

Abstract
The protein kinases ATM and DNA-PKcs play critical roles in the cellular response to DNA double strand breaks (DSBs). ATM and DNA-PKcs are activated in response to DSBs and play several important roles in propagation of the damage signal and for the repair of DNA damage. Recent work from several groups, including ours, has focused on studying the dynamics of each of these proteins at DSBs and the requirements and factors which play a role(s) in this process. The use of live cell imaging of fluorescently-tagged ATM and DNA-PKcs has allowed us to study the real-time response of these proteins to laser-generated DNA damage in vivo. Here, we will extensively discuss the behavior of the ATM and DNA-PKcs proteins at DSB sites.
Key words: ATM, DNA-PKcs, DNA double strand breaks, autophosphorylation, live cell imaging
Introduction
Human cells are exposed to at least 10,000 DNA lesions every day. Of the various types of DNA damage that can occur, DNA double strand breaks (DSBs) are the most lethal.1 Double strand breaks can form in response to endogenously generated agents, including reactive oxygen species and during normal V(D)J recombination, and exogenous agents such as ionizing radiation (IR) and radiomimetic drugs.2 The inability to repair DSBs can result in genomic instability, carcinogenesis or cell death. Because of the deleterious nature of these events, cells have developed multiple redundant mechanisms to repair DSBs. The two major pathways which are responsible for repairing DSBs are homologous recombination (HR) and non-homologous end-joining (NHEJ).3 The HR process mediates DSB repair by using a homologous DNA sequence as a template to guide proper restoration of the break. Because homologous templates are found on sister chromosomes, HR is thought to be only active during the S and G2 cell cycle phases. NHEJ is characterized by its ability to directly ligate the two ends of the broken DNA molecule. This process does not have the need for a homologous template and is therefore theoretically not restricted to a certain phase of the cell cycle. Finally, since the HR process utilizes a homologous DNA sequence, it is believed to be a relatively error-free process, whereas NHEJ, which only ligates the two DNA ends together, is potentially error-prone.
The cellular response to DNA DSBs is a complex process that includes recognition of DNA damage, activation of signaling pathways including cell cycle checkpoints, and repair of the DNA lesions. Two important proteins required for the cellular response to DSBs are the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and the ataxia telangiectasia mutated (ATM) protein.4–9 DNA-PKcs and ATM belong to the phosphatidylinositol 3-kinase-like family of serine/threonine protein kinases. Motif analysis shows that DNA-PKcs and ATM share many common features including a leucine zipper, a FAT domain (FRAP, ATM and TRRAP), the phosphoinositide 3,4-kinase domain and a FAT carboxy-terminal (FATC) domain. ATM and DNA-PKcs are both rapidly and specifically activated in response to DSBs and play important roles in transducing the damage signal and for the repair of the DNA damage. Once activated, ATM phosphorylates a number of substrates including other repair proteins and proteins which activate cell cycle checkpoints that are important for the repair of DSBs.8,9 ATM probably does not directly have a role in the repair of DNA DSBs, but it signals to the repair machinery to assist in the repair of DNA damage. Mutations in ATM can result in the genomic instability syndrome termed Ataxia-Telangiectasia (A-T), which is characterized by progressive cerebellar ataxia, immune deficiencies, radiation sensitivity and an increased risk of cancer.10 Cells isolated from A-T patients have defects in cell cycle checkpoint activation, radiosensitivity and an increased frequency of chromosome breakage.10 A-T cells do not have a gross defect in DNA DSB repair, but experiments have shown that a small subset (10%) of DSBs are not repaired in these cells.11 This subset of DNA damage requires processing by the nuclease Artemis and this processing requires Artemis phosphorylation by ATM. Later studies have shown that this subset of DNA damage is localized to the heterochromatin.12
DNA-PKcs is believed to be directly involved in the repair process via NHEJ. The NHEJ process repairs DSBs that are introduced by IR and reactive oxygen species but also by those that are generated during normal V(D)J recombination.3–6 NHEJ is initiated when the Ku protein binds to DNA ends. The Ku protein is a heterodimer consisting of the Ku70 and Ku80 proteins which forms a ring-like structure that has a high affinity for free DNA ends.13 Once bound to DNA, the Ku70/80-DNA complex functions as a scaffold to recruit other NHEJ factors to the DSB including DNA-PKcs. Upon binding the Ku70/80-DNA complex, the kinase activity of DNA-PKcs is activated. Although the kinase activity of DNA-PKcs is required for NHEJ, the primary role of DNA-PKs may be to tether the two ends of the broken DNA molecule and regulate the access of other repair enzymes to the DNA termini.4 Non-ligatable DNA ends are processed by polymerases and nucleases, including the Artemis protein. Finally, the DNA ends are ligated by the ligase IV/XRCC4 complex and may be enhanced by the presence of the XLF/Cernunnos protein. Recently a hypomorphic DNA-PKcs mutation was discovered in a patient with radiosensitive T−B− severe combined immunodeficiency.14 The causative mutation in DNA-PKcs is a missense mutation at the 3062 amino acid which results in insufficient Artemis activation and compromised V(D)J recombination, an essential process for the development of B and T lymphocytes.
Recruitment to DNA Double Strand Breaks
A concerted effort has been made to study the cellular response to DSBs including determining which proteins localize to DNA DSBs. The established method for studying the localization of proteins to DSBs was by visualization of repair proteins localizing into nuclear foci via immunofluorescence following induction of the damage.15 Unfortunately, in order to observe nuclear foci, cells must be fixed; therefore, the real-time observation of proteins localizing to DSBs remains challenging. Many labs, including our own, now use live cell imaging of fluorescently-tagged proteins to study DNA damage responses in vivo and in real time.16–19 Coupling irradiation via a micro-laser with fluorescent microscopy allows for continuous monitoring of real-time recruitment and retention of fluorescent-tagged proteins to DSBs in living cells. Previous studies have shown that DNA-PKcs and ATM are both recruited to laser-generated DSBs.17,19,20 To compare DNA-PKcs and ATM recruitment in the same cellular background to laser-induced DSBs, YFP-tagged DNA-PKcs and ATM were expressed in HT1080 cells and time-lapse imaging for each protein's recruitment to laser-generated DSBs were collected. Both proteins were shown to rapidly localize to DSBs (localization was observed as early as 60 seconds for each protein) (Fig. 1A), but DNA-PKcs reached its maximal level faster (10 minutes) than ATM (20 minutes) (Fig. 1B). The difference in reaching its maximal level between the two proteins may be because DNA-PKcs only localizes at the sites of DNA damage whereas ATM localizes to the DNA break and the flanking region of the break.21
Figure 1.

Accumulation and dissociation/retention kinetics of ATM and DNA-PKcs at laser-generated DNA double strand breaks. (A) Accumulation of YFP-tagged ATM and DNA-PKcs at laser-induced double strand breaks. Time-lapse imaging of exogenously expressed YFP-ATM and YFP-DNA-PKcs in HT1080 cells before and after micro-irradiation. Initial accumulation kinetics (180 seconds) of YFP-ATM and YFP-DNA-PKcs accumulating at the DSB site after micro-irradiation. (B) 2 hour time course after micro-irradiation, showing the kinetics of YFP-tagged ATM and DNA-PKcs in HT1080 cells. Each data point is the average of 10 independent measurements. Error bars represent the standard deviation.
Although ATM and DNA-PKcs can both directly interact with DNA, many studies have shown that each requires specific proteins to localize to DSBs.22,23 Many in vitro studies have shown that the Ku70/80 heterodimer directs DNA-PKcs to DNA ends and stabilizes the DNA-DNA-PKcs interaction.24–26 A previous study showed that the recruitment of DNA-PKcs to DSB sites is dependent on the Ku heterodimer in vivo.17 Furthermore, autophosphorylation of DNA-PKcs at serine 2056 was abrogated in the Ku deficient cells indicating that activation of DNA-PKcs' kinase activity is dependent on the Ku heterodimer in vivo. Similar to DNA-PKcs, a multimer protein complex recruits ATM to DSBs. Previous studies have shown that the MRE11/RAD50/NBS1 (MRN) complex is required for focus formation of endogenous ATM in response to radiation, radiomimetic agents and ATM's association to DSBs generated by endonucleases.27–30 Like the Ku heterodimer, the MRN complex is believed to be an initial sensor of DSBs. The MRE11/RAD50 complex binds to DNA ends via two DNA biding domains in MRE11 and the two termini of the broken DNA molecule are tethered by RAD50.31 MRE11 has exonuclease and endonuclease activity and the exonuclease activity is required for resection for the initiation of HR.32,33 Although the NBS1 protein does not have any known enzymatic activity, it does stimulate MRE11's endonuclease activity and is important for activating ATM.34 These studies were verified and expanded upon by showing that YFP-tagged ATM localization to laser-induced DSBs is abrogated in MRN knock-down cells and in a NBS-deficient cell line in living cells.19 Previous data suggests that the exonuclease activity of MRE11 is required for activation of ATM.29 To determine if the nuclease activity of MRE11 is required for localization of ATM to DSBs, MRE11 deficient cells were complemented with wild-type DsRed-tagged MRE11, DsRed-tagged MRE11-3 (nuclease dead mutation) or the DsRed empty vector and localization of YFP-ATM to laser-generated DSBs was determined.35 MRE11 and MRE11-3 both localize to laser-induced DSBs as does ATM (Fig. 2). These data show that the exonuclease activity of MRE11 is not required for localization of MRE11 or ATM to DSBs.
Figure 2.
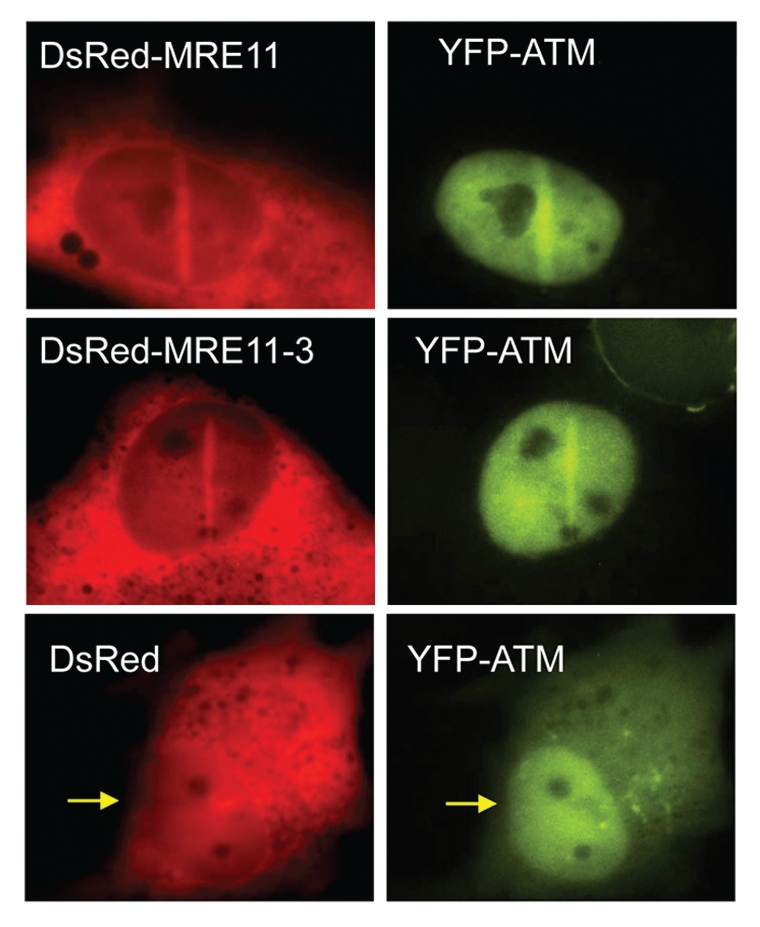
The exonuclease activity of MRE11 is not required for ATM localization to DNA double strand breaks. YFP-ATM expressing MRE11 deficient (ATLD) cells were transfected with DsRed-tagged full-length MRE11 and nuclease-dead MRE11-3 and micro-irradiated.
A previous study reported a conserved carboxy-terminal motif in human Ku80 and NBS1 that is required for their interaction with DNA-PKcs and ATM, respectively.28 This carboxy-terminal motif was shown to be required for the recruitment of each of these proteins to DSBs, signaling events and DNA repair. Further studies have shown that although this carboxy-terminal motif is important for ATM and DNA-PKcs activation, it is not absolutely required for it. For example, at low doses of irradiation, MRE11-RAD50 activates ATM in the absence of NBS1, but NBS1 cells expressing NBS1 with C-terminal motif deleted had attenuated levels of ATM activation.36 Furthermore, ATM localized to laser-generated DSBs in mouse knock-out cells which were complemented with NB1 with the C-terminus deleted and ATM was shown to interact with this mutant NBS1 protein.37 Lastly, ATM interacts with the MRN complex at multiple sites. Similar to this, further studies with DNA-PKcs and Ku80 have shown that the Ku80 C-terminal motif is not essential for DNA-PKcs kinase activity or its ability to localize to DSBs.38 However, deletion of the C-terminal portion of Ku80 results in radiosensitivity, which is probably due to its importance for phosphorylation of DNA-PKcs at the 2609 cluster and the DNA end processing by the Artemis endonuclease and ultimately end-joining. These data suggest that ATM and DNA-PKcs are both recruited to DSBs via multimeric complexes, but further detailed studies need to be performed to delineate the specific domains/residues required for localization of ATM and DNA-PKcs to DSBs.
Retention at and Dissociation from DNA Double Strand Breaks
Once DNA-PKcs and ATM are recruited to a DSB, the next question is what their dynamics at the DSB are after recruitment. Although DNA-PKcs and ATM both rapidly associate to DSBs, their retention/dissociation from DSBs is significantly different. Following reaching its maximal level, DNA-PKcs starts to rapidly dissociate from DSBs. The fluorescence intensity of the accumulation area in cells expressing DNA-PKcs decreased to 20% of its maximum level in a 2 hour period (Figs. 1C and 3). On the other hand, the fluorescence intensity of ATM at the accumulation area was still at 60% of its maximal level at 2 hours (Figs. 1C and 3). The non-homologous end-joining (NHEJ) factors Ku70, Ku80 and DNA-PKcs dissociate from DSBs rather quickly (80% is dissociated in 2 hours).17,39 The dissociation of NHEJ factors is similar to the resolution of γH2AX foci, which suggests that NHEJ factors dissociate once DSB repair is completed. To support this notion, DNA-PKcs showed persistent localization at DSBs in NHEJ deficient cells.17 ATM may be retained because it is not there to solely repair the break, but it also to assist in chromatin remodeling at DSBs and/or due to its role in signaling and halting the cell cycle.30,40,41 Furthermore, recent data also shows that ATM is required for repair of DNA damage in the heterochromatin and it is possible that prolonged localization of ATM is due to its requirement for repair of this subset of DNA.12 Interestingly, FRAP data shows that the YFP-ATM signal within the bleached regions in the micro-irradiated area rapidly recovered to the 80% of pre-bleached values (half-time is 9.69 seconds), while the recovery of YFP-DNA-PKcs proceeded with slower kinetics (half-time is 70.98 seconds) (Fig. 3). These results indicate that the DNA-PKcs-DSB interaction is much tighter than that of ATM-DSB. The reason for the tight interaction of DNA-PKcs-DSB could be the role of DNA-PKcs to tether the two broken ends.4 ATM phosphorylates its substrates not only at DSBs but also in the nucleoplasm (for example p53) and to do this activated ATM should be released from DSBs.9 This may be why the ATM-DSB interaction is not stable.
Figure 3.

Phosphorylation of ATM and DNA-PKcs influences their dynamics at DNA double strand breaks. (A) AT5BIVA cells expressing wild-type, 1981A or kinase-dead (KD) forms of YFP-ATM were micro-irradiated, incubated for 10 min, pre-extracted, fixed and immunostained with a phospho-specific antibody to ATM (S1981). (B) 2 hour time course after micro-irradiation, showing the kinetics of YFP-tagged wild-type, 1981A and KD forms ATM in AT5BIVA cells. Each data point is the average of 10 independent measurements. Error bars represent the standard deviation. ©Sairei So et al. 2009; Originally published in J Cell Biol doi:10.1083/jcb.200906064 (C) 2 hour time course after micro-irradiation, showing the kinetics of YFP-tagged wild-type, 7A and KD forms DNA-PKcs in V3 cells. Each data point is the average of 10 independent measurements. Error bars represent the standard deviation. ©Naoya Uematsu et al. 2007; Originally published in J Cell Biol doi:10.1083/jcb.200608077.
Impact of the Phosphorylation Status on the Dynamics at DSBs and Functions of ATM and DNA-PKcs
Studies have shown that the phosphorylation status of ATM and DNA-PKcs play an important role in the dynamics of each protein at DNA DSBs. In human cells, ATM is autophosphorylated on serines 367, 1893 and 1981 in response to IR.42–44 The best characterized of these sites is serine 1981. Autophosphorylation at serine 1981 leads to dissociation of ATM from a dimer into an active monomer and this active monomer is recruited to DNA breaks where it can phosphorylate a number of substrates.42 Previously, contradictory data has been produced in experiments which tested the requirement of serine 1981 autophosphorylation for ATM functions, including localization to DSBs. Studies with human ATM have shown that autophosphorylation of 1981 is required for association of ATM to chromatin.30 In contrast, studies in ATM knockout mice complemented with ATM-S1987A and ATM S367AS1899AS1987A (mouse homologue of human serine 367, 1893 and 1981) demonstrated normal localization of ATM to DSBs.20,45 Recent work from our laboratory has shown that autophosphorylation at serine 1981 is not required for localization of ATM to laser-generated DSBs.19 Furthermore, we found that other autophosphorylation sites are not required either, as the kinase dead (KD) form of ATM localizes to DSBs (Fig. 3A). Wild-type, S1981A and KD forms of YFP-tagged ATM had similar accumulation kinetics to DSBs during the first 10 minutes after micro-irradiation. But, time lapse imaging experiments showed that after its initial localization (first 10 minutes), S1981A and KD rapidly dissociated from DSBs. Two hours after irradiation, 65% of wild-type ATM was still present at laser-generated DSBs whereas only 20% of S1981A and KD were (Fig. 3A). Biochemical assays showed that ablation of ATM autophosphorylation resulted in a reduced interaction with MDC1 and that MDC1 is required for ATM retention at DSB sites.19
Similar to ATM, the phosphorylation status of DNA-PKcs regulates its dynamics at DNA DSBs. DNA-PKcs is phosphorylated at a number of sites including autophosphorylated at serine 2056 and phosphorylated on six sites by ATM between the threonine 2609 and threonine 2647 amino acids (T2609 cluster).46–49 Wild-type, KD and 7A (DNA-PKcs in which the six phoshphorylation sites of the T2609 cluster and serine 2056 have been mutated to alanine) forms of DNA-PKcs show similar accumulation kinetics to laser-generated DSBs.17 However, time-lapse imaging experiment for a period of 2 hours after laser micro-irradiation shows that the fluorescence intensity of the accumulation area in the wild-type cells decreased to 20% of the maximum levels at 2 hours, whereas the intensity of the accumulation area in the KD and 7A cells only dropped to 80% of the maximum level in the same time period (Fig. 3B). These results demonstrate that impairment of phosphorylation at these seven sites results in sustained retention of DNA-PKcs at the sites of DSBs. The retention of KD and 7A forms of DNA-PKcs at the laser-generated DSBs is most likely due to the inability of the cells to repair the introduced DSBs. This is supported by the experiments which show that KD or 7A expressing cells displays a deficiency in DSB repair.46,50,51 Furthermore, wild-type DNA-PKcs is also retained at DSBs in a similar fashion to the KD and 7A proteins when it is expressed in XR1 cells which are XRCC4 deficient and have been shown to not be able to repair DSBs.52
Comparing the two proteins, autophosphorylation does not play a role in the ability of either protein to localize to laser-generated DSBs. But, when it comes to retention/dissociation, ATM and DNA-PKcs are opposites. Ablation of autophosphorylation of ATM results in rapid dissociation of ATM following its normal association. On the other hand, ablation of phosphorylation of DNA-PKcs results in a sustained retention at DSBs. FRAP data shows that S1981A is a bit more mobile than wild-type ATM with the half-time to recover to 50% of maximum recovery value is 6.93 seconds to S1981A compared to 9.69 seconds for wild-type ATM (Fig. 4). The half-time to recover to 50% of maximum recovery value is 123.95 seconds for 7A compared to 70.98 seconds for wild-type DNA-PKcs (Fig. 4). Comparing the phosphorylation site mutant of ATM compared to DNA-PKcs, the numbers obviously show that ATM is much more mobile than DNA-PKcs at DSBs and that mutation of the phosphorylation sites make this difference even larger.
Figure 4.

Phosphorylation of ATM and DNA-PKcs influence the exchange rate between DNA-bound and free forms of each protein at DNA double strand breaks sites. (A and B) FRAP curves of wild-type ATM and 1981A in AT5BIVA cells (A) and wild-type DNA-PKcs and 7A in V3 cells (B) at DNA double strand break sites. The data show the normalized data from the average of three independent experiments, fitted to an Exponential Rise to Max model. Pre-bleach intensity levels were normalized to 1 and post-bleach intensity levels were normalized to 0. In each experiment at least 10 cells were measured.
Although it appears the phosphorylation status differentially regulates ATM and DNA-PKcs at DSBs, the ablation of the autophosphorylation sites similarly results in deficiency in DSB repair in both situations. A marked radiosensitivity in ATM deficient cells is observed when compared with the same cell that had been complemented with wild-type ATM.19,43 Complementation with S367A, S1893A or S1981A results in an intermediate sensitivity to IR.19,43 Furthermore, ablation of each of these phosphorylation sites results in defects in ATM signaling, radiation-induced DNA repair and a failure to correct cell cycle checkpoint defects. Interestingly, S1981A showed reduced phosphorylation of SMC1 and KAP1, proteins phosphorylated at DSBs, however, phosphorylation of p53 which is not phosphorylated at sites of damage was not affected, suggesting that defective phosphorylation of SMC1 and KAP1 is due to the premature dissociation of ATM from DSBs when ATM autophosphorylation at serine 1981 is ablated.19 Furthermore, ablation of the autophosphorylation site results in a weakened interaction with MDC1. These data suggest that autophosphorylation at serine 1981 promotes the interaction of ATM with MDC1 which serves to stabilize ATM at DSBs and thereby regulates phosphorylation of substrates and response to DNA damage in human cells. The importance of DNA-PKcs autophosphorylation for NHEJ is demonstrated by numerous studies which show that ablation of autophosphorylation sites results in an increase in radiosensitivity and a decrease in DSB repair.46,50,51 Biochemical studies showed that unphosphorylated DNA-PKcs block DNA ends, and thereby inhibits efficient ligation.50,53,54 Importantly, this inhibition can be cleared by autophosphorylation. Furthermore, solution structure of DNA-PKcs and Ku in complex with DNA revealed that autophosphorylation induced a large conformational change that opens DNA-PKcs.55 In addition, mutation of 2056 and the entire 2609 cluster results in tight binding of DNA-PKcs to DNA ends (Fig. 3 FRAP). Together, these data suggest that phosphorylation of DNA-PKcs promotes a conformational change to open the DNA-DNA-PKcs complex to let other NHEJ factors access, process, and ligate the DNA ends.
Concluding Remarks
The PI3K kinases ATM and DNA-PKcs are key proteins in the cellular response to DNA double strand breaks. Our data suggests that there are common mechanisms which regulate their association with DSBs with multimeric complexes recruiting and stabilizing the proteins at the damage site. FRAP analysis shows that the DNA-ATM and DNA-DNA-PKcs complexes are different with the DNA-DNA-PKcs complex being tighter than the DNA-ATM complex. The retention/dissociation of ATM and DNA-PKcs are markedly different with DNA-PKcs dissociating much quicker from DSBs than ATM. A key player in the retention/dissociation rate of both proteins is phosphorylation. Autophosphorylation of ATM is required for sustained retention of ATM to DSBs and autophosphorylation of ATM at serine 1981 promotes its interaction with MDC1 which serves to stabilize ATM at DSBs. On the other hand, DNA-PKcs phosphorylation is required for dissociation from the DNA damage site possibly by promoting a conformational change which allows dissociation from DSB sites following efficient repair of the DNA damage. Further delineating the mechanisms which regulate ATM and DNA-PKcs dynamics is an important area for further investigation.
Acknowledgements
We thank Eric Weterings and Kazi Fattah for their critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (CA50519, CA134991 and PO1-CA92584).
Abbreviations
- ATM
ataxia-telangiectasia mutated
- DNA-PKcs
DNA dependent protein kinase catalytic subunit
- DSBs
DNA double strand breaks
- HR
homologous recombination
- NHEJ
non-homologous end-joining
- MRN
MRE11/RAD50/NBS1
- YFP
yellow fluorescent protein
- FRAP
fluorescence recovery after photobleaching
- IR
ionizing radiation
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/12148
References
- 1.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 3.Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006;5:1042–1048. doi: 10.1016/j.dnarep.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 4.Weterings E, Chen DJ. The endless tale of nonhomologous end-joining. Cell Res. 2008;18:114–124. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 5.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 6.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 8.Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 9.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 10.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 11.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 12.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 14.Van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous endjoining. J Clin Invest. 2009;119:91–98. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukas C, Bartek J, Lukas J. Imaging of protein movement induced by chromosomal breakage: tiny ‘local’ lesions pose great ‘global’ challenges. Chromosoma. 2005;114:146–154. doi: 10.1007/s00412-005-0011-y. [DOI] [PubMed] [Google Scholar]
- 17.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177:219–229. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yano K, Morotomi-Yano K, Wang SY, Uematsu N, Lee KJ, Asaithamby A, et al. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9:91–96. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–990. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–225. doi: 10.1038/nature05112. [DOI] [PubMed] [Google Scholar]
- 21.You Z, Bailis JM, Johnson SA, Dilworth SM, Hunter T. Rapid activation of ATM on DNA flanking double-strand breaks. Nat Cell Biol. 2007;9:1311–1318. doi: 10.1038/ncb1651. [DOI] [PubMed] [Google Scholar]
- 22.Smith GC, Cary RB, Lakin ND, Hann BC, Teo SH, Chen DJ, et al. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci USA. 1999;96:11134–11139. doi: 10.1073/pnas.96.20.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammarsten O, Chu G. DNA-dependent protein kinase: DNA binding and activation in the absence of Ku. Proc Natl Acad Sci USA. 1998;95:525–530. doi: 10.1073/pnas.95.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drouet J, Delteil C, Lefrancois J, Concannon P, Salles B, Calsou P. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J Biol Chem. 2005;280:7060–7069. doi: 10.1074/jbc.M410746200. [DOI] [PubMed] [Google Scholar]
- 25.Hammarsten O, DeFazio LG, Chu G. Activation of DNA-dependent protein kinase by single-stranded DNA ends. J Biol Chem. 2000;275:1541–1550. doi: 10.1074/jbc.275.3.1541. [DOI] [PubMed] [Google Scholar]
- 26.Cary R, Peterson S, Wang J, Bear D, Bradbury E, Chen D. DNA looping by Ku and the DNA-dependent protein kinase. Proc Natl Acad Sci USA. 1997;94:4267–4272. doi: 10.1073/pnas.94.9.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 29.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 31.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 32.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 33.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arthur LM, Gustausson K, Hopfner KP, Carson CT, Stracker TH, Karcher A, et al. Structural and functional analysis of Mre11-3. Nucleic Acids Res. 2004;32:1886–1893. doi: 10.1093/nar/gkh343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerosaletti K, Concannon P. Independent roles for nibrin and Mre11-Rad50 in the activation and function of Atm. J Biol Chem. 2004;279:38813–38819. doi: 10.1074/jbc.M404294200. [DOI] [PubMed] [Google Scholar]
- 37.Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Reina San Martin B, Van Laethem F, et al. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol. 2005;7:675–685. doi: 10.1038/ncb1270. [DOI] [PubMed] [Google Scholar]
- 38.Weterings E, Verkaik NS, Keijzers G, Florea BI, Wang SY, Ortega LG, et al. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol Cell Biol. 2009;29:1134–1142. doi: 10.1128/MCB.00971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mari PO, Florea BI, Persengiev SP, Verkaik NS, Bruggenwirth HT, Modesti M, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci USA. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 42.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 43.Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006;25:3504–3514. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 45.Daniel JA, Pellegrini M, Lee JH, Paull TT, Feigenbaum L, Nussenzweig A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J Cell Biol. 2008;183:777–783. doi: 10.1083/jcb.200805154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333–2338. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, et al. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280:14709–14715. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- 48.Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, et al. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J. 2002;368:243–251. doi: 10.1042/BJ20020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Douglas P, Cui X, Block WD, Yu Y, Gupta S, Ding Q, et al. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol Cell Biol. 2007;27:1581–1591. doi: 10.1128/MCB.01962-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, et al. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol. 2003;23:5836–5848. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soubeyrand S, Pope L, Pakuts B, Hache RJ. Threonines 2638/2647 in DNA-PK are essential for cellular resistance to ionizing radiation. Cancer Res. 2003;63:1198–1201. [PubMed] [Google Scholar]
- 52.Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, et al. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83:1079–1089. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 53.Block WD, Yu Y, Merkle D, Gifford JL, Ding Q, Meek K, et al. Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Res. 2004;32:4351–4357. doi: 10.1093/nar/gkh761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol. 2005;25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]