

Abstract
Subcellular localization is an important protein property, which is related to function, interactions and other features. As experimental determination of the localization can be tedious, especially for large numbers of proteins, a number of prediction tools have been developed. We developed the PROlocalizer service that integrates 11 individual methods to predict altogether 12 localizations for animal proteins. The method allows the submission of a number of proteins and mutations and generates a detailed informative document of the prediction and obtained results. PROlocalizer is available at http://bioinf.uta.fi/PROlocalizer/.
Keywords: Protein localization prediction, Cell compartments, Mutations, Disease-causing mutations, Prediction method
Introduction
Cells have several membrane-enclosed compartments, which have different protein constituents and composition. The subcellular localization of a protein is important to know, as it is linked to protein function, interactions and activity in signaling pathways. Proteins are sorted to their destinations based on targeting signals that can appear anywhere in the protein sequence. During the protein translation in animals, proteins can enter the classical secretory pathway (SP) in which the protein is sorted via endoplasmic reticulum (ER), e.g., to the Golgi apparatus, plasma membrane, lysosome or to the extracellular matrix. Protein can also stay at ER. If the protein does not contain a signal peptide to the SP it can remain in the cytoplasm, enter non-classical secretory pathways (nSPs) or it can be sorted to the nucleus, peroxisomes or mitochondrion (Dönnes and Höglund 2004).
All details in protein sorting are not very well understood, but several SP targeting signals have been identified. These signals are usually N-terminal and their amino acid compositions are diverse as they are not strongly conserved (Stroud and Walter 1999). SP signal peptides have three part structure usually with polar and positively charged n-part, hydrophobic h-part and c-part, which often contains prolines and glycines to interfere with α-helix formation (Martoglio and Dobberstein 1998).
Mitochondrial proteins are targeted via N-terminal signal peptides as well. Another signal peptide can further sort the protein inside the mitochondrion. Nuclear localization signals are difficult to recognize as they are diverse and they can exist anywhere in the protein sequence. In addition, C-terminal signal peptides exist (Chou 2002). Of peroxisomal proteins, some are targeted with C-terminal sequences (Dönnes and Höglund 2004) and others with N-terminal signal peptide (Emanuelsson 2002).
Information on protein localization is scattered throughout publications and numerous databases and does not exist for many proteins. A protein can have several localizations, often depending on the state of the cell. Experimental determination of protein subcellular localization can be time consuming and expensive even though couple of high-throughput approaches have been developed (Davis 2004; Falk et al. 2007). However, in these methods, protein localization can be interfered with reporter genes or cell fractionation. Consequently, numerous computational tools have been developed for localization prediction. The very first methods identified the existence of signal sequence, which was soon followed by methods for cleavage site prediction (Chou 2002). Dozens of prediction methods for subcellular localization have been developed, mainly for single compartments and in recent years some multicompartment predictors have also been released (for a review see Dönnes and Höglund 2004; Emanuelsson 2002; Schneider and Fechner 2004; Sprenger et al. 2006).
Present methods differ in many details, such as biological information used, algorithms and reliability. Many methods do predictions based on the amino acid sequence while others may need some additional information such as gene expression data. Several methods utilize amino acid composition because proteins in different environments are known to have different amino acid composition (Dönnes and Höglund 2004). Searches for predetermined signal peptide motifs and homology-driven approaches are also common. Several methods combine different rules to achieve better performance. The algorithms behind the methods also vary largely. The rules of prediction can be readily determined or they can be learnt from the training data set by different machine learning approaches. Most often used machine learning methods are Hidden Markov models, neural networks, self-organizing maps and support vector machines (Schneider and Fechner 2004).
The performance of the subcellular localization predictors varies. The localization of SP containing proteins can be predicted quite well while the knowledge of many non-SP signal peptides is insufficient as targeting sequences are usually just few residues long and as also the protein conformation affects the recognition of individual sites (Schneider and Fechner 2004).
For the single compartment predictors, the accuracy of prediction can be high, around 90% (Klee and Ellis 2005). Besides these binary predictors, some multipredictors have also been introduced. One of the pioneers in this field has been PSORT family of tools (Nakai and Horton 1999). In addition, some other methods have been launched including Hum-mPLoc (Shen and Chou 2007), HSLPred (Garg et al. 2005), LOCtree (Nair and Rost 2005), pTarget (Guda and Subramaniam 2005), Cello (Yu et al. 2006) Proteome analyst (Lu et al. 2004) and Euk-mPLoc 2.0 (Chou and Shen 2009). These predictors utilize machine learning methods such as support vector machine and k-Nearest Neighbours classification.
Based on individual predictors, a protocol was presented for a large number of subcellular localizations (Emanuelsson et al. 2007). The Scandinavian protocol is not actually a computer program, but a scheme by which individual predictions are manually run and interpreted. We evaluated the accuracy of WoLF PSORT (Horton et al. 2007) and Scandinavian protocol and predicted whether among 22,000 disease-related missense mutations are changes that could affect protein localization (Laurila and Vihinen 2009). According to the results, large number of diseases may arise due to mislocalization of proteins. Here we present an automated implementation of the Scandinavian protocol, which can be applied to predict the cellular localization of proteins and changes introduced by mutations.
Materials and methods
PROlocalizer service implements the Scandinavian protocol of Emanuelsson et al. The method can be applied to animal proteins as the signals and localization machinery are well conserved in metazoans. The protocol can predict altogether 12 individual subcellular localizations, which are mitochondrial inner membrane, transmembrane; mitochondrial periplasmic space; mitochondrial matrix; Golgi, transmembrane; plasma membrane; secreted; ER lumen; nucleus; peroxisome; cytoplasmic; plasma membrane, GPI anchor; and plasma membrane, myristoylated. The prediction scheme is in Fig. 1. PROlocalizer is freely accessible for academic use at http://bioinf.uta.fi/PROlocalizer.
Fig. 1.
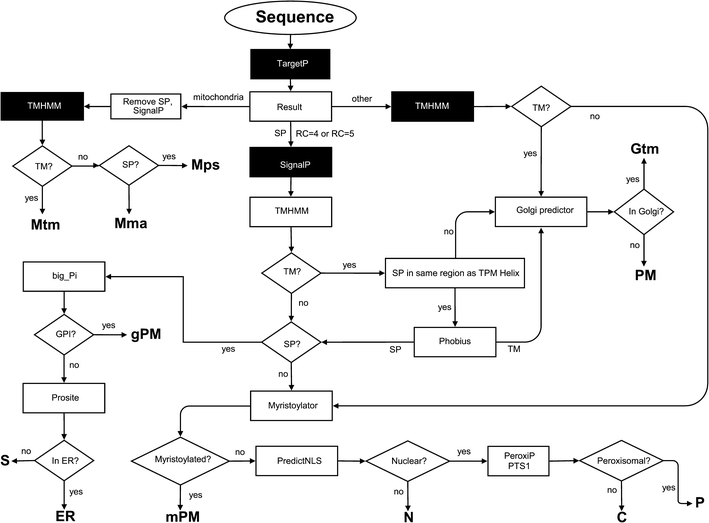
PROlocalizer prediction scheme. The service predicts proteins to 12 compartments: C cytosol; ER endoplasmic reticulum, lumen; Gtm Golgi, transmembrane; Mtm mitochondrial inner membrane (transmembrane); Mps mitochondrial periplasmic space; Mma mitochondrial matrix; PM plasma membrane; S secreted; N nucleus; P peroxisome; gPM plasma membrane, GPI anchor; and mPM plasma membrane, myristoylated. Programs run locally are indicated with black box. In some rare cases double localizations are predicted if the reliability coefficient (RC) in TargetP is high, i.e. poor
The protocol is based mainly on binary classifiers, which predict whether a protein is localized into a specific compartment or not. Of the 11 different programs in PROlocalizer, TargetP (Emanuelsson et al. 2000), SignalP (Bendtsen et al. 2004) and TMHMM (Krogh et al. 2001; Sonnhammer et al. 1998) were downloaded from http://www.cbs.dtu.dk/services/ and are run locally while programs Big-PI (Eisenhaber et al. 1998, 1999, 2000; Sunyaev et al. 1999) (http://mendel.imp.ac.at/sat/gpi/gpi_server.html), NMT (http://mendel.imp.ac.at/myristate/SUPLpredictor.htm), PeroxiP (Emanuelsson et al. 2003) (http://bioinfo.se/PeroxiP/), PredictNLS (Cokol et al. 2000) (http://cubic.bioc.columbia.edu/predictNLS/), PTS1 (Neuberger et al. 2003a, b (http://mendel.imp.ac.at/mendeljsp/sat/pts1/PTS1predictor.jsp), Golgi predictor (Yuan and Teasdale 2002) (http://ccb.imb.uq.edu.au/golgi/), Phobius (Käll et al. 2004) (http://phobius.sbc.su.se/), and Prosite (de Castro et al. 2006) (http://au.expasy.org/prosite/) are run over the Internet. If TargetP has a problem in sorting a protein to mitochondria with poor reliability coefficient (RC) (value 4 or 5) then also SignalP is used and the protein obtains two alternative localizations. Java and perl scripts were written to submit the protein sequence to prediction programs and to automatically interpret the results and to generate the report for the user.
Briefly, the prediction protocol works as follows (Fig. 1): TargetP assigns whether the protein goes to secretory pathway or mitochondia or not. Then, mitochondrial proteins are further sorted to transmembrane, periplasmic space or matrix based on the analysis of transmembrane and signal peptide sequences. Transmembrane proteins have two prediction routes and are finally classified into those going to Golgi transmembrane or plasma membrane. Proteins with signal peptide(s) are predicted to their compartments based on the targeting motifs.
PROlocalizer has a user-friendly graphic interface (Fig. 2). The user needs to provide only protein sequence(s) in fasta format. Submission can be done via a file or pasting the information to the submission form. PROlocalizer can also efficiently predict the subcellular localization for relatively large protein datasets. Furthermore, effects of one or more missense mutations per protein can be predicted by PROlocalizer just by providing the wild type sequence and the original and mutated residues along with position information.
Fig. 2.
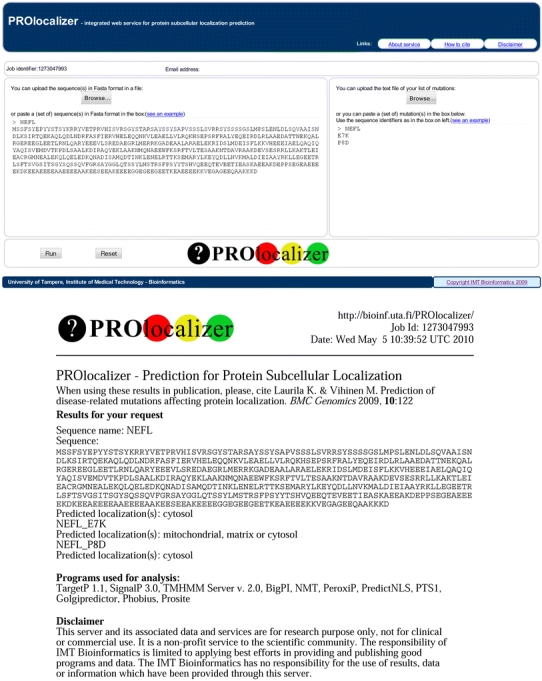
PROlocalizer home page and example of a prediction. Top PROlocalizer home page, Bottom example of the output for a prediction. Only part of the report is shown
The length of a single protein sequence cannot be longer than 4,000 amino acids, because of the limitations in some of the programs. As many of the classifiers are run outside our system and can take some time, the submitter is provided with search ID. In case any of the servers running outside our system is down the analysis cannot be performed. The user should then return to the service to rerun the analysis. An e-mail is sent to the submitter once the analysis is finished. The e-mail contains the results and a link to a web page where the same data are available with a number of links. The web page will be available for a limited period of time. A pdf format report lists the details of the query and analysis, used programs and their versions and predicted localization(s) (Fig. 2).
Results and discussion
Localization is a very important property for proteins, however, sometimes difficult to experimentally determine. A protein may have several localizations depending on the state of the cell and tissue. As novel protein sequences are identified at increasing pace it is beneficial to be able to predict properties of these molecules. PROlocalizer is based on the state-of-the-art binary predictors that have been combined to work as the Scandinavian protocol. We have automated the use, submission and interpretation of results in this protocol.
The performance of the Scandinavian protocol and the individual predictors that it is using has been previously evaluated with a dataset containing more than 1,500 proteins (Laurila and Vihinen 2009) and therefore we are not discussing the performance issue in detail here. The accuracy for different compartments varied from 0.55 to 0.97 being on average of 0.84. Additionally, the authors of the Scandinavian protocol discuss the performance in their article (Emanuelsson et al. 2007).
We have previously indicated that localization affecting mutations are likely involved in a number of disorders (Laurila and Vihinen. 2009). Due to the lack of sufficient number of known cases we were not able to provide statistical analysis for the performance when predicting mutation localization effects. As our previous study indicates, certain disease-causing mutations likely affect mutation signals just like other functionally and structurally crucial sites. Thus, analysis of variation effects may require localization predictions (Thusberg and Vihinen. 2009). PROlocalizer can also be used to analyze the mutation effects on protein subcellular localization. Compared with many other protein localization prediction services, the Scandinavian procedure behind the PROlocalizer does not utilize sequence homology searches or protein amino acid composition, instead the predictions are based on the identification of protein sorting signals. Thus, PROlocalizer is more likely to detect the point mutation effects on localization than several other prediction methods. Fully automated PROlocalizer has a user-friendly interface and it generates a detailed report of predictions. The service is freely available for academic, non-commercial use.
Conclusions
PROlocalizer is a tool for prediction of altogether 12 protein subcellular localizations. It is implemented in a user-friendly, automated service. The user can submit at a time, a number of proteins and/or variations, if necessary. The service automatically interprets the result(s) based on the complex prediction scheme. PROlocalizer provides a detailed report in several formats, which also include in addition to the prediction results details for the used methods. In the future, we plan to extend the system to allow predictions for plants, fungi and bacteria.
Acknowledgments
This work was supported by the Biocenter Finland, Finnish Academy (application number 213462, Finnish Programme for Centres of Excellence in Research 2006–2011), the Medical Research Fund of Tampere University Hospital, Sigrid Juselius Foundation, Tampere Graduate School in Information Science and Engineering (TISE), and Otto A. Malm Foundation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Chou KC. Prediction of protein signal sequences. Curr Protein Pept Sci. 2002;3:615–622. doi: 10.2174/1389203023380468. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. A new method for predicting the subcellular localization of eukaryotic proteins with both single and multiple sites: Euk-mPLoc 2.0. PLos One. 2009;5:e9931. doi: 10.1371/journal.pone.0009931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokol M, Nair R, Rost B. Finding nuclear localization signals. EMBO Rep. 2000;1:411–415. doi: 10.1093/embo-reports/kvd092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TN. Protein localization in proteomics. Curr Opin Chem Biol. 2004;8:49–53. doi: 10.1016/j.cbpa.2003.11.003. [DOI] [PubMed] [Google Scholar]
- de Castro E, Sigrist CJ, Gattiker A, Builliard V, Langendjik-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dönnes P, Höglund A. Predicting protein subcellular localization: past, present, and future. Genomics Proteomics Bioinformatics. 2004;2:209–215. doi: 10.1016/S1672-0229(04)02027-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Eisenhaber F. Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 1998;11:1155–1161. doi: 10.1093/protein/11.12.1155. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Eisenhaber F. Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999;292:741–758. doi: 10.1006/jmbi.1999.3069. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Yuan Y, Löffler G, Eisenhaber F. Automated annotation of GPI anchor sites: case study C. elegans. Trends Biochem Sci. 2000;25:340–341. doi: 10.1016/S0968-0004(00)01601-7. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O. Predicting protein subcellular localization from amino acid sequence information. Brief Bioinform. 2002;3:361–376. doi: 10.1093/bib/3.4.361. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Elofsson A, von Heijne G, Cristobal S. In silico prediction of the peroxisomal proteome in fungi, plants and animals. J Mol Biol. 2003;330:443–456. doi: 10.1016/S0022-2836(03)00553-9. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Falk R, Ramstöm M, Ståhl S, Hober S. Approaches for systematic proteome exploration. Biomol Eng. 2007;24:155–168. doi: 10.1016/j.bioeng.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Garg A, Bhasin M, Raghava GP. Support vector machine-based method for subcellular localization of human proteins using amino acid compositions, their order, and similarity search. J Biol Chem. 2005;280:14427–14432. doi: 10.1074/jbc.M411789200. [DOI] [PubMed] [Google Scholar]
- Guda C, Subramaniam S. pTARGET: A new method for predicting protein sub-cellular localization in eucaryotes. Bioinformatics. 2005;21:3963–3969. doi: 10.1093/bioinformatics/bti650. [DOI] [PubMed] [Google Scholar]
- Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Klee EW, Ellis LB. Evaluating eukaryotic secreted protein prediction. BMC Bioinformatics. 2005;6:256. doi: 10.1186/1471-2105-6-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Laurila K, Vihinen M. Prediction of disease-related mutations affecting protein localization. BMC Genomics. 2009;10:122. doi: 10.1186/1471-2164-10-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Szafron D, Greiner R, Lu P, Wishart DS, Poulin B, Anvik J, Macdonell C, Eisner R. Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics. 2004;20:547–556. doi: 10.1093/bioinformatics/btg447. [DOI] [PubMed] [Google Scholar]
- Martoglio B, Dobberstein B. Signal sequences: more than greasy peptides. Trends Cell Biol. 1998;8:410–415. doi: 10.1016/S0962-8924(98)01360-9. [DOI] [PubMed] [Google Scholar]
- Nair R, Rost B. Mimicking cellular sorting improves prediction of subcellular localization. J Mol Biol. 2005;348:85–100. doi: 10.1016/j.jmb.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Nakai K, Horton P. PSORT: a program for detecting the sorting signals of proteins and predicting their subcellular localization. Trends Biochem Sci. 1999;24:34–35. doi: 10.1016/S0968-0004(98)01336-X. [DOI] [PubMed] [Google Scholar]
- Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F. Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J Mol Biol. 2003;328:581–592. doi: 10.1016/S0022-2836(03)00319-X. [DOI] [PubMed] [Google Scholar]
- Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F. Motif refinement of the peroxisomal targeting signal 1 and evaluation of taxon-specific differences. J Mol Biol. 2003;328:567–579. doi: 10.1016/S0022-2836(03)00318-8. [DOI] [PubMed] [Google Scholar]
- Schneider GH, Fechner U. Advances in the prediction of protein targeting signals. Proteomics. 2004;4:1571–1580. doi: 10.1002/pmic.200300786. [DOI] [PubMed] [Google Scholar]
- Shen HB, Chou KC. Hum-mPLoc: an ensemble classifier for large-scale human protein subcellular location prediction by incorporating samples with multiple sites. Biochem Biophys Res Commun. 2007;355:1006–1011. doi: 10.1016/j.bbrc.2007.02.071. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. In: Proceedings of the 6th international conference on intelligent systems for molecular biology (ISMB-98), vol 6, 28 June–1 July, Montréal, pp 175–182 [PubMed]
- Sprenger J, Fink JL, Teasdale RD. Evaluation and comparison of mammalian subcellular localization prediction methods. BMC Bioinformatics. 2006;7(Suppl 5):S3. doi: 10.1186/1471-2105-7-S5-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud RM, Walter P. Signal sequence recognition and protein targeting. Curr Opin Struct Biol. 1999;9:754–759. doi: 10.1016/S0959-440X(99)00040-8. [DOI] [PubMed] [Google Scholar]
- Sunyaev SR, Eisenhaber F, Rodchenkov IV, Eisenhaber B, Tumanyan VG, Kuznetsov EN. PSIC: profile extraction from sequence alignments with position-specific counts of independent observations. Protein Eng. 1999;12:387–394. doi: 10.1093/protein/12.5.387. [DOI] [PubMed] [Google Scholar]
- Thusberg J, Vihinen M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum Mutat. 2009;30:703–714. doi: 10.1002/humu.20938. [DOI] [PubMed] [Google Scholar]
- Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins:Struct, Function Bioinformatics. 2006;64:643–651. doi: 10.1002/prot.21018. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Teasdale RD. Prediction of Golgi Type II membrane proteins based on their transmembrane domains. Bioinformatics. 2002;18:1109–1115. doi: 10.1093/bioinformatics/18.8.1109. [DOI] [PubMed] [Google Scholar]