regulation occurs via the transcriptional co-activation function of the Dube3a protein. It has previously been shown that at least in mouse neurons one form of Ube3a protein is exclusively nuclear, consistent with a role in transcriptional regulation (Dindot et al., 2008). The first task, therefore, was to establish if fly Dube3a-C/A is present in the nucleus at significant levels. Quantitative IR Western blot analysis of nuclear enhanced extracts indicate that both transgenic Dube3a and Dube3a-C/A can be detected at high levels in nervous system nuclear extracts, although endogenous Dube3a could not be detected at all in the nucleus (Fig. 3B). No Dube3a protein could be detected in the Dube3a15b homozygous mutants as predicted (Fig. 3B). It is also significant to note that both the wild type Dube3a and the Dube3a-C/A protein appear to be highly ubiquitinated in the nuclear fraction as indicated by the high molecular weight smear (Fig. 3B). Since UBE3A is known to trans-ubiquitinate itself in vitro, it is, perhaps, not surprising to find that the levels of the enzymatically defective Dube3a-C/A construct were more stable in vivo in both the cytoplasm and the nucleus (Fig. 3A lower and 3B).
Endogenous Dube3a protein can trans-ubiquitinate catalytically inactive Dube3a-C/A in vivo
We were interested in determining if the ubiquitination of both wild type and mutated Dube3a noted in both cytoplasmic and nuclear extracts (Fig. 3) was indeed caused by endogenous Dube3a acting on the transgenic Dube3a proteins in vivo. For these experiments we constructed transgenic fly lines that expressed the pan-neuronal GAL4 driver elav and another line containing an N-terminally FLAG epitope tagged Dube3a-C/A-FLAG construct under the control of UAS, both in a Dube3a15b mutant background. This allowed us to express the ubiquitination defective form of Dube3a in a Dube3a deficient background and also to distinguish this Dube3a-C/A-FLAG transgenic protein from endogenous Dube3a protein.
We found that Dube3a protein levels were elevated by 12-fold over wild type levels when we expressed Dube3a-C/A in a Dube3a+ background as determined by quantitative IR Western blot of cytoplasmic extracts (Fig. 4A). The amount of Dube3a protein detected when Dube3a-C/A-FLAG was expressed in the Dube3a15b mutant background was only 3-fold greater than wild type levels. This implies that the additional Dube3a protein detected in the elav>Dube3a-C/A lane is from an increase in endogenous Dube3a expression levels, since the Dube3a-C/A-FLAG genomic insertion site and the elav-GAL4 driver are the same in both wild type and Dube3a15b mutant backgrounds (Fig. 4A; far right lanes). This hypothesis was confirmed by using an α-FLAG antibody on the same membrane that revealed the expression levels for transgenic Dube3a-C/A-FLAG proteins in these two extracts differ by only ∼1.6 fold, well within the error range for quantitative detection (Fig. 4B). In addition, a putative poly-ubiquitin smear can be observed in the extracts from Dube3a+ animals but not Dube3a15b mutant background animals (Fig. 4B) indicating that endogenous Dube3a can indeed promote the trans-ubiquitination of transgenic Dube3a-C/A in vivo.
Figure 4. Cytoplasmic extracts from flies expressing N-terminal FLAG tagged Dube3a-C/A-F in a Dube3a mutant background.
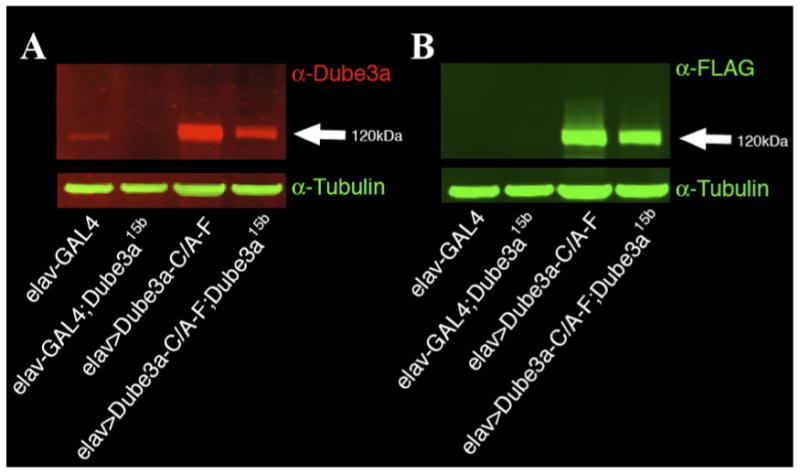
(A) Fly head extracts probed with α-Dube3a monoclonal antibody identify a ∼120kDa band in all lanes. This band is quite faint in the elav-GAL4 extract and absent in the elav-GAL4; Dube3a15b background, but clearly detected in both extracts where transgenic Dube3a-C/A-F is being expressed in neurons. Although there is a 3-fold increase in Dube3a protein from the transgene detected in the Dube3a15b mutant background over elav-GAL4 alone, there is a 12-fold increase in Dube3a protein levels when Dube3a-C/A-F is expressed in the Dube3a+ background indicating that the Dube3a-C/A transgene alone is not responsible for this signal. (B) Probing this same blot with α-FLAG reveals that the level of transgenic Dube3a-C/A-F being expressed in both extracts is less than 1.6-fold different. A putative poly-ubiquitin smear can also be observed in the Dube3a+ background indicating that endogenous Dube3a can trans-ubiquitinate the transgenic Dube3a-C/A-F protein, but a much fainter smear may also be visible in the Dube3a15b background as well.
Punch B transcript levels are modulated by changes in Dube3a
Since the ubiquitin defective, but presumably transcriptionally co-activating Dube3a-C/A construct was able to elevate Punch protein levels, we next investigated transcriptional changes for Punch transcripts as well as other monoamine pathway enzymes including pale (tyrosine hydroxylase), Ddc (dopamine decarboxylase and Tbh (tyroside beta-hydroxylase) (Fig. 5). There are three splice forms of Punch so we used qRT-PCR primers and probes for both shared regions and regions unique to Pu-RB or Pu-RA/RC. We noted a 3.6+1.6 fold increase in Pu-RB transcript in brains of flies expressing wild type Dube3a and a 15.7+7.6 fold increase in Pu-RB transcript in the brains of flies expressing Dube3a-C/A. In addition, we detected a 4.7+1.4 fold decrease in Pu-RB transcript levels in the brains of Dube3a15b homozygous mutant flies (Table 1). Despite this strong effect of Dube3a loss of function, Pu-RB transcript was detectable in the mutant, indicating that transcriptional regulation of Punch is not solely dependent on Dube3a levels. Interestingly, no significant changes were detected, in either the mutant or over-expression lines, in the levels of Pu-RA/RC transcripts, which are derived from an alternate promoter in the Punch locus (McLean et al., 1993). Therefore, the observed parallel elevation in the 36kDa and 38kDa Punch polypeptides derived from the RA/RC transcripts must arise by a mechanism distinct from that of the RB product, as discussed below. We detected no changes in transcription greater than 2-fold up or down for pale, Ddc, or Tbh transcripts in Dube3a15b mutants (Table 1). We did, however, see a marginal decrease in pale transcripts in the Dube3a-C/A expressing flies. This decrease should have no effect on the production of dopamine, however, since the rate limiting co-factor for monoamine synthesis, THB, is dependent on Punch protein levels. Our results suggest that the Pu-RB transcript can be regulated by changes in Dube3a expression levels through a mechanism distinct from its ubiquitination function since elevated Pu-RB transcripts are observed when either the wild type or ubiquitin ligase-deficient forms of Dube3a are expressed.
Figure 5. The key enzymes of the dopamine synthesis pathway.
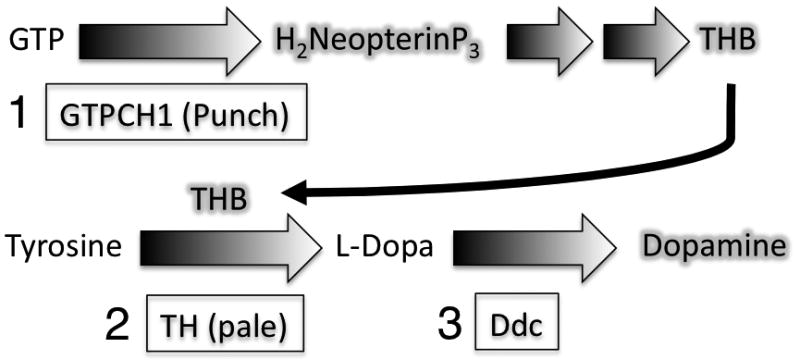
The rate-limiting step in monoamine synthesis is the production of a key cofactor, tetrahydrobiopterin (THB), which is regulated by the enzyme GTP cyclohydrolase I (Punch) that makes dihydroneopterin triphosphate from guanosine triphosphate (1). L-Dopa is then produced from tyrosine by the enzyme tyrosine hydroxylase (pale) in the presence of the THB co-factor (2). Finally, dopa decarboxylase produces dopamine from L-dopa (3).
Dube3a levels coincide with changes in dopamine levels in the brain
The observation that Dube3a can regulate expression levels of Punch-PB led us to the hypothesis that Dube3a might be able to regulate the production of monoamines in the adult fly brain through regulation of the activity of the rate-limiting enzyme GTP cyclohydrolase I (Punch). To test this hypothesis we manipulated Dube3a levels in the fly nervous system using the pan neural elav-GAL4 driver as well as in Dube3a15b homozygous mutant animals and Dube3a15b heterozygotes. We also tested an independently generated loss of function allele Dube3a80, which also deletes part of the upstream gene CG7600, to determine if monoamine precursors and dopamine levels respond as predicted if Dube3a can regulate Punch levels.
The rate-limiting step in monoamine synthesis is the production of a key co-factor known as tetrahydrobiopterin (THB) that is synthesized from the product of the GTP cyclohydrolase I reaction, dihydroneopterin triphosphate (Fig. 5). Expression of an RNAi construct designed to down-regulate Dube3a levels (UAS-RNAi-Dube3a) using the elav-GAL4 driver resulted in statistically significant decreases in both THB and neopterin levels in newly eclosed adult fly brains (Fig. 6A, C). As predicted, Dube3a15b and Dube3a80 mutant animals also had lower levels of these cofactor components compared to wild type controls (Fig. 6B, D). In contrast, expression of both wild type and the ubiquitination defective form of Dube3a caused statistically significant increases in both THB and neopterin (Fig. 6A, C). We also found a significant increase in dopamine levels when Dube3a constructs were expressed using elav-GAL4 and a significant decrease in dopamine levels when RNAi against Dube3a was expressed or in the Dube3a15b mutant animals (Fig. 6E, F).
Figure 6. Changes in Dube3a levels in the brain cause changes in the products of the THB-mediated dopamine synthesis pathway.
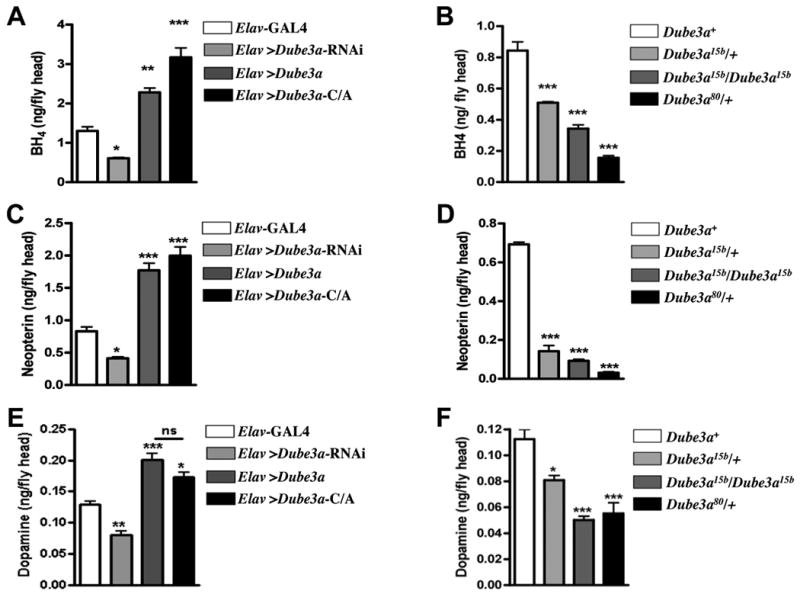
Heads were collected from adult Drosophila 3-5 days post-eclosion. Each Dube3a strain was maintained in a w- background. For each genotype, n=3-5, and each sample contained 80–150 flies with a male: female ratio of 1:1. (A) THB levels are reduced in flies expressing UAS-Dube3a-RNAi in all neurons via the elav-GAL4 driver, which results in a functional knockdown of Dube3a protein (data not shown). Over-expression of Dube3a leads to a significant elevation of THB, as does expression of the dominant negative Dube3a-C/A mutant, which can bind to but cannot ubiquitinate proteins. (B) In agreement with the diminished THB pools observed when Dube3a RNAi is expressed in neurons, both homozygous Dube3a15b and heterozygous Dube3a15b and Dube3a80 mutations result in decreased levels of THB. (C) In direct correlation with THB levels in over-expression and Dube3a RNAi heads, neopterin levels are reduced in flies expressing UAS-Dube3a-RNAi and elevated in flies with excess Dube3a, due to either over-expression of wild type Dube3a or the accumulation of stable Dube3a-C/A mutant protein. (D) Similarly, homozygous Dube3a15b and heterozygous Dube3a15b and Dube3a80 mutations result in depleted neopterin stores. (E) As predicted by the changes in THB levels, dopamine levels are significantly diminished by expression of UAS-Dube3a-RNAi in neurons and significantly elevated as a result of either wild type Dube3a expression or expression of the enzymatically defective Dube3a-C/A protein. (F) Homozygous Dube3a15b and heterozygous Dube3a15b and Dube3a80 mutations cause reductions in dopamine pools, consistent with the depletion of the THB cofactor required for dopamine synthesis. All data were analyzed by one-way Anova with a Bonferroni post-test for statistical analysis (*, p<0.05, **, p<0.01, ***, p<0.001).
To gauge the phenotypic significance of these changes in dopamine levels we measured activity levels of male flies expressing the various levels of Dube3a. We found that Dube3a-dependent changes in dopamine directly corresponded to changes in activity (Fig. 7), a behavioral measure of dopamine changes in the brain (Pendleton et al., 2002). Similar activity changes were observed in females (data not shown) and were therefore not related to mating activity (Liu et al., 2008). Taken together these data suggest that Dube3a can regulate both the production of dopamine though the regulation GTP cyclohydrolase I and the locomotor activity level of flies in both a positive (too much Dube3a) and negative (too little Dube3a) direction.
Figure 7. Dube3a expression levels have a direct effect on activity in adult flies.
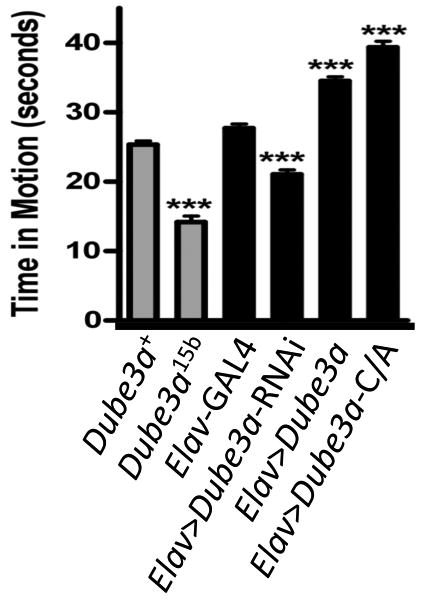
The changes in dopamine levels described in Figure 6 were directly correlated with changes in activity levels in adult male flies. Similar effects were observed in assays of female flies (data not shown). Decreasing dopamine levels in neurons by expression of UAS-Dube3a-RNAi or in the Dube3a15b mutant resulted in decreased activity, while elevation of dopamine levels via Dube3a or Dube3a-C/A over-expression caused an increase in activity. Activity was measured by determining the amount of time individual flies spent in motion over a period of 45 seconds, with n=20. All data were analyzed one-way Anova with a Bonferroni post-test for statistical analysis (*, p<0.05, **, p<0.01, ***, p<0.001).
Discussion
Finding proteins or genes regulated by UBE3A that result in neurological defects is a daunting task. Unlike the analysis of mutants for a developmental pathway which exhibit obvious phenotypic endpoints, it is clear from phenotypic variability in both AS and duplication 15q autism, that disruption of UBE3A pathway members may result in subtle synaptic or biochemical changes in the brain that are difficult to detect. For example, it has only recently been determined that loss of Ube3a results in a defect in neocortical plasticity, despite the fact that this mouse model was generated over ten years ago (Yashiro et al., 2009). Just generating these AS animal models is not enough, one must also take maximum advantage of the particular strengths of these models. For example, behavior and neuroanatomical studies are more suited to the mouse model while genetic pathway and biochemical analysis is better suited to the fly model. Here we have taken a strictly biochemical approach to the identification of Dube3a targets in Drosophila. We have identified a protein that not only changes expression as a result of changes in Dube3a but also has a direct effect on brain neurochemistry related to monoamine pools. We also have demonstrated, for the first time in neuronal tissues that this regulation is at the transcriptional level and is not dependent on the ubiquitin ligase function of Dube3a. We also demonstrated that endogenous Dube3a can ubiquitinate ectopically expressed Dube3a proteins in vivo, a phenomenon that was assumed from in vitro work but never actually demonstrated in animals. Our results with the FLAG-tagged form of Dube3a also suggest that this transgenic construct can increase expression at the endogenous Dube3a locus (i.e. Dube3a may regulate its own expression) (Fig. 4).
It is not surprising to find Dube3a in the nucleus, per se, since it has been known for some time that Ube3a antibodies show a nuclear signal and that at least two splice-forms of Ube3a localize to the nucleus in the mouse (Dindot et al., 2008; Reiter et al., 2006). In this case, we see changes in both transcript and protein levels in Punch-RB, as well as downstream up-regulation of dopamine when we over-express a ubiquitination-defective form of Dube3a in neurons. These results are bolstered by the observations that Punch transcription levels decrease in a homozygous Dube3a mutant, but are still detectable, suggesting that Dube3a may act as a transcriptional co-activator in fly neurons just as it does in cultured cells with regards to the human steroid hormone receptor (Ramamoorthy and Nawaz, 2008). A recent survey of gene expression changes in the cerebellum of Ube3a deficient mice also supports the argument that transcriptional regulation may play an important role the pathogenesis of AS. Low et al. found that 89% of transcripts that were differentially expressed in Ube3a deficient versus wild type mice were down-regulated in the Ube3a deficient brain consistent with the idea that transcriptional co-activation by Dube3a may be just as critical as ubiquitination (Low and Chen, 2010). The possibility that transcriptional change may be at least partially responsible for the human AS phenotype are bolstered by the identification of individuals with AS-like features who have mutations in TCF4 (Takano et al., 2010), which encodes a transcription factor protein, and MeCP2 (Milani et al., 2005; Watson et al., 2001), which encodes a transcriptional repressor protein. The possibility that UBE3A is a transcriptional co-activator in conjunction with TCF4 in humans has not yet been investigated, but the exploration of the interaction between Dube3a and the fly orthologue to TCF4, the daughterless transcription factor, could be an interesting avenue of research in flies, leading to a better understanding of the cadre of genes regulated at the transcriptional co-activation level by UBE3A in humans.
In principle, the elevation in transcription of the Punch locus could occur through one of two mechanisms. The ubiquitination function of Dube3a could act to remove a transcriptional silencer of Punch, indirectly stimulating elevated Punch expression. Alternatively, the transcriptional co-activator function of Dube3a could directly, or indirectly, lead to stimulation of Punch transcription. Since over-expression of the ubiquitination-defective Dube3a-C/A mutant led to elevated transcription (and subsequent elevation of translation) of Punch mRNA, we conclude that the ubiquitination function of the Dube3a enzyme plays no detectable role in the regulation of GTP cyclohydrolase expression in flies.
The modulation of GTP cyclohydrolase synthesis has direct consequences for the production of the monoamines, dopamine and serotonin, and therefore, in synaptic function and downstream behaviors. The GTP cyclohydrolase catalytic function, the conversion of GTP to the pteridine dihydroneopterin triphosphate, is the rate-limiting step in the production of THB. THB is a redox cofactor that is absolutely required by the rate-limiting enzyme in dopamine biosynthesis, tyrosine hydroxylase (TH), for the conversion of tyrosine to 3, 4-dihydroxyphenylalanine (L-Dopa), which is subsequently converted to dopamine (Axelrod, 1971; Nagatsu et al., 1964). Drosophila TH, encoded by pale (ple) (Neckameyer and White, 1993), shares 60% amino acid similarity with human TH (Neckameyer et al., 2005). TH catalytic activity in Drosophila, is tightly regulated by availability of the THB cofactor, and therefore by GTP cyclohydrolase modulation, as it is in mammals. In heterozygous Punch mutants, reductions in THB pools are closely mirrored by similar deficits in TH activity and in dopamine pools (Chaudhuri et al., 2007; Krishnakumar et al., 2000; Mackay et al., 1985). Similarly, mutations in the human GCH1 locus lead to the hereditary diseases hyperphenylalaninemia and Dopa-responsive dystonia (reviewed in (Thony et al., 2000)). This protein, like TH, is also highly conserved: the human and Drosophila GTPCH proteins share 80% similarity within the catalytic core, diverging only in N-terminal domains that serve to regulate catalytic activity (Funderburk et al., 2006; McLean et al., 1993).
The human GCH1 gene encodes several isoforms of GTP cyclohydrolase I, only one of which is enzymatically active. The remaining forms are truncated at the C-terminus and are thought to have regulatory functions. In contrast, the Punch locus of Drosophila encodes at least 3 isoforms of GTP cyclohydrolase, all sharing identical C-terminal catalytic domains and therefore, all are catalytically active. Each isoform has a unique N-terminal domain originating through a combination of alternative RNA splicing and alternative promoter use. Interestingly, Pu-RB (originally designated as GTPCH isoform A in McLean et al., 1993) is transcribed from a different promoter than the remaining forms, and this promoter must therefore possess target sequences for a transcription factor capable of functionally interacting with Dube3a or that is itself regulated by Dube3a.
While there is concordance between the effects of varying Dube3a expression on the Pu-RB transcript and protein isoform levels, the levels of Punch isoforms RA and RC appear to be elevated in parallel with the RB isoform, despite the apparent lack of RA/RC transcriptional response when the human UBE3A or the ubiquitination-defective form of Dube3a is expressed. Since the levels of Transcripts RA and RC do not change, one explanation for this observation is that the elevated levels of Isoform RB serve to stabilize the remaining isoforms in the GTP cyclohydrolase homodecamer complex. All isoforms have identical catalytic and homomultimer interaction domains, differing only in their N-terminal regulatory domains. Therefore, the excess RB polypeptides have the capacity to associate with RA and RC isoforms, and in consequence, could slow the turnover of isoforms that are normally highly sensitive to neural signaling. In principle, such hetero-isoform assemblies could be detected in native electrophoresis gels, but with a molecular mass approaching 500kDa it would be exceptionally challenging. The consequence of these complex interactions is that we cannot conclude that the observed elevation in THB pathway products or dopamine are due solely to the action of Dube3a in regulating RB transcription. These complex relationships between isoforms may also contribute to the enhanced Dube3a over-expression phenotype in the adult eye. We expected to observe suppression of the Dube3a eye phenotype in Punch mutant backgrounds, but instead found that the eye phenotype was enhanced. This result may be due to uncoordinated expression of the various Punch isoforms in the over-expression background.
Another unexpected outcome of our experiments is that pan-neuronal over-expression of wild type Dube3a results in a 3.6 + 1.6 fold elevation in Punch-RB transcript (Table 1), while the wild type form has a modest effect on Punch RB protein levels (Fig. 3). Since we observe elevation of both the THB pathway components and dopamine pools in our neurochemical analysis and observe a functional consequence of these modulations in levels of dopamine, we infer that the immunoblots are perhaps not as sensitive in quantifying expression levels. We do not observe, nor did we expect, a precise correspondence between the transcriptional effects of over-expressing the wild type and ubiquitination-defective forms of Dube3a and the THB and dopamine endpoints. Under normal conditions, the expression of Punch is rate-limiting for THB and dopamine production, but under over-expression conditions it is expected that other components of these biosynthesis pathways will become limiting to some extent. Moreover, the THB and dopamine pathways are very sensitively regulated by post-translational mechanisms that include end-product feedback inhibition and phosphorylation or dephosphorylation of both GTP cyclohydrolase and tyrosine hydroxylase (Axelrod, 1971; Funderburk et al., 2006; Neckameyer et al., 2005; Thony et al., 2000). These homeostatic mechanisms can be over-ridden by over-expression of Punch only to a point, as sensitive regulation of these pathways is critical for neuronal function.
The consequences of mutations in the Punch locus are varied as expected for the rate-limiting step in the biosynthesis of a cofactor that is not only required for dopamine synthesis, but for the synthesis of serotonin and nitric oxide, as well (Thony et al., 2000). Serotonin deficits associated with Punch mutations have been linked to developmental abnormalities including failure of ectodermal cell movements during gastrulation and in cuticular patterning (Colas et al., 1999), while diminished production of dopamine results in aberrant tracheal cell migration in Drosophila embryos (Hsouna et al., 2007). Subsequently, abnormalities in dopamine pools lead to variations in activity/locomotion, as well as to altered stress responses (Chaudhuri et al., 2007). There are clear parallels in these functions with those ascribed to these neurotransmitters in mammals, and suggest that the effect of changes in Dube3a expression in Drosophila will be an important model for identifying the underlying molecular framework of syndromes associated with altered Ube3a gene dosage in humans.
There is at least some evidence that selective serotonin reuptake inhibitors can dampen the hyperactivity and anxiety in both AS deletion (Pelc et al., 2008) and duplication 15q autism individuals (Hogart et al., 2010) indicating that altered serotonin levels contribute to the phenotype in these conditions. Significantly, associations of dopamine-related variation such as dopamine D1 receptor haplotypes, in ASD families (Canitano and Scandurra, 2008) have been reported, and deficits in dopamine-dependent behaviors have been recently in a mouse Ube3a knock-out model of AS (Mulherkar and Jana, 2010). It is likely that both monoamine classes, which are both dependent upon GCHI activity, are altered is ASD individuals. This study is the first step in connecting UBE3A levels with changes in brain neurochemistry, but subsequent studies of THB levels in cerebrospinal fluid from both AS and duplication 15q autism subjects will be required in the future to establish the regulation of GCH1 by UBE3A in the brain extends to humans.
Conclusions
Here we show that Drosophila Dube3a is able to regulate dopamine levels through GCH1 with direct behavioral consequences even in the absence of ubiquitin ligase function. We conclude that this regulation may occur through the transcriptional co-activation domain of the Dube3a protein.
Research Highlights.
Dube3a regulates GCH1 in a non-ubiquitin ligase dependent manner.
Expression of a Dube3a mutant that cannot ubiquitinate can regulate GCH1.
Endogenous Dube3a can ubiquitinate ectopically expressed Dube3a proteins in vivo.
Activity is dependent on Dube3a levels and directly proportional to dopamine levels.
The transcriptional co-activation function of Dube3a may be regulating GCH1 activity.
Supplementary Material
Acknowledgments
We are grateful for primary antibodies supplied by J. Fischer and the Developmental Studies Hybridoma Bank. We also thank the UTHSC Molecular Resource Center for use of the Roche real-time PCR machine and the UTHSC Cancer Center for use of the Odessy IR imaging system. This work was funded in part by grants from Autism Speaks, the Angelman Syndrome Foundation and NIH NINDS R01NS059902 to L.T.R. Additional support was provided to J.M.O. by the University of Alabama. The National Institutes of Health and the University of Alabama had no role in the design of experiments, interpretation of data or submission of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albrecht U, et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- Axelrod J. Noradrenaline: fate and control of its biosynthesis. Science. 1971;173:598–606. doi: 10.1126/science.173.3997.598. [DOI] [PubMed] [Google Scholar]
- Baron CA, et al. Genomic and functional profiling of duplicated chromosome 15 cell lines reveal regulatory alterations in UBE3A-associated ubiquitin-proteasome pathway processes. Hum Mol Genet. 2006;15:853–69. doi: 10.1093/hmg/ddl004. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Canitano R, Scandurra V. Risperidone in the treatment of behavioral disorders associated with autism in children and adolescents. Neuropsychiatr Dis Treat. 2008;4:723–30. doi: 10.2147/ndt.s1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone MA, et al. Phenotypic variation and natural selection at catsup, a pleiotropic quantitative trait gene in Drosophila. Curr Biol. 2006;16:912–9. doi: 10.1016/j.cub.2006.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A, et al. Interaction of genetic and environmental factors in a Drosophila parkinsonism model. J Neurosci. 2007;27:2457–67. doi: 10.1523/JNEUROSCI.4239-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas JF, et al. Maternal and zygotic control of serotonin biosynthesis are both necessary for Drosophila germband extension. Mech Dev. 1999;87:67–76. doi: 10.1016/s0925-4773(99)00140-9. [DOI] [PubMed] [Google Scholar]
- Cummings ED, et al. High-throughput proteomics processing of proteins in polyacrylamide in a multiwell format. J Proteome Res. 2007;6:1603–8. doi: 10.1021/pr060472y. [DOI] [PubMed] [Google Scholar]
- Descheemaeker MJ, et al. Pervasive developmental disorders in Prader-Willi syndrome: the Leuven experience in 59 subjects and controls. Am J Med Genet A. 2006;140:1136–42. doi: 10.1002/ajmg.a.31235. [DOI] [PubMed] [Google Scholar]
- Dindot SV, et al. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17:111–8. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- Duffy JB. GAL4 system in Drosophila: a fly geneticist's Swiss army knife. Genesis. 2002;34:1–15. doi: 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- Funderburk CD, et al. A typical N-terminal extensions confer novel regulatory properties on GTP cyclohydrolase isoforms in Drosophila melanogaster. J Biol Chem. 2006;281:33302–12. doi: 10.1074/jbc.M602196200. [DOI] [PubMed] [Google Scholar]
- Haas KF, Broadie K. Roles of ubiquitination at the synapse. Biochim Biophys Acta. 2008;1779:495–506. doi: 10.1016/j.bbagrm.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger JA, et al. A DRD1 haplotype is associated with risk for autism spectrum disorders in male-only affected sib-pair families. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:628–36. doi: 10.1002/ajmg.b.30655. [DOI] [PubMed] [Google Scholar]
- Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- Hogart A, et al. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010 doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsouna A, et al. Drosophila dopamine synthesis pathway genes regulate tracheal morphogenesis. Dev Biol. 2007;308:30–43. doi: 10.1016/j.ydbio.2007.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, et al. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol Cell Biol. 1993;13:4918–27. doi: 10.1128/mcb.13.8.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, et al. Genetics of Angelman syndrome. Am J Hum Genet. 1999;65:1–6. doi: 10.1086/302473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino T, et al. UBE3A/E6-AP mutations cause Angelman syndrome. Nature Genetics. 1997;15:70–3. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Kolevzon A, et al. Selective serotonin reuptake inhibitors in autism: a review of efficacy and tolerability. J Clin Psychiatry. 2006;67:407–14. doi: 10.4088/jcp.v67n0311. [DOI] [PubMed] [Google Scholar]
- Krishnakumar S, et al. Functional interactions between GTP cyclohydrolase I and tyrosine hydroxylase in Drosophila. J Neurogenet. 2000;14:1–23. doi: 10.3109/01677060009083474. [DOI] [PubMed] [Google Scholar]
- Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64:947–60. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, et al. Increased dopamine level enhances male-male courtship in Drosophila. J Neurosci. 2008;28:5539–46. doi: 10.1523/JNEUROSCI.5290-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low D, Chen KS. Genome-wide gene expression profiling of the Angelman syndrome mice with Ube3a mutation. Eur J Hum Genet. 2010 doi: 10.1038/ejhg.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay WJ, O'Donnell JM. A genetic analysis of the pteridine biosynthetic enzyme, guanosine triphosphate cyclohydrolase, in Drosophila melanogaster. Genetics. 1983;105:35–53. doi: 10.1093/genetics/105.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay WJ, et al. Tissue-specific and complex complementation patterns in the Punch locus of Drosophila melanogaster. Genetics. 1985;111:885–904. doi: 10.1093/genetics/111.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkonen I, et al. Serotonin and dopamine transporter binding in children with autism determined by SPECT. Dev Med Child Neurol. 2008;50:593–7. doi: 10.1111/j.1469-8749.2008.03027.x. [DOI] [PubMed] [Google Scholar]
- McClung C, Hirsh J. The trace amine tyramine is essential for sensitization to cocaine in Drosophila. Curr Biol. 1999;9:853–60. doi: 10.1016/s0960-9822(99)80389-3. [DOI] [PubMed] [Google Scholar]
- McLean JR, et al. Multiple mRNAs from the Punch locus of Drosophila melanogaster encode isoforms of GTP cyclohydrolase I with distinct N-terminal domains. J Biol Chem. 1993;268:27191–7. [PubMed] [Google Scholar]
- Milani D, et al. Another patient with MECP2 mutation without classic Rett syndrome phenotype. Pediatr Neurol. 2005;32:355–7. doi: 10.1016/j.pediatrneurol.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Mulherkar SA, Jana NR. Loss of dopaminergic neurons and resulting behavioural deficits in mouse model of Angelman syndrome. Neurobiol Dis. 2010;40:586–92. doi: 10.1016/j.nbd.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, et al. Tyrosine Hydroxylase. The Initial Step in Norepinephrine Biosynthesis. J Biol Chem. 1964;239:2910–7. [PubMed] [Google Scholar]
- Neckameyer WS, et al. Biochemical conservation of recombinant Drosophila tyrosine hydroxylase with its mammalian cognates. Biochem Genet. 2005;43:425–43. doi: 10.1007/s10528-005-6781-3. [DOI] [PubMed] [Google Scholar]
- Neckameyer WS, White K. Drosophila tyrosine hydroxylase is encoded by the pale locus. J Neurogenet. 1993;8:189–99. doi: 10.3109/01677069309083448. [DOI] [PubMed] [Google Scholar]
- Pelc K, et al. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. 2008;4:577–84. doi: 10.2147/ndt.s2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton RG, et al. Effects of tyrosine hydroxylase mutants on locomotor activity in Drosophila: a study in functional genomics. Behav Genet. 2002;32:89–94. doi: 10.1023/a:1015279221600. [DOI] [PubMed] [Google Scholar]
- Peterson A, Reiter L. Genetic screen for potential protein substrates of the Drosophila E3 ubiquitin ligase gene (dube3a). 47th Annual Drosophila Research Conference; Houston, TX. 2006. [Google Scholar]
- Ramamoorthy S, Nawaz Z. E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl Recept Signal. 2008;6:e006. doi: 10.1621/nrs.06006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, et al. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum Mol Genet. 2006;15:2825–35. doi: 10.1093/hmg/ddl225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeulle C, et al. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain [letter] Nat Genet. 1997;17:14–5. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- Schroer RJ, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet. 1998;76:327–336. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Takano K, et al. Two percent of patients suspected of having Angelman 724 syndrome have TCF4 mutations. Clin Genet. 2010;78:282–288. doi: 10.1111/j.1399-0004.2010.01380.x. [DOI] [PubMed] [Google Scholar]
- Tani Y, et al. Decrease in 6R-5,6,7,8-tetrahydrobiopterin content in cerebrospinal fluid of autistic patients. Neurosci Lett. 1994;181:169–72. doi: 10.1016/0304-3940(94)90586-x. [DOI] [PubMed] [Google Scholar]
- Thony B, et al. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347(Pt 1):1–16. [PMC free article] [PubMed] [Google Scholar]
- Veltman MW, et al. Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet. 2005;15:243–54. doi: 10.1097/00041444-200512000-00006. [DOI] [PubMed] [Google Scholar]
- Watson P, et al. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet. 2001;38:224–8. doi: 10.1136/jmg.38.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg EP, O'Donnell JM. Purification and characterization of GTP cyclohydrolase I from Drosophila melanogaster. J Biol Chem. 1986;261:1453–8. [PubMed] [Google Scholar]
- Wu Y, et al. A Drosophila model for Angelman syndrome. Proc Natl Acad Sci U S A. 2008;105:12399–404. doi: 10.1073/pnas.0805291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. 2009;12:777–83. doi: 10.1038/nn.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.