


Abstract
Transfection of the human malaria parasite Plasmodium falciparum is currently performed with circularised plasmids that are maintained episomally in parasites under drug selection but which are rapidly lost when selection pressure is removed. In this paper, we show that in instances where gene targeting is not favoured, transfected plasmids can change to stably replicating forms (SRFs) that are maintained episomally in the absence of drug selection. SRF DNA is a large concatamer of the parental plasmid comprising at least nine plasmids arranged in a head-to-tail array. We show as well that the original unstable replicating forms (URFs) are also present as head-to-tail concatamers, but only comprise three plasmids. Limited digestion and γ irradiation experiments revealed that while URF concatamers are primarily circular, as expected, SRF concatamers form a more complex structure that includes extensive single-stranded DNA. No evidence of sequence rearrangement or additional sequence was detected in SRF DNA, including in transient replication experiments designed to select for more efficiently replicating plasmids. Surprisingly, these experiments revealed that the bacterial plasmid alone can replicate in parasites. Together, these results imply that transfected plasmids are required to form head-to-tail concatamers to be maintained in parasites and implicate both rolling-circle and recombination-dependent mechanisms in their replication.
INTRODUCTION
Plasmodium falciparum is the cause of the most severe form of human malaria. Currently, there is no vaccine to control this devastating disease and the emergence of parasites resistant to commonly used anti-malarial drugs is a major problem. There is an urgent need to better understand the function of P.falciparum genes (the sequence of all of which will soon be known via the Malaria Genome Project) to assist in the development of new and effective anti-malarial strategies. The development of systems for transient (1) and stable (2–6) transfection of blood-stage Plasmodium parasites has provided critically important tools for the dissection of malaria parasite gene function. In P.falciparum, gene disruption, gene replacement, allelic exchange and foreign gene expression experiments have been described (7–11). Despite these advances, much remains to be learned about the rules that govern the transfection of bacterial plasmids into parasites, particularly in the areas of plasmid entry, replication, segregation and integration. The need to learn more about the process is particularly relevant here as transfection efficiency in P.falciparum remains extremely poor, a factor that greatly limits the technical options available to those researching malaria.
Transfection of P.falciparum blood-stage parasites currently involves the electroporation of parasites with a circular (super-coiled) plasmid. Under drug selection (usually pyrimethamine or other DHFR-inhibitors), a resistant population is established and the majority of parasites in the population maintain the transfected plasmid episomally. It is not known how these plasmids are replicated or segregated during mitosis; however, it is well established that in the absence of drug selection, plasmids are lost in transfected parasite populations. In the rodent malaria parasite Plasmodium berghei, there is evidence to suggest that this occurs because of uneven segregation between daughter merozoites (12).
The unstable nature of the episomally replicating plasmids can be exploited to isolate rare parasites in a transfected population that possess integrated forms. For this, transfected parasite populations are subjected to a drug cycling procedure whereby drug pressure is removed for 3–4 weeks before being reapplied. Drug-resistant populations generally take some time to re-establish and may contain integrated forms. Integration generally occurs via a homologous, single-crossover recombination event provided that an appropriate target sequence is present in the vector. Non-homologous integration is rare but has been reported (3). Generally, if appropriate target sequences are not provided in the plasmid vector there is a tendency for the plasmid to be maintained episomally for an extended period (7).
Our laboratories have been attempting to disrupt a series of genes encoding molecules that are predicted to play a role in parasite invasion of red blood cells. Not surprisingly, most of the plasmids designed for this purpose do not integrate (13), presumably because disruption of these genes affects parasite invasion and is therefore deleterious to blood-stage growth. In these instances, plasmids were maintained episomally for extended periods, usually in the range of 4–6 months. We noted on a number of occasions with different plasmid vectors, that the nature of the episomally maintained plasmid changed over time. This change is from the normally observed unstable replicating form (URF) to an apparently stably replicating form (SRF). In this paper we characterise these forms and show that both URF and SRF have a concatameric structure with plasmids arranged in a head-to-tail orientation. URF concatamers appear to include three plasmid copies and are circular whereas SRF concatamers are much larger (more than nine plasmid copies) and have a complex structure that includes extensive single-stranded DNA. We conclude that it is the formation of this complex structure, rather than the modification of primary sequence, that allows even segregation between daughter merozoites.
MATERIALS AND METHODS
Parasite culture and transfection procedures
Plasmodium falciparum line D10 was cultivated and synchronised as per standard procedures (14,15). Ring stage parasites were transfected with either 50 or 100 µg of CsCl-purified plasmid DNA for transient and stable transfection, respectively, as described previously (3,7). Briefly, for stable transfection, parasites were cultured in 150 cm Petri dishes for 48 h post-transfection prior to drug selection. Pyrimethamine (0.2 µM) was then added for 48 h before reducing to 0.1 µM thereafter. At day 11, parasites were subcultured to a 50 cm Petri dish and one-third of the culture discarded. For transient transfections, transfected parasites were cultured in the absence of selective pressure and genomic DNA (gDNA) prepared at 4 days post-transfection.
Plasmid construction
The plasmid pΔMSP1#2 was constructed by the insertion of a 900 bp XhoI fragment comprising an internal region of the P.falciparum merozoite surface protein-1 (MSP-1) gene into the unique XhoI site of pHC1 (7). This fragment was obtained by PCR amplification of D10 gDNA using the oligonucleotides Pf#1 5′-ATTTCTCGAGAATCCGAAGATAATGACG-3′ and Pf#2 5′-ATTGCTCGAGATCGATGTTTAACATATCTTGGAATTT-3′ into which XhoI sites (bold type) were inserted to facilitate cloning. An additional 300 bp fragment corresponding to the 3′-end of the Plasmodium chabaudi MSP-1 gene was ligated into a unique ClaI site (italicised in Pf#2) located at the 3′-end of the 900 bp fragment. The original purpose of pΔMSP1#2 was to construct an MSP-1 chimera similar to one described previously (10); however, due to a frame-shift mutation in the 300 bp P.chabaudi fragment, pΔMSP1#2 is effectively an MSP-1 ‘knockout’ plasmid as homologous integration would result in disruption of the MSP-1 gene.
Nucleic acids and pulsed-field gel electrophoresis (PFGE)
gDNA was extracted from mixed trophozoite/schizont stage parasites as described (16). Manipulation of recombinant DNA and analysis of nucleic acids by Southern blot hybridisation was carried out using standard procedures (17). Agarose blocks containing intact parasite chromosomes were prepared according to Kemp et al. (18) and chromosomes were separated by PFGE using contour clamped homogeneous electric-field apparatus (CHEF) (19) at 4.2 V/cm and a pulse time of 225 s for 70 h.
For partial restriction endonuclease digest analysis, chromosome blocks were first equilibrated in Tris–EDTA then digestion buffer [containing 1× Multi-Core buffer (Promega), 1 mM dithiothreitol, 0.01% Triton X-100 and 10 µg/ml bovine serum albumin] before incubation at 25°C overnight with 0–3 U of SmaI (Promega). Digestion was terminated by the addition of 50 mM EDTA. Restricted chromosome blocks were separated via PFGE with CHEF apparatus set at 5.6 V/cm and a pulse time of 3 s for 18 h. Molecular weight markers used were λHindIII (Progen) and λ ladder (Bio-Rad).
For irradiation experiments PFGE was carried out in a custom-built CHEF apparatus (20) using gels containing 1.0% chromosomal grade agarose (Gibco BRL) in 0.25× TBE (0.025 M Tris, 0.025 M boric acid, 0.0625 M EDTA). The electrophoresis regime throughout comprised a linear ramp of 90–300 s at 3.8 V/cm for 32 h followed by a second linear ramp of 300–750 s at 3.4 V/cm for 32 h. This regime provides a complete spread of P.falciparum chromosomes. Markers used were the 350 mid-range marker from New England Biolabs (a λ ladder plus HindIII λ fragments) and yeast strain YP148 (courtesy of P. Hieter). The latter strain had been engineered to separate chromosome 15 into two fragments of ~120 and 1050 kb, respectively, both of which carry targets that hybridise to pUC19. On completion, the molecules were transferred by alkali blotting to charged nylon membranes (Hybond N+, Amersham) and hybridised using standard methods to 32P-labelled pUC19 prepared by nick translation (21).
Recovery and retransfection of SRF
gDNA extracted from cycle 2 D10-ΔMSP1#2 parasite (SRF DNA) was treated with DpnI overnight to destroy any of the originally transfected Escherichia coli-derived plasmid that may still have been in the preparation. Following transformation into E.coli, DNA was prepared from individual colonies for restriction analysis. To derive a plasmid pool, 10 000 colonies were scraped into medium, cultured overnight and plasmid DNA purified by CsCl density gradient fractionation. Plasmid DNA (100 µg) was then transfected into D10 parasites and subjected to pyrimethamine selection as described above.
Transient replication assay
In a separate approach to identify sequences that promote plasmid replication in parasites a transient replication assay was established. Briefly DpnI-treated SRF gDNA was linearised with SmaI and religated to obtain monomerised plasmids before recovery in E.coli as described above. Approximately 5000 colonies were scraped into medium overnight for CsCl DNA preparations. DNA (50 µg) was transiently transfected into parasites and gDNA extracted on day 4 post-transfection. The gDNA was treated exhaustively with DpnI, resistant plasmids were recovered in E.coli and the transient transfection procedure repeated.
A random P.falciparum genomic library was similarly examined for sequences that promote replication. Plasmodium falciparum (D10 line) gDNA was digested with RsaI and fragments (generally 0.5–3.0 kb) were purified (BANDPURE™ DNA purification kit, Progen) and ligated into the SmaI site of pGEM4Z (Promega). The ligation mix was transformed into E.coli and approximately 8000 colonies pooled into medium and cultured overnight. DNA prepared from this culture was treated in two ways: (i) it was retransformed into E.coli to calculate the ratio of plasmids containing insert; (ii) it was transiently transfected into parasites where on day 4 the gDNA was extracted and recovered in E.coli as detailed above for recovery of SRF. Analysis involved DNA extraction from E.coli and restriction with EcoRI–PstI to release any insert before electrophoresis. The 1 kb PLUS DNA Ladder™ (Life Technologies) was used as a marker in Figures 6 and 7.
Figure 6.
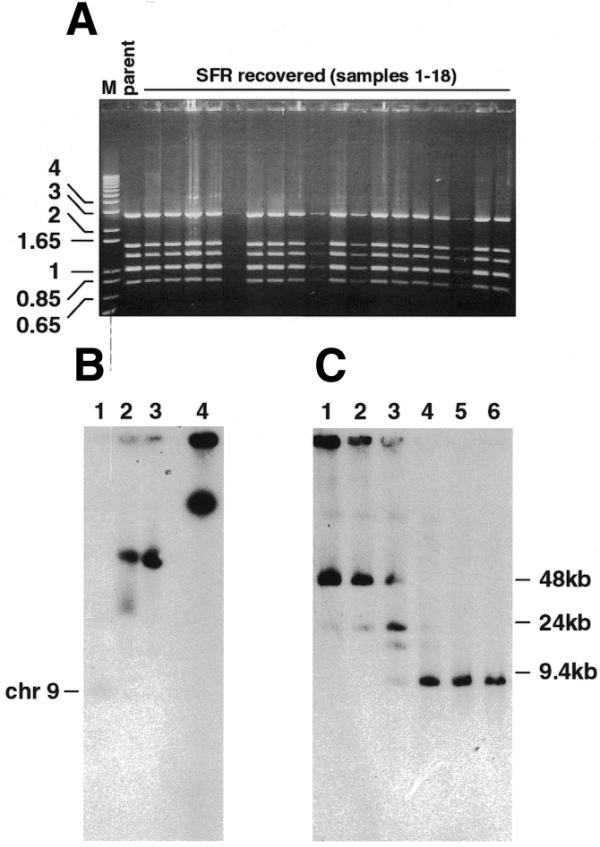
Recovery in E.coli and retransfection in P.falciparum of SRF plasmids. (A) Ethidium bromide-stained agarose gel showing plasmid DNA prepared from individual E.coli colonies obtained following transformation with DpnI-treated SRF gDNA and digested with a combination of BamHI and EcoRI. The pΔMSP1#2 parent plasmid is included (parent). DNA markers in kb (M) are indicated to the left. (B) PFGE of recSRF chromosomes obtained from retransfection of an E.coli recovered SFR plasmid pool. Lane 1, parental D10; lane 2, URF; lane 3, recSRF; lane 4, SRF. (C) recSRF chromosomes were digested at 25°C overnight with varying units of SmaI: lane 1, 0 U; lane 2, 0.01 U; lane 3, 0.1 U; lane 4, 1 U; lane 5, 2 U; lane 6, 3 U. The DNA was transferred to a nitrocellulose membrane and hybridised with the MSP-1 probe.
Figure 7.

Plasmid backbone is sufficient for plasmid replication in parasites. An RsaI plasmid library was constructed from P.falciparum gDNA and introduced into both E.coli (left) and P.falciparum (right). In the case of P.falciparum, transfected parasites were cultured for 4 days before preparation of DpnI-treated gDNA and recovery in E.coli. DNA was extracted from 18 random colonies in each case and digested with a combination of EcoRI and PstI. The restricted plasmids were electrophoresed on 1% agarose gels containing ethidium bromide and visualised via UV transilluminator. DNA markers in kb (M) are indicated to the left.
RESULTS
Continual drug cycling of transfected parasites can lead to the formation of stably maintained episomal plasmids
We have described previously the construction of a plasmid, pΔMSP1, designed to disrupt the MSP-1 gene and our inability to obtain a parasite population with this gene disrupted (10). Transfection of another MSP-1 ‘knockout’ plasmid, pΔMSP1#2, which, following homologous integration, would also disrupt the MSP-1 gene, is described here. This plasmid is identical to pΔMSP1 except that in pΔMSP1#2 the MSP-1 target sequence is slightly modified at the 3′-end (Fig. 1).
Figure 1.

Schematic representation pΔMSP1#2. This plasmid includes a 1.2 kb XhoI (X) insert that contains MSP-1 targeting sequence (MSP-1 KO). The first 900 bp of this insert are P.falciparum MSP-1 sequence and this region was used as a probe throughout these studies (MSP-1 probe). The location of the selectable marker cassette (TgDHFR cassette), the TgDHFR-TS probe and the HSP86 3′-UTR (HSP3′) are shown. A representation of the P.falciparum MSP-1 gene is shown below the plasmid. The locations of relevant restriction enzyme XmnI (Xm), XbaI (Xb), BglII (B), SpeI (S) and EcoRI (E) are indicated. All sizes are to scale with the exception of the plasmid backbone (dashed line).
Following transfection of super-coiled plasmid DNA, parasites were selected with pyrimethamine and a drug-resistant parasite population was established at ∼3 weeks post-transfection. Transfected populations were subjected to drug cycling where pyrimethamine was removed from the culture medium for 3 weeks and then reintroduced until a drug-resistant population was re-established. Parasites were drug-cycled three times and chromosome blocks prepared at various time points and subjected to PFGE and Southern blot analysis. Profiles of drug-resistant parasites obtained before cycling and following one cycle were characteristic of episomes normally seen in transfected P.falciparum parasites (3,7). These show a diffuse hybridisation pattern with a major band independent of chromosome location when compared to the original ethidium bromide-stained agarose gel (Fig. 2A, lanes 2 and 3). The hybridisation pattern observed after two and three cycles in the D10-ΔMSP1#2 population was very different with a dominant slowly migrating species, which also did not correspond to any chromosome (Fig. 2A, lanes 4 and 5). This suggested that the plasmid, now termed SRF, had been modified and was still being maintained extra-chromosomally.
Figure 2.
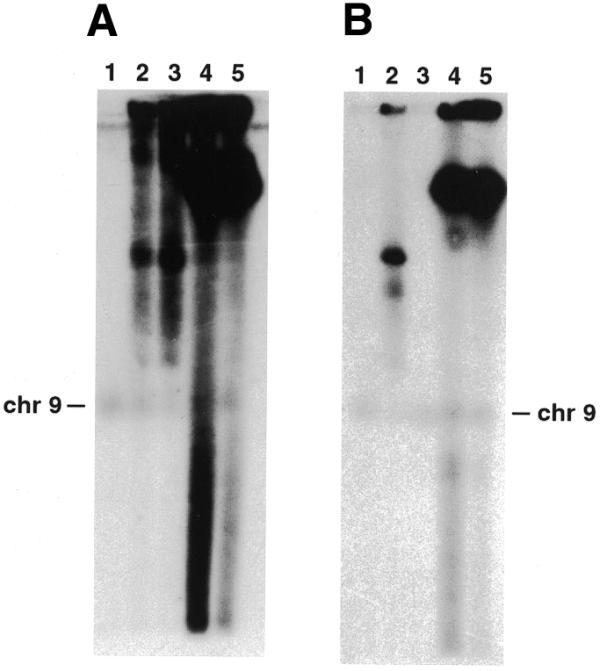
Analysis of transfected D10-ΔMSP1#2 parasite populations at different time points. (A) PFGE separating chromosomes from parental D10 (lane 1) and transfected parasite populations of D10-ΔMSP1#2 after no (lane 2) or one to three cycles in culture (lanes 3–5). Following transfer to a nitrocellulose membrane, the filter was hybridised with the 900 bp MSP-1 probe. Chromosome 9, the location of the endogenous MSP-1 gene, is indicated (chr9). (B) PFGE separating chromosomes from parental D10 (lane 1) and transfected parasite populations of D10-ΔMSP1#2 after no and two drug cycles either on drug selection (lanes 2 and 4, respectively) or maintained in the absence of pyrimethamine selection for 5 weeks (lanes 3 and 5, respectively). Southern blotting and hybridisation was as described in (A).
Following the reintroduction of pyrimethamine in the first and second drug cycle, most of the parasites in the D10-ΔMSP1#2 population died and, consequently, a significant delay (2–3 weeks) was observed before a drug-resistant population was once again established. This was expected due to the known instability of URFs and their loss from parasites in the absence of selection. However, this delay was not observed in subsequent cycles suggesting that SRFs were being stably replicated and evenly segregated. To confirm this, non-cycled parasites (D10-ΔMSP1#2/0) and second cycle parasites (D10-ΔMSP1#2/2) were cultured in the absence of pyrimethamine for 5 weeks and then analysed by PFGE (Fig. 2B). As expected, no hybridisation was detected in D10-ΔMSP1#2/0 parasites maintained in the absence of drug due to the loss of URF DNA (Fig. 2B, lane 3). In contrast, no corresponding reduction in hybridisation intensity was observed in D10-ΔMSP1#2/2 parasites indicating that SRFs were efficiently replicated and evenly segregated between daughter merozoites (Fig. 2B, lane 5). Similarly, in several independent experiments in our laboratories where other target sequences have been used, apparently stably replicating plasmids have been obtained (data not shown). One of these lines, transfected with a plasmid containing a target sequence of rhoptry-associated protein 2, was shown to have an SRF-like profile following PFGE and Southern blotting (R.A.O’Donnell and B.S.Crabb, unpublished data). This indicates that the stability of SRF DNA replication is probably not specific to the target sequence.
Both URF and SRF DNA are concatamers of the parent plasmid but differ dramatically in size and structure
To examine the integrity of the transfected plasmids, gDNA containing URF or SRF DNA from D10-ΔMSP1#2 parasites was subjected to Southern blot analysis (Fig. 3A). gDNA was digested with either XmnI or EcoRI, electrophoresed on a 0.6% agarose gel and transferred to a nitrocellulose membrane. The filter was hybridised with a TgDHFR-TS probe (Fig. 3A) and separately with an MSP-1 target sequence probe (data not shown). The hybridisation patterns of both URF and SRF DNA were identical and as expected for pΔMSP1#2, including when these blots were overexposed, indicating that SRF DNA did not possess major rearrangements or insertions that may explain its stable maintenance.
Figure 3.
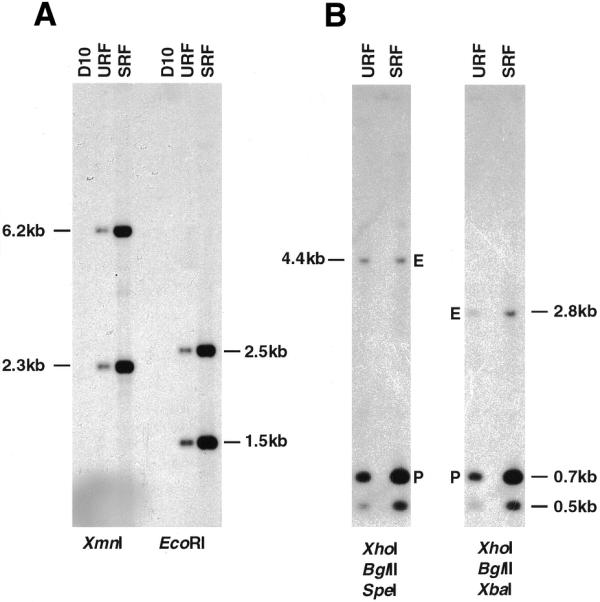
Southern blot analysis of D10-ΔMSP1#2 gDNA following no (URF) or three cycles (SRF). (A) Parental D10, URF and SRF gDNA was digested with XmnI or EcoRI separated on 0.6% agarose gel and transferred to a nylon membrane. The filter was then hybridised with a TgDHFR-TS probe. (B) URF or SRF gDNA was digested with a combination of either XhoI, BglII, SpeI or XhoI, BglII, XbaI as indicated, electrophoresed on 0.6% agarose gel and transferred to nitrocellulose membrane. The filter was hybridised with the 900 bp MSP-1 probe and the signal quantitated by phosphorimager analysis. The larger band (4.4 or 2.8 kb) represents the single endogenous copy of the MSP-1 target sequence (E) while the 0.7 kb species corresponds to the XhoI–BglII fragment derived from the transfected plasmid (P).
It appeared from these blots that SRF DNA was present in greater copy number per parasite than URF DNA. To determine copy number, gDNA was digested with several enzymes to generate fragments of the endogenous MSP-1 locus that were well separated from fragments derived from the MSP-1 insert in pΔMSP1#2 (Fig. 1). These digests were subjected to Southern blot analysis and probed with the MSP-1 internal probe (Fig. 3B). The slowly migrating bands in each panel represent either the 4.4 kb SpeI–BglII or the 2.8 kb XbaI–BglII fragments (labelled E in Fig. 3B) derived from the single copy MSP-1 endogenous gene. The larger of the two lower bands represents the 0.7 kb XhoI–BglII fragment (labelled P in Fig. 3B) derived from the MSP-1 target region of pΔMSP1#2. This sequence is also contained completely within the 4.4 kb SpeI–BglII and the 2.8 kb XbaI–BglII endogenous MSP-1 fragments and also within the MSP-1 hybridisation probe. The intensity of the bands was determined by phosphorimager analysis and the P:E (a measure of plasmid copy number per parasite) was calculated. This analysis revealed that approximately 6 and 18 plasmid copies are present per URF- and SRF-containing parasite, respectively.
Using standard agarose gel electrophoresis and Southern blot analysis, we have observed that when undigested both URF and SRF DNA migrate with the limit mobility (data not shown). We also observed this previously with other plasmids episomally replicating in P.falciparum and suggested that, in general, URFs are probably concatamers (7). To explore the structure of parasite-replicated plasmids further, chromosome blocks were subjected to limited digestion with SmaI (unique in pΔMSP1#2) before separation by PFGE, Southern blotting and hybridisation with the MSP-1 probe. Under these conditions, undigested URF DNA migrates to ∼50 kb whereas SRF DNA remains at the limit mobility (Fig. 4A and B, lanes 1). As expected, when fully-digested both URF and SRF DNA migrate to 8.5 kb, the size of monomeric pΔMSP1#2 (Fig. 4A and B, lanes 4–6). In contrast, following partial digestion, a ladder of bands separated by ∼8.5 kb was observed for both URF and SRF DNA (Fig. 4A and B, lanes 2 and 3). This is characteristic of both concatamers being comprised of a head-to-tail arrangement of pΔMSP1#2 plasmid. For URF, it would appear that the concatamers are mostly circular 3mers because an ∼25 kb species, which probably represents linearised URF concatamer, is the major most slowly migrating species observed following partial SmaI digestion. This is addressed further below. In contrast, the SRF concatamer is comprised of at least nine copies as nine bands of 8.5 kb can be seen before reaching the limit mobility. In lane 3 of Figure 4B, partial digestion has progressed such that there is a decrease in hybridisation as copy number increases from one to nine, indicating that the vast majority of SRF concatamers have been digested at least once. Therefore, the relatively strong hybridisation at the limit mobility in this lane indicates that the concatamer is probably larger than nine plasmid copies.
Figure 4.
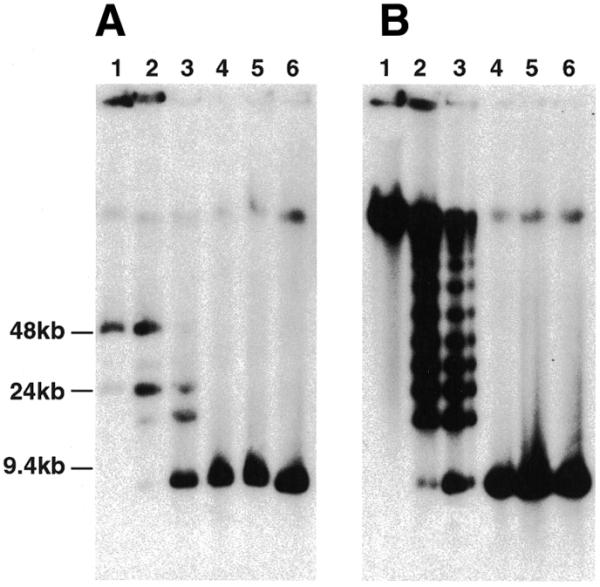
Limited SmaI digestion of URF (A) or SRF (B) chromosome blocks. Blocks were digested at 25°C overnight with varying units of SmaI: lane 1, 0 U; lane 2, 0.01 U; lane 3, 0.1 U; lane 4, 1 U; lane 5, 2 U; lane 6, 3 U, and subjected to an 18 h PFGE procedure as described in the Materials and Methods. The separated DNA was transferred to a nitrocellulose membrane and hybridised with the MSP-1 probe. Molecular weight markers are indicated to the left of (A).
In a separate approach to characterising the structure of URF and SRF DNA, we used varying doses of γ radiation to introduce increasing numbers of single-strand breaks into the DNA molecules. These samples were subjected to PFGE, using different conditions to those described above, Southern blotted and hybridised to a pUC19 probe (Fig. 5). Prior to irradiation (Fig. 5A), most of the URF DNA remained in the slot, with only small amounts entering the gel at X, C and L (the latter barely visible in Fig. 5A). Even the smallest dose of radiation (25 Krad) had an effect, with migration of material to the positions at C, C1, C2 and L. Increasing dosage led to progressive accumulation of band L and finally to its fragmentation, while the group C bands continued to be represented, largely unchanged. The apparent radiation resistance of the molecules in these bands was almost certainly an artefact due to mobilisation of material in the slots (marked P in Fig. 5), balanced by transfer from the group C bands to the fast-moving material at L. This was confirmed by a two-dimensional (2D) PFGE experiment in which we first irradiated a plug with 25 Krad and, following the first PFGE, subjected the track to a further 100 Krad and ran it in a second PFGE at 90° to the first. In this gel (Fig. 5C), material was evidently liberated from the slot by the second radiation dose, forming a smear down to the position of C, while molecules originally at C (and C1, also visible) failed to form a continuous smear, but clearly generated faster running molecules at L.
Figure 5.

PFGE analysis of the effect of ionising radiation from a 60Co source on transfected plasmids. The gels were electrophoresed, Southern blotted and hybridised to a pUC19 probe as described in the Materials and Methods. (A) URF and (B) SRF agarose blocks (P) exposed to 0–400 Krad γ-rays. (C) URF DNA 2D PFGE. An agarose block was first irradiated with 25 Krad and after PFGE, the resulting track was removed, exposed to 100 Krad and electrophoresed at 90° under the same conditions. (D) SRF DNA 2D PFGE. Conditions were the same as for (C), except that the initial agarose block was unirradiated before the first-dimension electrophoresis. Other labels are referred to in the text. Molecular weight markers are indicated to the right of (A) and (B).
These results can be partially understood by reference to the known effects of ionising radiation and PFGE behaviour of differently structured DNA molecules. By their fragmentation at higher doses, the molecules at L are clearly linear. The size of this species is consistent with the presence of a linear trimer as observed by limited digestion (Fig. 4A). The release of small numbers of these molecules by even 25 Krad may reflect their entrapment by radiation-sensitive single-stranded DNA molecules, which could also partly account for retention of signal in the slots (22). In this event, the relatively weak smears running from the slots down to the C position suggest that some of the slot-bound molecules ultimately destined for the bands in this position are released as single-stranded DNA is increasingly degraded. The fact that the C group molecules themselves failed to smear but generated the linear trimers discontinuously (i.e. with a gap between the target molecule and the products) indicates that they consist largely of circular forms of these trimers.
Irradiation of the SRF cells revealed a rather different pattern. As shown in Figure 5B, prior to irradiation most of the target molecules left the slots and migrated mainly as a large slow-moving blob (X), roughly in the same position as the material at C in the URF gel of Figure 5A. However, the unirradiated SRF also displayed a distinct downward trail (XL) ending in a long lens-shaped smear (L) with a front somewhere in the 23–48 kb region of the gel (compare with track 4 in Fig. 2A). The molecules in the SRF were highly sensitive to radiation, 25 Krad completely disrupting the material at X in the control track and generating a long lens-shaped smear. This indicates extensive involvement of radiation-sensitive molecules, most likely single-stranded DNA. The structure of the molecules in the lens-shaped smears resulting from dosage in the lower ranges is not obvious. A 2D PFGE analysis, in which the initial plug was unirradiated and the first-dimension track exposed to 100 Krad prior to the second PFGE (Fig. 5D), showed that a 100 Krad exposure of the long weak trail marked XL in unirradiated cells resulted discontinuously in molecules migrating in the same range as those generated by disruption of the major complex at X. It seems likely that both sets of molecules are similarly structured. Higher doses of radiation do not result in a discrete, relatively radiation-resistant band that would represent a single linear double-stranded DNA species as observed for URF. At 400 Krad, however, such a species does appear to be present at >48 kb, with the downward smear representative of degradation of the double-stranded material. Although it is difficult to be precise about the molecular weight of this species, it would appear to be smaller than the size predicted for a molecule that is at least a 9mer concatamer (∼77 kb) as seen in Figure 4. It is reasonable to suppose that the molecules liberated by radiation of the SRF material comprised linear duplexes held together by single-stranded DNA that was removed with increasing irradiation. Possibilities will be discussed, but in any event, the clear structural differences between the URF and SRF forms are most likely to be relevant to their different segregating properties.
Recovery and retransfection of SRF DNA leads to a reversion to URF DNA
SRF DNA from D10-ΔMSP1#2 parasites was recovered in E.coli. Colonies transformed with this DNA maintained plasmids with restriction enzyme patterns indistinguishable from the parent vector (Fig. 6A). We presume that these plasmids were no longer as large as SRFs but had recombined into smaller forms capable of replication in E.coli. Approximately 10 000 colonies were pooled and cultured overnight, and DNA purified from this culture was transfected into parasites. A drug-resistant parasite population was observed after ∼3 weeks, which is the normal time period for this to occur. PFGE analysis revealed that plasmid DNA from these parasites (now termed recSRF) had reverted to a URF-like hybridisation pattern (Fig. 6B). Partial digestion of recSRF chromosome blocks with SmaI confirmed that plasmid DNA from these parasites was arranged as a head-to-tail circular concatamer of three plasmids (Fig. 6C).
Sequences within the plasmid backbone alone promote replication in P.falciparum parasites
In order to further explore the possibility that SRFs possess an altered or (presumably small) introduced sequence that may explain their improved stability, a transient transfection assay was established in an attempt to select for these forms. SRF gDNA was digested with SmaI and religated to obtain monomeric forms. This ligation mix was transformed into E.coli. About half of the resulting colonies possessed plasmid DNA with a restriction pattern identical to pΔMSP1#2, while the remainder of the colonies possessed plasmid backbone sequence only (data not shown). These latter forms were presumably recombination artefacts as they were smaller than the original plasmid backbone and had deleted the poly-cloning site (data not shown). DNA prepared from a pool of approximately 5000 colonies was transfected into parasites in an attempt to select for forms that replicated more efficiently than the original plasmid. At day 4 post-transfection, gDNA was prepared and treated with DpnI. Following recovery in E.coli, only the recombined form of the vector comprising the vector backbone alone was observed during this process (data not shown). This indicated that there was no dominantly replicating plasmid that contained parasite DNA and suggested that the plasmid backbone alone possesses all the necessary signals required for replication in parasites.
We examined this further in a separate experiment that was designed to search for autonomously replicating sequences (ARSs) in the Plasmodium genome. Here, a random library of RsaI-restricted D10 gDNA was constructed. Plasmid DNA from this library was transformed back into E.coli to determine the proportion of plasmids containing inserts (Fig. 7). It was determined that the vast majority of plasmids contained inserts of 0.5–3.0 kb although a few appeared to possess only the plasmid backbone. The identical DNA preparation was also transiently transfected into parasites as described above and the extracted DNA was recovered in E.coli. The majority of the recovered plasmids appeared to contain only the pGEM4Z backbone (Fig. 7). This experiment was repeated on two further occasions for an identical result. These experiments support our assertion that sequence within the plasmid backbone alone is sufficient for plasmid replication in Plasmodium parasites. However, our inability to isolate an ARS by this approach does not suggest an absence of such sequences in P.falciparum.
DISCUSSION
At the time of transfection, the supercoiled, E.coli-derived plasmids are presumably monomeric. However, our results show that the initial transformants contain plasmids that are head-to-tail trimeric concatamers arranged predominantly as circles. No monomers or dimers were found, implying that concatamerisation is essential for maintenance of these forms. The requirement for concatamerisation may contribute to the substantial delay observed from the time of transfection to the appearance of parasites, and perhaps is one reason as to why transfection in P.falciparum is relatively inefficient and somewhat unreliable. In addition to the reorganisation of the plasmid, our results indicate that URFs fail to survive in the absence of selection, presumably due to ineffective segregation. This is the usual circumstance for plasmids replicating in P.falciparum, and has been shown to be a property of plasmid-borne DNA in genetically transformed rodent malaria parasites (12). In contrast to URF, SRF DNA is maintained as a much larger concatameric species that is effectively segregated between daughter merozoites even in the absence of drug selection. Concatamerisation may be a minimal requirement for segregation of both URF and SRF DNA.
Clearly neither form is maintained as simple linear concatamers. Both migrate by PFGE to positions that are superficially consistent with circular molecules. However, both forms were sensitive to low levels of ionising radiation, the SRF exquisitely so, which would not have generated double-strand breaks. In the case of URF, such a low dose (25 Krad) gave rise to two main species of molecules that had a gap between the target (C, C1 and C2) and product DNA (L), the latter revealed by increasing doses to be linear trimeric duplexes. At first glance this suggests that some of the molecules in URF are linear trimers linked together by single-stranded sequences, necessarily radiation-sensitive. However, the slow migration of the unirradiated URF molecules and the fact that irradiation failed to produce molecules migrating between the large material and the linear trimers clearly indicates that much of this material is in fact circular. A unifying explanation would be that at least some of the molecules are trimeric circles involved in rolling circle replication and carry linear tails with small single-stranded regions, commonly seen in molecules that replicate in this way. Rolling circle replication is usually attributed to organellar or prokaryotic DNAs, but is not unknown in malaria, having been reported as being one element in the replication of the mitochondrial DNA of P.falciparum (23). If the URF molecules replicate in this way, it would account for their initial radiation sensitivity since rolling circle tails frequently carry single-stranded regions. In addition, the increasing accumulation of trimeric linears with dosage would be a direct consequence of double-strand breakage of trimeric circular elements.
The SRF DNA presents a much more complex picture. The partial restriction digests showed that it comprises oligomers up to at least 9mers, but uncomplicated linear molecules would have to be several megabase pairs in length to migrate as slowly as this material does in a chromosomal PFGE (Fig. 2). Furthermore, upon irradiation it did not show the fragmentation pattern characteristic of simple linear duplexes. Further complications include its exquisite sensitivity to ionising radiation, the discontinuous lens-shaped smears produced by the lower levels of radiation and the fact that the higher doses of radiation ultimately liberated linear duplex molecules of at least 48 kb (i.e., at least six plasmid copies). One possibility that partially reconciles these properties of the SRF molecules is that the long linear duplexes we detected by partial digests replicate by a recombination-dependent replication process normally considered the preserve of certain bacteriophage and plasmids (24,25). This is a highly complex process involving multiple recombinational interactions between constituent molecules and can lead to the formation of massive complexes that would certainly account for the slow migration of SRF molecules in PFGE. It may be relevant that this mechanism was detected in malarial mitochondria (23) and in this case appeared to involve recombination between linear tandem arrays originally produced by the rolling circles referred to above.
Applying this model to the transfected plasmid, one might suppose that the first replication event produces linear trimers that for some reason are not heritable in the absence of selection. However, a few cells produce long linear oligomers (9mers or more) which then enter into the recombination-dependent mode and that, unaccountably, are effectively transmitted to daughter merozoites even in the absence of selection. At least one round of drug cycling subsequent to the initial establishment of the URF would be both necessary and sufficient for establishing and selecting the stable SRF forms. This highly speculative model would account for some of the structural complexities of the SRF DNA. Single-stranded molecules are a notable feature of recombination-dependent replication and their abundance in the recombinational aggregates would explain the extreme radiation sensitivity of this material. Increasing radiation would be expected to release linear molecules of various sizes, often featuring single-stranded material or other impediments to migration that would be trimmed off with increasing exposure. This would explain the long discontinuous trails resulting from irradiation of the SRF, and also the breadth of the band formed by the molecules accumulating in and around the 48 kb position, prior to their final fragmentation (seen at the highest dose of radiation used here). The apparent absence of single-stranded material in restricted gDNA shown in Figure 3 probably relates to the vigorous extraction procedure used for the isolation of gDNA, which most likely results in extensive shearing of single-stranded material. All other procedures (Figs 2, 4, 5 and 6) were performed using DNA extracted gently from within an agarose block. This model does not offer any explanation for the different segregational properties of the two forms of DNA, nor does it suggest a specific origin of replication. We found no evidence for the acquisition of sequences in SRF DNA that could aid in either segregation or replication. On the contrary, transient replication assays suggested, surprisingly, that plasmid backbone alone can replicate in parasites apparently more efficiently than plasmids containing P.falciparum sequence.
One of our main reasons for investigating the stability of the transfected plasmids was so that it could be used as a shuttle vector. Shuttle vectors enable elevated expression of foreign and native genes with the benefit of easy recovery and/or removal of a given plasmid. A similar study to ours has been carried out in Trypanosoma cruzi parasites, where the parent plasmid was a monomer and the resultant transfectants contained stable large extra chromosomal circular elements composed of head-to-tail tandem vector repeats (26). In addition, the vector could express foreign genes at elevated levels and be introduced into T.cruzi, Leishmania mexicana or Leishmania donovani. We were interested in utilising SRF as a shuttle vector and addressed the ability of the large stable concatamer to be recovered in E.coli before reintroduction into P.falciparum. Interestingly, we found that upon reintroduction into Plasmodium, the plasmid was no longer stable, but had reverted to the trimer form of URF. This suggests that the complex aggregate that is SRF contains (or readily gives rise to) smaller circular forms that are preferentially replicated by E.coli; the SRF complex itself presumably being too large to be replicated or even transmitted to the bacterium.
Further analysis of transfected bacterial plasmids and their modes of replication in parasites requires detection of single-stranded DNA, and might also profit from the application of the Brewer/Fangman 2D-gel electrophoretic technique for detection of replication intermediates (27), which is especially useful for detecting extensive recombination.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the Australian Red Cross Blood Bank for the provision of human blood and serum. This work was supported by the National Health and Medical Research Council of Australia and the Medical Research Council of Great Britain. R.A.O. is the recipient of an Australian Postgraduate Research Award and a scholarship from the CRC for Vaccine Technology, Australia. B.S.C. is a Howard Hughes Medical Institute International Research Scholar.
References
- 1.Wu Y., Sifri,C.D., Lei,H.H., Su,X.Z. and Wellems,T.E. (1995) Transfection of Plasmodium falciparum within human red blood cells. Proc. Natl Acad. Sci. USA, 92, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fidock D.A. and Wellems,T.E. (1997) Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl Acad. Sci. USA, 94, 10931–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crabb B.S. and Cowman,A.F. (1996) Characterization of promoters and stable transfection by homologous and nonhomologous recombination in Plasmodium falciparum. Proc. Natl Acad. Sci. USA, 93, 7289–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Dijk M.R., Waters,A.P. and Janse,C.J. (1995) Stable transfection of malaria parasite blood stages. Science, 268, 1358–1362. [DOI] [PubMed] [Google Scholar]
- 5.van Dijk M.R., Janse,C.J. and Waters,A.P. (1996) Expression of a Plasmodium gene introduced into subtelomeric regions of Plasmodium berghei chromosomes. Science, 271, 662–665. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y., Kirkman,L.A. and Wellems,T.E. (1996) Transformation of Plasmodium falciparum malaria parasites by homologous integration of plasmids that confer resistance to pyrimethamine. Proc. Natl Acad. Sci. USA, 93, 1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crabb B.S., Triglia,T., Waterkeyn,J.G. and Cowman,A.F. (1997) Stable transgene expression in Plasmodium falciparum. Mol. Biochem. Parasitol., 90, 131–144. [DOI] [PubMed] [Google Scholar]
- 8.Crabb B.S., Cooke,B.M., Reeder,J.C., Waller,R.F., Caruana,S.R., Davern,K.M., Wickham,M.E., Brown,G.V., Coppel,R.L. and Cowman,A.F. (1997) Targeted gene disruption shows that knobs enable malaria-infected red cells to cytoadhere under physiological shear stress. Cell, 89, 287–296. [DOI] [PubMed] [Google Scholar]
- 9.Lobo C.A., Fujioka,H., Aikawa,M. and Kumar,N. (1999) Disruption of the Pfg27 locus by homologous recombination leads to loss of the sexual phenotype in Plasmodium falciparum. Mol. Cell, 3, 793–798. [DOI] [PubMed] [Google Scholar]
- 10.O’Donnell R.A., Saul,A., Cowman,A.F. and Crabb,B.S. (2000) Functional conservation of the malaria vaccine antigen MSP-119 across distantly related Plasmodium species. Nat. Med., 6, 91–95. [DOI] [PubMed] [Google Scholar]
- 11.Triglia T., Wang,P., Sims,P.F., Hyde,J.E. and Cowman,A.F. (1998) Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves the role of dihydropteroate synthase in sulfadoxine-resistant malaria. EMBO J., 17, 3807–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Dijk M.R., Vinkenoog,R., Ramesar,J., Vervenne,R.A., Waters,A.P. and Janse,C.J. (1997) Replication, expression and segregation of plasmid-borne DNA in genetically transformed malaria parasites. Mol. Biochem. Parasitol., 86, 155–162. [DOI] [PubMed] [Google Scholar]
- 13.Cowman A., Baldi,D., Healer,J., Mills,K., O’Donnell,R., Reed,M., Triglia,T., Wickham,M. and Crabb,B. (2000) Functional analysis of proteins involved in Plasmodium falciparum merozoite invasion of red blood cells. FEBS Lett., 476, 84–88. [DOI] [PubMed] [Google Scholar]
- 14.Lambros C. and Vanderberg,J.P. (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol., 65, 418–420. [PubMed] [Google Scholar]
- 15.Trager W. and Jensen,J.B. (1976) Human malaria parasites in continuous culture. Science, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- 16.Coppel R.L., Bianco,A.E., Culvenor,J.G., Crewther,P.E., Brown,G.V., Anders,R.F. and Kemp,D.J. (1987) A cDNA clone expressing a rhoptry protein of Plasmodium falciparum. Mol. Biochem. Parasitol., 25, 73–81. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbour Laboratory Press, Cold Spring Harbor, NY.
- 18.Kemp D.J., Corcoran,L.M., Coppel,R., Stahl,H., Bianco,A., Brown,G. and Anders,R. (1985) Size variation in chromosomes from independent cultured isolates of Plasmodium falciparum. Nature, 315, 347–350. [DOI] [PubMed] [Google Scholar]
- 19.Chu G., Vollrath,D. and Davis,R.W. (1986) Separation of large DNA molecules by contour-clamped homogeneous electric fields. Science, 234, 1582–1585. [DOI] [PubMed] [Google Scholar]
- 20.Vollrath D. and Davis,R.W. (1987) Resolution of DNA molecules greater than 5 megabases by contour-clamped homogeneous electric fields. Nucleic Acids Res., 15, 7865–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinberg A. and Vogelstein,B. (1983) A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem., 132, 6–13. [DOI] [PubMed] [Google Scholar]
- 22.Maleszka R. (1993) Single-stranded regions in yeast mitochondrial DNA revealed by pulsed- field gel electrophoresis. Appl. Theor. Electrophor., 3, 259–263. [PubMed] [Google Scholar]
- 23.Preiser P.R., Wilson,R.J., Moore,P.W., McCready,S., Hajibagheri,M.A., Blight,K.J., Strath,M. and Williamson,D.H. (1996) Recombination associated with replication of malarial mitochondrial DNA. EMBO J., 15, 684–693. [PMC free article] [PubMed] [Google Scholar]
- 24.Luder A. and Mosig,G. (1982) Two alternative mechanisms for initiation of DNA replication forks in bacteriophage T4: priming by RNA polymerase and by recombination. Proc. Natl Acad. Sci. USA, 79, 1101–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viret J.F., Bravo,A. and Alonso,J.C. (1991) Recombination-dependent concatemeric plasmid replication. Microbiol. Rev., 55, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly J.M., Ward,H.M., Miles,M.A. and Kendall,G. (1992) A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Res., 20, 3963–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brewer B.J. and Fangman,W.L. (1987) The localization of replication origins on ARS plasmids in S. cerevisiae. Cell, 51, 463–471. [DOI] [PubMed] [Google Scholar]