

Abstract
Checkpoint kinase 2 (CHK2) is a major effector of the DNA damage response pathway and although its mechanism of activation has been well studied, the attenuation of its activity following DNA damage has not been explored. Here, we identify the B’α subunit of protein phosphatase 2A (PP2A) as a CHK2 binding partner and show that their interaction is modulated by DNA damage. B’α binds to the SQ/TQ repeat region of CHK2, which is a target of ATM phosphorylation. The induction of DNA double-strand breaks by gamma irradiation as well as treatment with doxorubicin causes dissociation of the B’α and CHK2 proteins. This dissociation correlates with an increase in the ATM-dependent phosphorylation of CHK2 at serines 33 and 35 in the SQ/TQ region. Indeed, mutating these sites to mimic phosphorylation increases the dissociation after irradiation. PP2A negatively regulates CHK2 phosphorylation at multiple sites, as well as its kinase activity. These data reveal a novel mechanism for PP2A to keep CHK2 inactive under normal conditions while also allowing for a rapid release from this regulation immediately following DNA damage. This is followed by a subsequent reconstitution of the PP2A/CHK2 complex in later time points after damage, which may help to attenuate the signal.
Keywords: CHK2, PP2A, DNA damage, phosphorylation, signaling
Introduction
Maintenance of genomic integrity is an essential part of cellular physiology. Genotoxic insults that induce DNA breaks must be repaired in order to prevent the propagation of mutations that can contribute to malignant transformation. The primary role of the DNA damage response pathway is to induce cell cycle arrest, repair the incurred DNA damage, or, in cases where the extent of damage is beyond repair, signal for the initiation of apoptosis.1
The CHK1 and CHK2 kinases, the main effectors of the DNA damage response pathway, are activated after genotoxic insults by the ataxia telangiectasia mutated (ATM) and ATM-Rad3-related (ATR) kinases.2 CHK2 contains three major domains (Fig. 1A): an SQ/TQ region (amino acid residues 19–69), an FHA domain (amino acid residues 112–175), and a kinase domain (amino acid residues 220–486), all of which play a role in the activation of CHK2. In response to DNA damage, ATM phosphorylates CHK2 on threonine 68 in the SQ/TQ region.3-5 The FHA domain of one molecule of CHK2 binds to the phosphorylated threonine 68 of another, allowing autophosphorylation of threonines 383 and 387 within the kinase domain,6,7 as well as serine 516.8 CHK2 then dissociates into active monomers9 and phosphorylates a number of downstream targets including BRCA1, p53, E2F1 and Cdc25,10 which work to arrest the cell cycle and activate factors required for the proper repair of the DNA breaks. CHK2 is also phosphorylated at serines 19, 33 and 35 in the SQ/TQ region by ATM11,12 and these phosphorylation events enhance CHK2 activity.13
Figure 1.
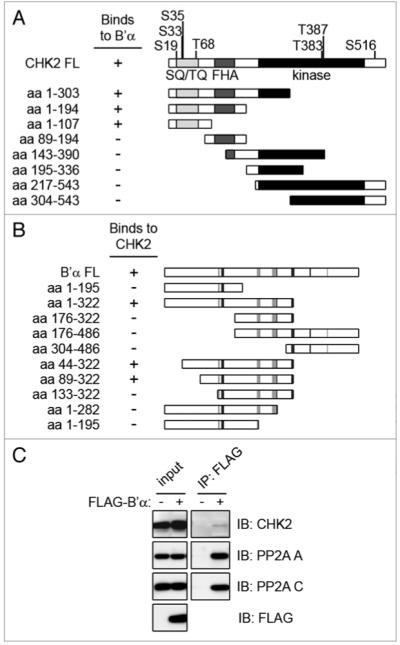
CHK2 binds to the B’α regulatory subunit of pp2A. (A) the full length CHK2 protein is shown with phosphorylation sites and domains highlighted. the SQ/TQ domain is displayed in light gray, the FHA domain in dark gray, and the kinase domain in black. Using the yeast two-hybrid system, deletion mutants of CHK2 were co-transformed into yeast with full-length B’α. the minimal CHK2 binding region for B’α includes amino acid residues 1–107. (B) Full length B’α is shown with the residues responsible for binding to the pp2A A subunit in light gray and the C subunit in dark gray.23 Using the yeast two-hybrid system, deletion mutants of B’α were co-transformed with full-length CHK2. the minimal B’α binding region for CHK2 is comprised of amino acid residues 89–322. (C) Cell lysates of 293T cells expressing FLAG-B’α were immunoprecipitated (IP) using the FLAG antibody and immunoblotted (IB) for the indicated proteins. Cell lysates that were not expressing FLAG-B’α were used as a negative control.
Mutations in CHEK2, the gene encoding human CHK2, have been found in various cancers. The first mutation found in CHEK2 was 1100delC, originally identified in families with Li-Fraumeni syndrome that did not have mutations in TP53.14 Later, this mutation was also found to be significantly associated with breast cancer in familial cases without a BRCA1 or BRCA2 mutation.15 This CHEK2 allele seems to act as a low-penetrance tumor suppressor gene16 and may be an adverse indicator of prognosis.17 Other CHK2 mutations have been discovered as well which cause mislocalization of the protein, diminished kinase activity, or inability to interact with substrates.18,19
Protein Phosphatase 2A (PP2A) is one of the major phosphatases of the cell and it participates in diverse cellular processes such as differentiation, cell cycle regulation and signal transduction.20 It is comprised of three subunits: the structural A subunit (PR65), the catalytic C subunit, and one of many regulatory B subunits that confer to PP2A its functional specificity.21 The B subunits can be divided into four major families: the B (PR55), B’ (PR61/B56), B” (PR72) and B”’ (PR93/PR110). These regulatory subunits are transcribed from 15 genes that encode at least 26 differentially spliced isoforms.22 The crystal structure of the holoenzyme containing PP2A A, C and B’γ1 was solved, revealing the active enzyme structure as well as binding sites between the three subunits.23,24
Several independent lines of evidence suggest a role for the PP2A holoenzyme as a tumor suppressor. The SV40 small T antigen can bind to the PP2A A-C complex, inhibiting PP2A activity,25,26 and this interaction is required for SV40-mediated transformation.27 Okadaic acid, a known PP2A inhibitor, was found to promote tumor formation.28 Interestingly, the PP2A A subunit was found to be mutated in primary lung and colon tumors, as well as in lung cancer cell lines.29 Mutations in PP2A A that prevent binding to the B’ or B and C subunits have been found in breast cancer tissue and cell lines.30,31 These data indicate the importance of inhibiting PP2A function in cellular transformation.
While much is currently known about the induction of the DNA damage response, there still remain questions about the negative regulation of the pathways when there is no damage present as well as the recovery from the checkpoint when the DNA breaks are resolved. In this paper, we present a novel mechanism for the negative regulation of CHK2 via PP2A in the absence of DNA damage as well as attenuation of the signal after damage.
Results
CHK2 interacts with the B’α subunit of PP2A
In order to identify novel interacting proteins of CHK2, we screened a human mammary gland cDNA library using full length CHK2 as bait in the yeast two-hybrid system. We identified two independent clones of the B’α regulatory subunit of PP2A (data not shown), also known as B56α and PR61α (official symbol: PPP2R5A). The B’ family was originally cloned in 1995 and is comprised of five genes,32 two of which are differentially spliced to form three isoforms each. In this screening we also identified MUS81, a DNA damage tolerance protein, and karyopherin KPNA2, that were previously described as interactors to CHK2,33,34 indicating that our CHK2 construct was properly folded.
To further investigate the interaction between B’α and CHK2, we mapped the B’α interacting domain in CHK2. Multiple deletion constructs of CHK2 were constructed and each was co-transformed with the full length B’α. Binding was determined using the yeast-two hybrid system. The minimal CHK2 fragment that bound to full length B’α was comprised of amino acid residues 1–107. This region contains the entire SQ/TQ region (Fig. 1A), an important site of ATM phosphorylation.
To determine the CHK2 binding region in B’α, multiple deletion constructs were constructed of B’α and each was co-expressed with full length CHK2. Binding was determined using the yeast-two hybrid system. The minimal binding region in B’α was found to contain amino acid residues 89–322 (Fig. 1B). Although this region spans almost half of the protein, the crystal structure of the PP2A holoenzyme containing B’γ1, which is closely related to B’α, reveals unique features of this enzyme.23,24 B’γ1 contains eighteen α helices which form eight HEAT-like repeats and the interaction to the A and C subunits is mediated by many residues scattered throughout the B’γ1 protein,23 as noted in Figure 1B. Therefore, it is probable that there are select residues within this span of amino acid residues 89–322 that are particularly important for the interaction to CHK2.
We next wanted to verify the binding of B’α and CHK2 in a mammalian system. 293T cells were transiently transfected to express FLAG-B’α. Whole cell extracts were immunoprecipitated using a FLAG antibody and immunoblotted for endogenous CHK2, demonstrating that B’α and CHK2 interact in vivo (Fig. 1C). Immunoblotting also detected the endogenous PP2A A and C subunits, indicating that FLAG-B’α is able to function as a member of the heterotrimeric PP2A complex.
B’α and CHK2 dissociate upon DNA damage
Since CHK2 is activated upon DNA damage, we examined the binding between CHK2 and B’α for any changes upon the induction of DNA double strand breaks (DSBs) by exposure to irradiation (IR). Whole cell extracts of 293T cells transiently expressing FLAG-B’α were immunoprecipitated using a FLAG antibody and immunoblotted for endogenous CHK2, PP2A A and PP2A C (Fig. 2A). Interestingly, when cells were exposed to 20 Gy IR and incubated for 1 h, this markedly reduced the binding of CHK2 to B’α without affecting the binding of B’α to the PP2A A and C subunits. Phosphorylation of CHK2 serine 33 and serine 35 is recognized by the same antibody and therefore we will refer to them as serines 33/35. Immunoblotting of the lysate input verifies that IR causes phosphorylation of CHK2 at serine 19, serines 33/35 and threonine 68 as previously described.3-5,13
Figure 2.

Binding between CHK2 and PP2A is dissociated by IR-induced DNA damage. (A) 293T cells transiently transfected with FLAG-B’α or empty vector (FLAG) were irradiated with 20 Gy and incubated for 1 h. Whole cell lysates were immunoprecipitated using a FLAG antibody and immunoblotted for the indicated proteins. (B) 293T cells transiently transfected with HA-tagged B’α, B’β, B’δ, B’ε or B’γ3 were irradiated with 20 Gy and incubated for 1 h. Whole cell lysates were immunoprecipitated with the HA antibody and immunoblotted for the indicated proteins. (C) 293T cells were irradiated with 20 Gy and incubated for 1 h. Whole cell lysates were immunoprecipitated with an antibody to CHK2 and immunoblotted for PP2A C. (D) 293T cells transiently transfected with FLAG-B’α were exposed to either 20 Gy IR and incubated for 1 h, 50 J/m2 UV and incubated for 2 h, 2 mM hydroxyurea (HU) for 24 h, 0.5 μg/mL mitomycin C (MMC) for 24 h, or 1 mM doxorubicin (DoX) for 2 h. Whole cell lysates were immunoprecipitated using the FLAG antibody and immunoblotted for the indicated proteins.
CHK2 binds to many members of the B’ family
We have determined that CHK2 binds to one member of the PP2A B’ family, namely B’α. The B’ family of regulatory subunits is comprised of at least nine members encoded from five genes. To determine the specificity of CHK2 for additional B’ proteins, HA-tagged B’α, B’β, B’δ, B’ε and B’γ3 proteins were transiently expressed in 293T cells. Endogenous CHK2 was efficiently immunoprecipitated by all of the B’ proteins tested (Fig. 2B), in agreement with a previous study that demonstrated CHK2’s ability to bind to B’ proteins using an in vitro binding assay.35 In our study, CHK2 had the strongest interaction to B’γ3 and the weakest to B’β, with binding to B’α, B’δ and B’ε residing within this range. Additionally, the CHK2-B’ complexes were dissociated after the cells were exposed to IR. It should be noted that the ability of the B’β protein to bind to the PP2A A and C subunits was compromised compared to the other B’ proteins, suggesting there may be a problem with the conformation of this overexpressed B’β.
In order to determine whether CHK2 can bind to an active PP2A complex, we immunoprecipitated CHK2 from whole cell lysates of 293T cells and immunoblotted for the PP2A C catalytic subunit (Fig. 2C). Indeed, they were found to bind, suggesting that CHK2 can associate with an active PP2A complex through one of the B’ regulatory subunits. After irradiating the cells, the binding between CHK2 and PP2A C is severely diminished as was seen with the B’ subunits.
B’α and CHK2 are dissociated after DNA damage caused by ionizing radiation and doxorubicin, but not B’α and CHK1
CHK2 can also be activated by other types of DNA damage in addition to that caused by IR. Therefore, other DNA damaging agents were also tested for any effects on CHK2 and B’α binding. 293T cells transiently expressing FLAG-B’α were exposed to IR, UV, hydroxyurea (HU), mitomycin C (MMC), or doxorubicin (Dox). FLAG-B’α was immunoprecipitated and immunoblotted for endogenous CHK2, PP2A A and PP2A C. In a different case from the one observed with IR, treatment with UV, HU or MMC did not have any effect on the ability of CHK2 to bind to FLAG-B’α (Fig. 2D). However, treatment with Dox did decrease the binding between the two proteins, but not to the extent that IR did. By examining the phosphorylation of CHK2 in the lysate input via immunoblotting, it can be seen that the pattern of phosphorylation was different after each treatment. After IR and Dox, serine 19, serines 33/35 and threonine 68 were all phosphorylated; after UV and MMC, threonine 68 was primarily phosphorylated and serine 19 was weakly phosphorylated; and after HU, serine 19 and threonine 68 were phosphorylated. The dissociation between CHK2 and B’α occurs after IR and Dox: the only conditions that resulted in serine 33/35 phosphorylation at the time points studied.
Since CHK1 is functionally related to CHK2, we also tested whether CHK1 could bind to PP2A B’α, and whether this binding was disrupted by DNA damage (Fig. 2D). CHK1 was able to bind to B’α, but this binding was not disrupted by treatment with IR, UV, HU, MMC or Dox. CHK1 was highly phosphorylated at serine 317 after IR, UV and HU, which is in agreement with previous studies.36,37 This demonstrates that the modulation of the interaction by DNA damage is specific to the CHK2-B’α complex.
Dissociation is an early event and occurs in a dose-dependent manner
Since we have observed that IR causes the most dissociation of B’α and CHK2, we next ascertained which doses of IR provide maximal dissociation. 293T cells transiently expressing FLAG-B’α were exposed to 2, 5, 10, 20 or 50 Gy, and FLAG-B’α was immunoprecipitated and immunoblotted (Fig. 3A). Maximal dissociation of endogenous CHK2 and B’α reached a peak after exposure to 10 Gy IR. By examining the phosphorylation of CHK2 in the lysate input, it can be seen that serine 19 phosphorylation increased with the increasing doses of IR. Threonine 68 phosphorylation was maximal after 2 and 5 Gy, and then decreased with the increasing IR doses. However, we observed the phosphorylation of serines 33/35 to be maximal after 10 Gy of IR. This corresponds to the IR dose that induces maximal dissociation between CHK2 and B’α.
Figure 3.

Kinetics of CHK2 and PP2A B’α dissociation after exposure to IR. 293T cells were transiently transfected with FLAG-B’α and (A) irradiated with 2, 5, 10, 20 or 50 Gy and incubated for 1 h, (B) irradiated with 20 Gy and incubated for 0.5, 1.0, 1.5, 2.0 or 2.5 h, (C) irradiated with 6 Gy and incubated for 1, 4, 8 or 12 h, and (D) irradiated with 50 Gy and incubated for 1, 4, 8 or 12 h. Whole cell lysates were immunoprecipitated with the FLAG antibody and immunoblotted for the indicated proteins.
In order to determine the precise timing of B’α and CHK2 dissociation after irradiation, 293T cells transiently expressing FLAG-B’α were irradiated with 20 Gy IR and collected after different time points. FLAG-B’α was immunoprecipitated and it was found that CHK2 binding was minimal 0.5 h after IR, and the binding was slightly restored over the remainder of the time course (2.5 h) (Fig. 3B). Significantly, the dissociation at 0.5 h correlated with maximal phosphorylation of CHK2 at serines 33/35, but did not correlate with phosphorylation of serine 19 or threonine 68.
Re-association of CHK2 and B’α does not correlate with resolution of DNA damage
CHK2 plays an important role in activating cell cycle checkpoints after DNA damage.2 Since we have established that PP2A B’α and CHK2 dissociate after IR, we investigated whether re-association at a later time was correlated to the resolution of DNA damage. We used two different doses of IR for comparison. First, we exposed cells to a sub-lethal dose of 6 Gy, the lowest dose after which we could detect a significant dissociation between CHK2 and B’α and which would also allow for recovery. We also exposed the cells to 50 Gy, which is considered lethal. 293T cells expressing FLAG-B’α were collected at different time points 1 h–12 h after exposure to both doses of IR (Fig. 3C and D). Interestingly, the kinetics of dissociation after 50 Gy was similar to that after 6 Gy. CHK2 was dissociated from B’α after 1 h and began to re-associate approximately 4 h after IR with a subsequent increase through the remaining time points. After both doses, the phosphorylation of serine 19, serines 33/35 and threonine 68 followed similar patterns. Serines 33/35 had maximal phosphorylation after 1 h, again correlating with the most dissociation between CHK2 and B’α. There does seem to be some difference in CHK2 phosphorylation between the two doses, since after 6 Gy IR, the CHK2 that was bound to B’α after 4–12 h existed as a doublet, suggesting the existence of hyper and hypophosphorylated forms (Fig. 3C).
In order to assess how dissociation and re-association correlated with the resolution of DNA damage, we determined the levels of histone H2AX phosphorylated at serine 139, also known as γ-H2AX, which is a well characterized marker of DSBs.38 Thus, higher levels of γ-H2AX denote the presence of DSBs. When the breaks are repaired, γ-H2AX levels return to levels found before DNA damage (in some cases it can no longer be detected by western blot or immunofluorescence). In our experiments, after 6 Gy IR, the maximal amount of γ-H2AX appeared after 4 h, returning to pre-damage levels by 12 h (Fig. 3C). On the other hand, after 50 Gy IR, maximal levels of γ-H2AX were detected after 1 h and did not decline throughout the remainder of the time points (Fig. 3D). Since B’α and CHK2 were shown to re-associate throughout the time course, and DNA DSBs were present during these times as shown by the presence of γ-H2AX, the re-association does not correlate with the resolution of the DSBs.
In summary, dissociation of CHK2 and B’α is induced in a dose-dependent manner and is an early event after IR. CHK2 and B’α re-association after DNA damage does not require resolution of the DNA damage. Moreover, dissociation and re-association correlate with phosphorylation of serines 33/35 of CHK2.
The activity of ATM, but not PP2A, is necessary for the dissociation of CHK2 and B’α
We next investigated the importance of PP2A activity on the binding and dissociation of CHK2 and B’α. Okadaic acid (OA) is a phosphatase inhibitor that, when used at a specific concentration, blocks the PP2A catalytic subunit’s activity without affecting protein phosphatase 1.39 293T cells transiently expressing FLAG-B’α were treated with OA prior to IR exposure. FLAG-B’α was immunoprecipitated using an antibody to the FLAG tag and immunoblot analysis was performed (Fig. 4A). In the mock treated cells, CHK2 was able to bind to FLAG-B’α, and this binding was unaffected by OA treatment alone. After IR, CHK2 and B’α dissociated and the presence of OA did not affect the dissociation. The inhibition of PP2A by OA also had no effect on the ability of FLAG-B’α to bind to the PP2A A and C subunits, indicating that it can still form the holoenzyme complex. Therefore, PP2A activity is not required for B’α binding to, or dissociation from, CHK2.
Figure 4.
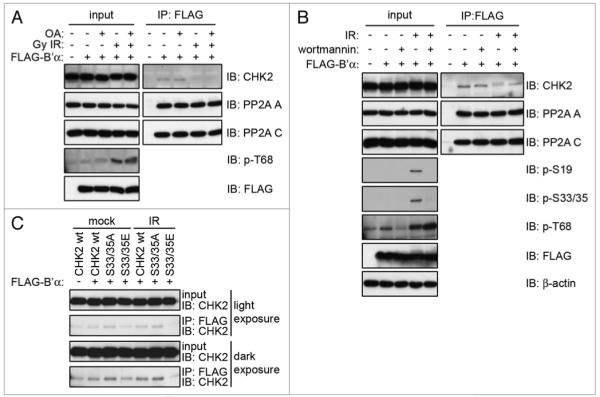
Dissociation of CHK2 and PP2A B’α is dependent on ATM activity, but not PP2A activity. (A) 293T cells were transiently transfected with FLAG-B’α, treated with 0.5 μM oA for 1 h and then irradiated with 20 Gy IR and incubated for 1 h. Whole cell lysates were immunoprecipitated with the FLAG antibody and immunoblotted for the indicated proteins. (B) 293T cells were transiently transfected with FLAG-B’α, treated with 20 μM wortmannin for 1 h then irradiated with 10 Gy IR and incubated for 1 h. Whole cell lysates were immunoprecipitated with the FLAG antibody and immunoblotted for the indicated proteins. (C) HCT116 CHK2−/− cells were transfected with FLAG-B’α and either wild-type CHK2 or the S33/35A or S33/35E mutants. Whole cell lysates were immunoprecipitated with the FLAG antibody and immunoblotted for CHK2.
As we have seen thus far, the most pronounced dissociation between CHK2 and B’α occurs when CHK2 is phosphorylated on serines 33/35, which are phosphorylated in an ATM-dependent manner.11-13 ATM belongs to the phosphoinositide 3-kinase related kinase (PI3KK) family along with the other DNA damage response proteins ATR and DNA-PK. To determine whether the CHK2-B’α dissociation is a PI3KK-dependent event as is the phosphorylation of CHK2 serines 33/35, the PI3KK inhibitor wortmannin was utilized at a concentration that inhibits ATM and DNA-PK, but not ATR activity in vivo.40 293T cells transiently expressing FLAG-B’α were incubated with wortmannin prior to exposure to IR. FLAG-B’α was immunoprecipitated using a FLAG antibody and immunoblot analysis was performed (Fig. 4B). Pre-incubation with wortmannin prevented the dissociation of CHK2 from FLAG-B’α after IR. This treatment also inhibited the phosphorylation of serines 19 and 33/35, indicating that the phosphorylation of CHK2 is required for the dissociation of CHK2 and B’α.
Mutation of CHK2 serines 33 and 35 affects binding to B’α
To further investigate the importance of the phosphorylation of CHK2 serines 33/35 on the binding and dissociation between CHK2 and B’α, we made two CHK2 mutants. The first substituted alanines for serines 33/35 to prevent phosphorylation (S33/35A) and the second substituted glutamic acids to mimic phosphorylation (S33/35E). Wild-type CHK2 (CHK2 WT), S33/35A and S33/35E were co-transfected along with FLAG-B’α into HCT116 CHK2−/− cells, which were then mock-irradiated or irradiated with 20 Gy. FLAG-B’α was immunoprecipitated and immunoblot analysis was performed (Fig. 4C). In mock treated cells, we saw an increase in S33/35A binding to B’α and a decrease in S33/35E binding as compared to CHK2 WT. Alternatively, after irradiation, CHK2 WT and S33/35A bound at the same levels as mock treated CHK2 WT, however, S33/35E binding is completely abrogated.
CHK2 can phosphorylate B’α in vitro, yet PP2A activity is unaffected by irradiation and is not influenced by CHK2
Since CHK2 is a kinase that is activated after irradiation, we determined whether B’α could be used as a substrate for CHK2 in an in vitro kinase assay. 293T cells were irradiated to activate CHK2 which was then immunoprecipitated and incubated in a kinase reaction with GST-B’α (Suppl. Fig. 1A). Indeed, phosphorylated B’α was detected, indicating that in an in vitro system, CHK2 can phosphorylate B’α.
Since CHK2 was able to phosphorylate B’α in vitro, we next determined if this had an effect on PP2A localization. Immunoblot analysis of the cytoplasm, nucleoplasm and chromatin fractions of 293T cells shows that CHK2, PP2A C and PP2A A are located in the cytoplasm and nucleoplasm. FLAG-B’α is located in all three cellular compartments. Cells were also exposed to IR to activate CHK2; however, this did not change the localization of CHK2 or any of the PP2A subunits (data not shown).
If CHK2 is indeed able to phosphorylate B’α in vivo, it may have an effect on PP2A activity. We first examined the total PP2A activity in the cell. The C subunit is responsible for all of the PP2A catalytic activity in the cell, regardless of which regulatory subunit is bound to the complex. Therefore, 293T cells were either exposed to IR or mock treated and immunoprecipitated using an antibody to the C subunit to assess total PP2A activity. Phosphatase activity of the immunoprecipitates was measured using an in vitro phosphatase assay (Suppl. Fig. 1B). The levels of PP2A activity were not changed by exposure to IR.
Next, the activity of the PP2A complexes specifically containing FLAG-B’α was determined. 293T cells transiently expressing FLAG-B’α were either exposed to IR or mock treated and immunoprecipitated using a FLAG antibody to measure only the activity of PP2A complexes containing B’α. Phosphatase activity was measured using an in vitro phosphatase assay and was unaffected by exposure to IR (Suppl. Fig. 1C). Since the assay measures activity against a small peptide, this may not accurately reflect changes in phosphatase activity toward specific substrates, however, it is a measure of the total activity of PP2A containing the B’α subunit.
In order to determine whether CHK2 has an effect on PP2A activity, the HCT116 wild-type (WT) cells and the isogenic HCT116 CHK2−/− cell lines were utilized. Both cell lines were either exposed to IR or mock treated and immunoprecipitated using an antibody for the PP2A C subunit to measure total PP2A activity (Suppl. Fig. 1D). The in vitro phosphatase assay revealed no difference in PP2A activity in the mock treated or irradiated samples in either cell line. Also, there was no difference observed between the HCT116 WT or CHK2−/− cell lines, suggesting that CHK2 has no effect on PP2A activity under these conditions. Taken together, this data indicates that PP2A activity is not affected by IR exposure or by CHK2.
Changes in CHK2 phosphorylation due to irradiation
In order to determine the extent to which PP2A has an effect on the phosphorylation of CHK2, the PP2A inhibitor OA was utilized. 293T cells were treated with OA in the presence and absence of irradiation, and the amount of phosphorylation of CHK2 at specific sites was measured via immunoblotting (Fig. 5A). Pre-treatment with OA increased the phosphorylation of CHK2 after IR, primarily at serine 19, although there were also slight increases in the phosphorylation of serines 33/35 and threonine 68, demonstrating that PP2A can dephosphorylate CHK2 at multiple sites.
Figure 5.
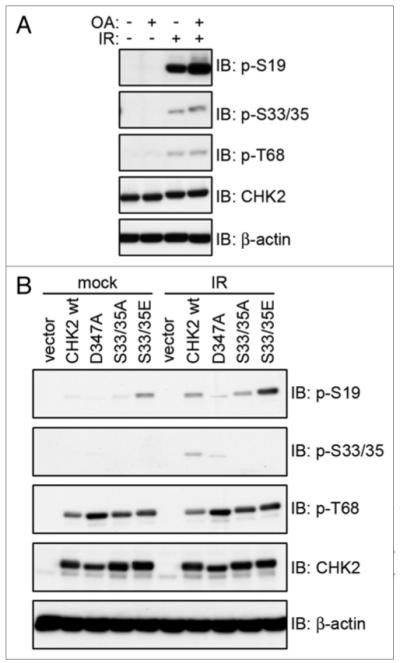
Effects of irradiation on CHK2 phosphorylation. (A) 293T cells were incubated with 0.5 μM oA for 1 h then irradiated with 20 Gy and incubated for 1 h. Whole cell extracts were examined for phosphorylation levels of CHK2 via immunoblot. (B) HCT15 cells transiently transfected with pCMV-Tag3B empty vector (vector) or myc-tagged CHK2 WT, D347A, S33/35A or S33/35E were exposed to 20 Gy IR or mock treated and incubated for 1 h. Whole cell extracts were examined for CHK2 phosphorylation via immunoblot.
Since we have shown that mutation of serines 33/35 can influence the binding between B’α and CHK2, this led us to investigate any effect phosphorylation at these sites has on CHK2 function. Therefore, we examined the ability of the S33/35A and S33/35E mutants to be phosphorylated at other CHK2 sites. HCT15 cells (which lack functional CHK2) transiently expressing myc-tagged CHK2 wild-type (WT), kinase dead (D347A), S33/35A or S33/35E were either exposed to IR or mock treated. The phosphorylation status of each CHK2 protein was determined via immunoblotting (Fig. 5B). In the mock treated samples, CHK2 WT and S33/35A showed a very faint phosphorylation of serine 19. However, S33/35E displayed a much stronger phosphorylation, even in the absence of DNA damage. After exposure to IR, CHK2 WT was phosphorylated on serine 19, with levels comparable to the mock treated S33/35E. The phosphorylation of S33/35A at serine 19 was slightly compromised. Threonine 68 phosphorylation was slightly higher in the S33/35A and S33/35E in both conditions as compared to CHK2 WT. Therefore, the S33/35 mutations primarily affect serine 19 phosphorylation, the same site that was affected most by PP2A inhibition.
Changes in CHK2 kinase activity
To directly measure the effect of PP2A on CHK2 activity, we transfected 293T cells with FLAG-B’α and measured CHK2 kinase activity using an in vitro kinase assay with GST-Cdc25C (amino acid residues 200–256) as a substrate (Fig. 6A and B). Cells were either mock treated or irradiated, CHK2 was immunoprecipitated, and the kinase activity was measured. In the mock treated samples, B’α did not have any effect on CHK2 kinase activity. However, after irradiation, overexpression of B’α prevented the full activation of CHK2.
Figure 6.
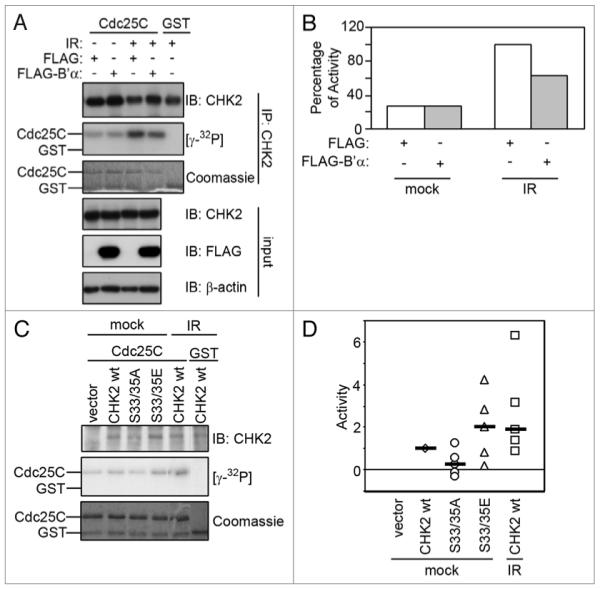
Effects of irradiation on CHK2 kinase activity. (A) 293T cells expressing FLAG-B’α or FLAG (as a negative control) were irradiated with 20 Gy and incubated for 1 h. Whole cell lysates were immunoprecipitated using a CHK2 antibody and incubated in an in vitro kinase assay using GST-Cdc25C (amino acids 200–256) as a substrate. (B) Autoradiographs in (A) were normalized against the levels of CHK2 protein per sample and represented by graphs. 100% activity was defined by the amount in the negative control sample after IR. (C) HCT15 cells were transiently transfected with empty vector (vector), CHK2 WT, S33/35A or S33/35E and mock-irradiated or exposed to 20 Gy IR and incubated for 1 h. Cell lysates were immunoprecipitated and incubated in an in vitro kinase assay using GST-Cdc25C (amino acids 200–256) as a substrate. (D) Five experiments as done in (C) were plotted on a single graph. Activity of 1 was defined as the activity in the CHK2 WT mock-treated sample. The median of each set of data points is shown with a black bar.
Since mutation of serines 33/35 to glutamic acid positively affects CHK2 phosphorylation primarily on serine 19, the effect on CHK2 kinase activity was also examined since phosphorylation of this site has been shown to enhance CHK2 activity.13 HCT15 cells expressing CHK2 WT, S33/35A or S33/35E were mock treated and cells expressing CHK2 WT were also irradiated to use as a measure of the full activation of CHK2. CHK2 activity was determined using an in vitro kinase assay with GST-Cdc25C (amino acid residues 200–256) as a substrate. Figure 6D displays five independent experiments with the medians noted and these experiments are also shown individually in Supplemental Figure 2. By comparing the medians in Figure 6D, it can be seen that in the mock treated samples, S33/35E had higher kinase activity than CHK2 WT, whereas S33/35A had lower kinase activity. In fact, the S33/35E mutant had similar kinase activity to CHK2 WT after IR. Figure 6C shows a representative experimentv. These data suggest that phosphorylation of serines 33/35 is important for CHK2 activity and that mimicking this phosphorylation can increase CHK2 activity even in the absence of DNA damage.
Discussion
After DNA damage, the cell must activate cell cycle checkpoints to allow time to repair the breaks, or if the damage is too extensive, trigger apoptosis signaling.1 ATM phosphorylates CHK2 on multiple sites in the SQ/TQ domain,3-5,11-13 leading to full CHK2 activation.6,7 Here we propose a novel mechanism for CHK2 regulation in which ATM phosphorylation causes a dissociation of PP2A from CHK2, thus allowing CHK2 activation. PP2A can then bind to CHK2 hours later, providing a mechanism for CHK2 downregulation after DNA damage.
We have identified the B’α subunit of PP2A as a CHK2-interacting protein via a yeast two-hybrid screen and have confirmed the interaction in mammalian cells. Full length CHK2 was found to bind to B’α amino acids 89–322. Our inability to further narrow down the interacting region suggests that residues that mediate the interaction are scattered within this region. Interestingly, the structure of the PP2A holoenzyme indicates that the majority of interactions between the B’ subunit and the A and C subunits are mediated by amino acid residues located at intervening loops between the 18 alpha helices.23,24 Thus, it is likely that this may also be the case for the interaction with CHK2. Identification of these residues could reveal the basis for substrate specificity of PP2A enzymes containing different B’ subunits. This large portion of B’α that binds to CHK2 is a highly conserved region between the B’ family members.22 Indeed, when we tested the specificity of CHK2 for the B’ family, we found that CHK2 could bind to B’β, B’δ, B’ε and B’γ3 as well and all of the interactions were dissociated upon exposure to irradiation.
In mammalian cells, binding between B’α and CHK2 is markedly decreased by exposure to IR. However, other types of damage caused by UV, hydroxyurea or mitomycin C did not disrupt binding. Doxorubicin had an effect, but not to the extent to which IR did although comparisons of agents with different mechanisms of action are challenging. B’α can also bind CHK1, though the interaction is not disrupted by any of the DNA damage-inducing agents tested. This demonstrates a unique aspect of the relationship between CHK2 and B’α: there is binding under normal conditions, but this is disrupted only after DSB induced by IR, when serines 33/35 are phosphorylated. CHK2 and B’α are able to re-associate hours after IR, and this correlates with the dephosphorylation of serines 33/35, but not with repair of the bulk of damaged DNA as measured by phosphorylation of histone H2AX.
We have found that maximal dissociation between CHK2 and B’α correlates with maximal phosphorylation of CHK2 serines 33/35, which are phosphorylated in an ATM-dependent manner.13 When we inhibited ATM with wortmannin, the phosphorylation of serines 33/35 was prevented as well as the dissociation between CHK2 and B’α. The region in CHK2 that binds to B’α is comprised of amino acids 1–107, which contains the entire SQ/TQ domain. Serines 33/35 are located in this domain, suggesting that the phosphorylation of these sites could directly contribute to the dissociation of the two proteins. We therefore mutated CHK2 serines 33/35 to alanine to prevent phosphorylation (S33/35A) or to glutamic acid to mimic phosphorylation (S33/35E) and tested the effects on binding to B’α. Our data indicates that phosphorylation at serines 33/35 is necessary for the dissociation of B’α and CHK2, but it is not sufficient on its own; other phosphorylation sites are probably involved as well. Interestingly, wild-type CHK2 did not dissociate from B’α after IR when they were both overexpressed. This may be because when CHK2 is overexpressed, it can become activated without DNA damage,4,6 allowing for dissociation in the absence of DNA damage.
In mock treated cells, S33/35E had a level of phosphorylation at serine 19 that was comparable to CHK2 WT after IR. Pharmacologic PP2A inhibition with OA also increases phosphorylation at serine 19. Alternatively, the S33/35A mutant had slightly less phosphorylation after IR compared to CHK2 WT. The S33/35E mutant also had increased kinase activity in mock treated conditions compared to CHK2 WT, whereas, the S33/35A mutant had decreased kinase activity, in agreement with a previous study.13 CHK2 kinase activity is decreased when B’α is overexpressed as well.
PP2A has already been demonstrated to negatively regulate other DNA damage response proteins under various conditions, including ATM,39 CHK1,41,42 p53,43 ATM and ATR,44,45 and H2AX,46 signifying the importance of PP2A in the DNA damage response pathway. In addition, two previous papers have implicated a role for PP2A in regulating CHK2. It was demonstrated that CHK2 can phosphorylate B’γ in vitro, increasing PP2A activity towards CHK2 in vitro.35 In an in vitro kinase assay, we found that CHK2 could phosphorylate B’α, however this did not have an effect on PP2A activity or localization. It has also been revealed that treating cells with okadaic acid or incubating CHK2 protein with PP2A affected CHK2 phosphorylation at threonine 68,47 in agreement with our results.
Another mode of CHK2 regulation has been characterized. The Wip1 phosphatase has been shown to dephosphorylate threonine 68 of CHK2 which leads to a decrease in its kinase activity and ability to induce apoptosis.48,49 Interestingly, Wip1 binding to CHK2 is enhanced by DNA damage,49 in opposition to the relationship between PP2A and CHK2, which are dissociated after IR. Interestingly, both Wip1 and PP2A B’α bind to the SQ/TQ domain of CHK2.50
Negative regulation of the DNA damage response pathway is important for the cell to prevent inappropriate checkpoint signaling and to return the cell to the cell cycle after the damage is repaired. Indeed, the fact that two different phosphatases regulate CHK2 may highlight the importance of these processes.
Materials and Methods
Cloning
CHEK2 (the gene encoding CHK2) was cloned into the pGBKT7 vector via PCR of a human mammary gland cDNA library (CLONTECH Laboratories). PCR was used to generate B’α fragments containing amino acid residues 1–486 (full length), 1–195, 1–322, 176–322, 176–486, 304–486, 44–322, 89–322, 133–322, 1–282 and 1–195 from the clone that was identified in the yeast-two hybrid to bind to CHK2. These fragments were all cloned into the pACT2 vector and the full-length was cloned into pCMV2-FLAG. PCR was also used to generate fragments of CHK2 containing amino acid residues 1–303, 1–194, 1–107, 89–194, 143–390, 195–336, 217–543 and 304–543. These were cloned into the pGBKT7 vector. Full length CHK2 was subcloned into the pCMV-Tag3B vector. The D347A, S33/35A and S33/35E mutant forms of CHK2 were constructed via site-directed mutagenesis and cloned into the pCMV-Tag3B and pcDNA3 vectors.
Yeast two-hybrid
The yeast-two hybrid was performed via the MATCHMAKER Two-Hybrid System 3 (CLONTECH Laboratories), briefly described here. First, full length CHEK2 in pGBKT7 was transformed into AH109 yeast and selection was made on SD-Trp media. Next, the human mammary gland library (CLONTECH Laboratories) in the pACT2 vector was transformed into the same yeast and selection was made on SD-Leu-Trp-Ade-His minimal media to verify binding between CHK2 and any prey from the library. For the deletion fragment analysis, the full length gene encoding B’α in pACT2 was co-transformed with each CHEK2 fragment in pGBKT7 or full length CHEK2 in pGBKT7 was transformed with each B’α gene fragment in pACT2 into AH109 yeast and selection was made on minimal media.
Cell culture, transfection and reagents
293T cells were grown in DMEM media (Sigma) supplemented with 7.5% Fetal Bovine Serum (SAFC), 1% Penicillin Streptomycin (Gibco), and 0.5% Amphotericin-B (Sigma). HCT15 cells were grown in RMPI media (Gibco; Sigma) supplemented with 10% Fetal Bovine Serum, 1% Penicillin Streptomycin and 0.5% Amphotericin-B. HCT116 wild type (WT) and HCT116 CHK2−/− cells were a gift from Bert Vogelstein and they were maintained in McCoy’s 5A media (Gibco) supplemented with 10% Fetal Bovine Serum, 1% Penicillin Streptomycin and 0.5% Amphotericin-B. 293T cells were transfected using Fugene 6 (Roche) and the HCT15 and HCT116 CHK2−/− cells were transfected using Lipofectamine 2000 (Invitrogen). When indicated, cells were treated with wortmannin, okadaic acid, hydroxyurea, mitomycin C or doxorubicin (all from Sigma) at the indicated concentrations.
Immunoprecipitations
For endogenous co-immunoprecipitations, 293T cells were lysed with RIPA buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA pH 8.0, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS), incubated with an antibody to CHK2 and Protein A/G PLUS-Agarose (Santa Cruz) in modified RIPA buffer (made without sodium deoxycholate and SDS). Samples were eluted via boiling in sample buffer and analyzed by immunoblotting. For co-immunoprecipitations with overexpressed proteins, 293T cells were transfected with a pCEP-4HA plasmid containing B’α, B’β, B’δ, B’ε or B’γ3 (obtained from Addgene; originally deposited by D. Virshup), a pCMV2-FLAG plasmid containing B’α, or co-transfected with pCMV2-FLAG-B’α and wild-type CHK2 or a mutated CHK2 construct in pcDNA3. For immunoprecipitations via the HA tag, cells were lysed with NETN buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1X protease inhibitors (Sigma), 1 mM PMSF, 0.5 mM Sodium Vanadate, 0.5 mM Sodium Molybdate) and incubated overnight with monoclonal anti-HA agarose conjugate (Clone HA-7; Sigma). Samples were eluted via boiling in sample buffer and analyzed by immunoblotting. For immunoprecipitations via the FLAG tag, lysates were incubated with anti-FLAG M2 Affinity Gel (Sigma). Samples were eluted with 150 ng/μL 3X FLAG peptide (Sigma) and analyzed by immunoblotting.
Immunoblotting
Cell lysis was performed with NETN buffer. Samples were incubated on ice for 15 min and then centrifuged for 20 min at 13,000 rpm at 4°C. Lysates were separated by SDS-PAGE gel and transferred to PVDF membranes. Membranes were immunoblotted using the following antibodies: CHK2 (Clone 7; Upstate), PP2A A (Clone H-300; Santa Cruz), PP2A C (Clone 1D6; Upstate), PP2A B’ (Upstate), CHK1 (Clone G4; Santa Cruz), CHK2 phospho-serine 19 (Cell Signaling), CHK2 phospho-serines 33/35 (Cell Signaling), CHK2 phospho-threonine 68 (Cell Signaling), CHK1 phospho-serine 317 (Cell Signaling), Talin (Clone TA205; Upstate), PARP (BD PharMingen), H2AX (Upstate), FLAG (M2, peroxidase conjugate; Sigma), β-actin (Clone AC-74; Sigma), HA (Clone 3F10; Roche). Immobilon western Chemiluminescent HRP Substrate (Millipore) or Amersham ECL Plus western blotting Detection Reagent (GE Healthcare) was used for detection.
Kinase assays
293T cells, 293T cells transiently transfected with pCMV2-FLAG empty vector or one containing FLAG-B’α, or HCT15 cells transiently transfected with empty vector (pCMV-Tag3B or pcDNA3) or one containing CHK2 WT, S33/35A or S33/35E were irradiated with 20 Gy IR or mock treated. Cells were lysed with NETN buffer and immunoprecipitated using an antibody to CHK2 (H-300, Santa Cruz; Clone 7, Upstate) or the c-Myc tag (Clone 9E10, Santa Cruz) and Protein A/G PLUS-Agarose (Santa Cruz). Immunoprecipitates were washed with NETN or RIPA followed by kinase assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM dithiothreitol, 1X protease inhibitors (Sigma), 1 mM PMSF, 0.5 mM sodium vanadate, 0.5 mM sodium molybdate). Kinase reactions were carried out at 30°C for 30 min in kinase assay buffer containing 25 μM ATP and 10 μCi [γ-32P]ATP with either GST-B’α or GST-Cdc25C (amino acids 200–256) as a substrate using equivalent amounts of GST as a negative control. Reactions were stopped by addition of sample buffer, boiled and separated by SDS-PAGE gels which were then immunoblotted for CHK2 and autoradiographed. The plasmid containing GST-Cdc25C (amino acids 200–256) was a gift from Larry Karnitz.51
PP2A phosphatase activity assay
Phosphatase activity was measured using the PP2A Immunoprecipitation Phosphatase Assay Kit (Upstate) following the manufacturer’s instructions. Briefly, cells were lysed with NETN buffer and immunoprecipitated with an antibody to PP2A C (for total PP2A activity) or FLAG (for activity of PP2A containing FLAG-B’α). Immunoprecipitates were incubated with a phosphopeptide for 10 min at 30°C and free phosphate was measured by addition of Malachite Green Detection Solution and the absorbance levels were read at 650 nm.
Supplementary Material
Acknowledgements
We would like to thank Bert Vogelstein for the HCT116 and HCT116 CHK2−/− cell lines and Larry Karnitz for the GST-Cdc25C (amino acids 200–256) plasmid. We would also like to thank Vesna Dapic for constructing the CHEK2 clone in pGBKT7. This work was funded by the Florida Breast Cancer Coalition Research Foundation pre-doctoral grant to A.F. and by NIH award CA116167 and has been supported in part by the Molecular Imaging Core at the H. Lee Moffitt Cancer Center.
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ATM-Rad3-related
- CHK2
checkpoint kinase 2
- Dox
doxorubicin
- DSB
DNA double-strand break
- HU
hydroxyurea
- IB
immunoblot
- IP
immunoprecipitation
- IR
ionizing radiation
- MMC
mitomycin C
- OA
okadaic acid
- PP2A
protein phosphatase 2A
Footnotes
Note
Supplementary materials can be found at: www.landesbioscience.com/supplement/FreemanCC9-4-Sup.pdf
References
- 1.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 3.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA. 2000;97:10389–94. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–6. [PubMed] [Google Scholar]
- 5.Melchionna R, Chen XB, Blasina A, McGowan CH. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol. 2000;2:762–5. doi: 10.1038/35036406. [DOI] [PubMed] [Google Scholar]
- 6.Xu X, Tsvetkov LM, Stern DF. Chk2 activation and phosphorylation-dependent oligomerization. Mol Cell Biol. 2002;22:4419–32. doi: 10.1128/MCB.22.12.4419-4432.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee CH, Chung JH. The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J Biol Chem. 2001;276:30537–41. doi: 10.1074/jbc.M104414200. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Chen J. Autophosphorylation of checkpoint kinase 2 at serine 516 is required for radiation-induced apoptosis. J Biol Chem. 2003;278:36163–8. doi: 10.1074/jbc.M303795200. [DOI] [PubMed] [Google Scholar]
- 9.Ahn JY, Li X, Davis HL, Canman CE. Phosphorylation of threonine 68 promotes oligomerization and autophosphorylation of the Chk2 protein kinase via the forkhead-associated domain. J Biol Chem. 2002;277:19389–95. doi: 10.1074/jbc.M200822200. [DOI] [PubMed] [Google Scholar]
- 10.Bartek J, Falck J, Lukas J. CHK2 kinase—a busy messenger. Nat Rev Mol Cell Biol. 2001;2:877–86. doi: 10.1038/35103059. [DOI] [PubMed] [Google Scholar]
- 11.Mochan TA, Venere M, DiTullio RA, Jr, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003;63:8586–91. [PubMed] [Google Scholar]
- 12.Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004;279:53272–81. doi: 10.1074/jbc.M406879200. [DOI] [PubMed] [Google Scholar]
- 13.Buscemi G, Carlessi L, Zannini L, Lisanti S, Fontanella E, Canevari S, et al. DNA damage-induced cell cycle regulation and function of novel Chk2 phosphoresidues. Mol Cell Biol. 2006;26:7832–45. doi: 10.1128/MCB.00534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286:2528–31. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- 15.Vahteristo P, Bartkova J, Eerola H, Syrjakoski K, Ojala S, Kilpivaara O, et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet. 2002;71:432–8. doi: 10.1086/341943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–9. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 17.de Bock GH, Schutte M, Krol-Warmerdam EM, Seynaeve C, Blom J, Brekelmans CT, et al. Tumour characteristics and prognosis of breast cancer patients carrying the germline CHEK2*1100delC variant. J Med Genet. 2004;41:731–5. doi: 10.1136/jmg.2004.019737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, Webster SR, Chen J. Characterization of tumor-associated Chk2 mutations. J Biol Chem. 2001;276:2971–4. doi: 10.1074/jbc.M009727200. [DOI] [PubMed] [Google Scholar]
- 19.Falck J, Lukas C, Protopopova M, Lukas J, Selivanova G, Bartek J. Functional impact of concomitant versus alternative defects in the Chk2-p53 tumour suppressor pathway. Oncogene. 2001;20:5503–10. doi: 10.1038/sj.onc.1204811. [DOI] [PubMed] [Google Scholar]
- 20.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–39. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–45. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009;1795:1–15. doi: 10.1016/j.bbcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, et al. Structure of the protein phosphatase 2A holoenzyme. Cell. 2006;127:1239–51. doi: 10.1016/j.cell.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 24.Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445:53–7. doi: 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- 25.Pallas DC, Cherington V, Morgan W, DeAnda J, Kaplan D, Schaffhausen B, et al. Cellular proteins that associate with the middle and small T antigens of polyomavirus. J Virol. 1988;62:3934–40. doi: 10.1128/jvi.62.11.3934-3940.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang SI, Lickteig RL, Estes R, Rundell K, Walter G, Mumby MC. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol Cell Biol. 1991;11:1988–95. doi: 10.1128/mcb.11.4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–23. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suganuma M, Fujiki H, Suguri H, Yoshizawa S, Hirota M, Nakayasu M, et al. Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc Natl Acad Sci USA. 1988;85:1768–71. doi: 10.1073/pnas.85.6.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J, et al. Alterations of the PPP2R1B gene in human lung and colon cancer. Science. 1998;282:284–7. doi: 10.1126/science.282.5387.284. [DOI] [PubMed] [Google Scholar]
- 30.Ruediger R, Pham HT, Walter G. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene. 2001;20:10–5. doi: 10.1038/sj.onc.1204059. [DOI] [PubMed] [Google Scholar]
- 31.Esplin ED, Ramos P, Martinez B, Tomlinson GE, Mumby MC, Evans GA. The glycine 90 to aspartate alteration in the Abeta subunit of PP2A (PPP2R1B) associates with breast cancer and causes a deficit in protein function. Genes Chromosomes Cancer. 2006;45:182–90. doi: 10.1002/gcc.20284. [DOI] [PubMed] [Google Scholar]
- 32.McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem. 1996;271:22081–9. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- 33.Zannini L, Lecis D, Lisanti S, Benetti R, Buscemi G, Schneider C, et al. Karyopherin-alpha2 protein interacts with Chk2 and contributes to its nuclear import. J Biol Chem. 2003;278:42346–51. doi: 10.1074/jbc.M303304200. [DOI] [PubMed] [Google Scholar]
- 34.Boddy MN, Lopez-Girona A, Shanahan P, Interthal H, Heyer WD, Russell P. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol. 2000;20:8758–66. doi: 10.1128/mcb.20.23.8758-8766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dozier C, Bonyadi M, Baricault L, Tonasso L, Darbon JM. Regulation of Chk2 phosphorylation by interaction with protein phosphatase 2A via its B’ regulatory subunit. Biol Cell. 2004;96:509–17. doi: 10.1016/j.biolcel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003;278:14806–11. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- 38.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 39.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, et al. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–61. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–82. [PubMed] [Google Scholar]
- 41.Leung-Pineda V, Ryan CE, Piwnica-Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol. 2006;26:7529–38. doi: 10.1128/MCB.00447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li G, Elder RT, Qin K, Park HU, Liang D, Zhao RY. Phosphatase type 2A-dependent and -independent pathways for ATR phosphorylation of Chk1. J Biol Chem. 2007;282:7287–98. doi: 10.1074/jbc.M607951200. [DOI] [PubMed] [Google Scholar]
- 43.Mi J, Bolesta E, Brautigan DL, Larner JM. PP2A regulates ionizing radiation-induced apoptosis through Ser46 phosphorylation of p53. Mol Cancer Ther. 2009;8:135–40. doi: 10.1158/1535-7163.MCT-08-0457. [DOI] [PubMed] [Google Scholar]
- 44.McConnell JL, Gomez RJ, McCorvey LR, Law BK, Wadzinski BE. Identification of a PP2A-interacting protein that functions as a negative regulator of phosphatase activity in the ATM/ATR signaling pathway. Oncogene. 2007;26:6021–30. doi: 10.1038/sj.onc.1210406. [DOI] [PubMed] [Google Scholar]
- 45.Petersen P, Chou DM, You Z, Hunter T, Walter JC, Walter G. Protein phosphatase 2A antagonizes ATM and ATR in a Cdk2- and Cdc7-independent DNA damage checkpoint. Mol Cell Biol. 2006;26:1997–2011. doi: 10.1128/MCB.26.5.1997-2011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20:801–9. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 47.Liang X, Reed E, Yu JJ. Protein phosphatase 2A interacts with Chk2 and regulates phosphorylation at Thr-68 after cisplatin treatment of human ovarian cancer cells. Int J Mol Med. 2006;17:703–8. [PubMed] [Google Scholar]
- 48.Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–80. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 49.Oliva-Trastoy M, Berthonaud V, Chevalier A, Ducrot C, Marsolier-Kergoat MC, Mann C, et al. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene. 2007;26:1449–58. doi: 10.1038/sj.onc.1209927. [DOI] [PubMed] [Google Scholar]
- 50.Yoda A, Xu XZ, Onishi N, Toyoshima K, Fujimoto H, Kato N, et al. Intrinsic kinase activity and SQ/TQ domain of Chk2 kinase as well as N-terminal domain of Wip1 phosphatase are required for regulation of Chk2 by Wip1. J Biol Chem. 2006;281:24847–62. doi: 10.1074/jbc.M600403200. [DOI] [PubMed] [Google Scholar]
- 51.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–82. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.