

Abstract
Tissue stem cells form the cellular base for organ homeostasis and repair. Stem cells have the unusual ability to renew themselves over the lifetime of the organ while producing daughter cells that differentiate into one or multiple lineages. Difficult to identify and characterize in any tissue, these cells are nonetheless hotly pursued because they hold the potential promise of therapeutic reprogramming to grow human tissue in vitro, for the treatment of human disease. The mammalian skin epithelium exhibits remarkable turnover, punctuated by periods of even more rapid production after injury due to burn or wounding. The stem cells responsible for supplying this tissue with cellular substrate are not yet easily distinguishable from neighboring cells. However, in recent years a significant body of work has begun to characterize the skin epithelial stem cells, both in tissue culture and in mouse and human skin. Some epithelial cells cultured from skin exhibit prodigious proliferative potential; in fact, for >20 years now, cultured human skin has been used as a source of new skin to engraft onto damaged areas of burn patients, representing one of the first therapeutic uses of stem cells. Cell fate choices, including both self-renewal and differentiation, are crucial biological features of stem cells that are still poorly understood. Skin epithelial stem cells represent a ripe target for research into the fundamental mechanisms underlying these important processes.
The skin is the first line of defense to protect the body from dehydration, injury, and infection. To meet these needs, the skin has evolved an elaborate differentiation process that results in a tough, water-impermeable outer covering that is constantly renewable. Mammalian skin consists of both dermal and epidermal components; this discussion will be restricted to the epidermal cells, referred to as keratinocytes. The mammalian epidermis is a stratified tissue, anchored to a basement membrane (Fig. 1A). The layer of cells directly contacting the basement membrane, termed the basal layer, contains proliferating cells. Like all keratinocytes, cells of the basal layer possess a network of 10-nm keratin intermediate filaments (IFs), but they are otherwise relatively undifferentiated. As the population of basal cells expands because of division, some cells detach from the basement membrane and begin to move outward toward the skin surface. The first change to occur is a strengthening of the IF network to increase the tensile strength of each cell. Cells achieve this by synthesizing large numbers of new sets of keratins, which assemble into IFs that aggregate into more resilient bundles or cables of IFs. IF cables anchor to cell-cell junctions called desmosomes, thus distributing force not over individual cells but over the entire tissue (reviewed in ref. 1). As the suprabasal cells, now connected by desmosomes, move in tandem toward the skin surface, they deposit and enzymatically cross-link proteins beneath the plasma membrane to form the cornified envelope. These cells also make lamellar granules filled with lipids, which are extruded onto the cornified envelope scaffold, providing a water-impermeable seal that prevents the unregulated escape of fluids (2). After production of all materials is complete, the cells cease transcriptional and metabolic activity and undergo a programmed cell death that shares some similarities with apoptosis (3). The cells (squames) that are sloughed from the skin surface consist largely of dead protein-aceous sacs of IF cables; these cell remnants are continually replaced by inner cells moving outward.
Fig. 1.
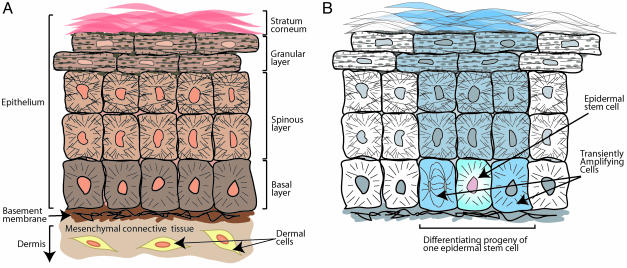
(A) Diagrammatic representation of skin epithelial histology. Cells of the basal layer attach to an underlying basement membrane. Basal cells are mitotically active, but they lose this potential when they detach from the basement membrane and embark on the outward trek toward the skin surface. As basal cells enter the spinous layer, they strengthen their cytoskeletal and intercellular connections, gaining resilience to mechanical stress. Once this task is completed, the cells enter the granular layer, where they produce the epidermal barrier. The barrier precursors consist of two major components: (i) glutamine- and lysine-rich cornified envelope precursor proteins, which are synthesized and deposited beneath the plasma membrane, and (ii) lamellar granules, which are filled with lipid bilayers. As the cells enter the final phases of terminal differentiation, a flux of calcium activates the enzyme transglutaminase, which biochemically cross-links the cornified envelope proteins through ε-(γ-glutamyl)lysine isopeptide bonds and which activates the extrusion of the lipid bilayers onto this scaffold. Cell death ensues, leaving dead, flattened squames at the skin surface, the end-process of terminal differentiation. These squames of the stratum corneum eventually slough from the skin surface, to be replenished continually by inner layer cells moving outward. (B) Diagram of the epidermal proliferative unit. A putative slow-cycling epidermal stem cell occasionally divides, giving rise to a stem cell daughter and a transiently amplifying daughter. The transiently amplifying cell divides two to four times, and these progeny then leave the basal layer and execute a program of terminal differentiation. This model is based on retroviral transduction of a β-galactosidase gene into cultured keratinocytes, which were then used in engraftments onto nude mice to trace stem cell lineages (9).
In mouse skin, as measured by autoradiography, the entire differentiation process from basal layer to squame takes 10-14 days (4). Human epidermis turns over more slowly; however, the proliferative reserve of human skin epithelial stem cells, which supply sufficient progeny to maintain 1-2 m2 of skin for decades, must be enormous.
An early observation in the field of skin biology was that epidermal keratinocytes could be grown in culture. As opposed to many other cell types that require transformation to be cultured effectively, epithelial cells taken directly from the skin can be passaged for many generations when cultured in the presence of a fibroblast feeder layer (5). When grown in the presence of an epidermal growth factor (EGF) receptor ligand such as EGF or transforming growth factor α (TGFα), human keratinocytes can be expanded by a factor of 1016 (6). A careful analysis of the growth potential of human skin keratinocytes revealed three different types of cells based on the size of the clones they are capable of generating in a single plating (7). Holoclones, which have the greatest proliferative potential, contain cells that almost all (95%) go on to form proliferative colonies on passaging. Meroclones have intermediate proliferative potential, and paraclones abort and differentiate after very few passages. Holoclone cells can transition to meroclone and paraclone cells, but the reverse transition was not observed in this study. It is tempting to speculate that holoclone-generating cells in vitro might be stem cells in vivo.
Where Are the Skin Epithelial Stem Cells Located?
On initial histological evaluation of mammalian skin, there is no obvious morphologically distinct region, or niche, of the basal layer where stem cells might be located. It has been known from the 1970s that the epidermis is organized into columns of maturing cell layers ≈10 cells wide (see Fig. 1B) (8). It was initially hypothesized that the entire basal layer consisted of stem cells, then later that the Langerhans cells were stem cells. Radiation dose-survival studies suggested that stem cells might comprise 2-7% of basal layer cells (reviewed in ref. 8). One method of retrospectively demonstrating the presence of stem cells in epidermal cultures is to label the population of cells and then use them to reconstitute epidermal tissue in vivo. Thus, when retrovirally tagged murine epidermal cultures expressing the β-galactosidase reporter gene were grafted onto a mouse, the reconstituted skin exhibited clonal columns of β-galactosidase-expressing epidermal cells in the host animal (Fig. 1B) (9). The size of the columns over the 12-week study period suggested that as many as 10-12% of murine basal layer cells might be “stem cells” capable of generating a single maturing column of cells.
Another method of identifying tissue stem cells makes use of their slow cycling nature. In a pulse-chase experiment, all dividing cells of a tissue incorporate nucleotide analogs such as bromodeoxyuridine (BrdUrd) or tritiated [3H]thymidine into newly synthesized DNAs. When the label is chased, only those cells that divide rarely and still reside within the tissue over time will retain their label. In oral epithelium, so-called label-retaining cells, or LRCs, are located in discrete regions of tongue and palatal papillae (10); in murine ear epidermis, LRCs reside in the basal layer, near the periphery of differentiating cell columns (11). Therefore, a model of skin epithelial maintenance emerged in the 1980s in which the periodic division of slow-cycling stem cells in the basal layer gives rise to transiently amplifying cells that populate most of the basal layer, dividing two or three times and then moving upward while differentiating into mature skin cells (Fig. 1B).
In the 1990s, researchers using [3H]thymidine to evaluate label retention in murine haired epidermis discovered that the majority of LRCs in the skin reside in the “bulge” region of the hair follicle, with only a small fraction of LRCs in the basal layer of interfollicular epidermis (12, 13). The hair follicle is an epidermal appendage that consists of an upper, permanent portion, and a lower, cycling portion that produces the hair (Fig. 2; reviewed in refs. 14 and 15). The outer root sheath (ORS) is contiguous with and biochemically similar to the basal layer of the epidermis. The inner layers of the hair follicle include three concentric layers of inner root sheath (IRS) and three concentric layers of hair-producing cells. At the base of the hair follicle is the germinative matrix, which contains rapidly proliferating “matrix” cells that differentiate to populate all of the layers of the IRS and the hair shaft itself. The hair follicle bulge, which contains the LRCs, resides within the ORS in a small niche just below the sebaceous gland, at or near the site of insertion of the arrector pili muscle. At first glance, the bulge is an interesting place for stem cells to live; it is down below the surface of the skin, protected by a column of cells in the upper portion of the hair follicle, as well as by the heavily keratinized hair shaft itself. Across the basement membrane, it is surrounded by a supportive dermal pocket that is richly vascularized and innervated.
Fig. 2.
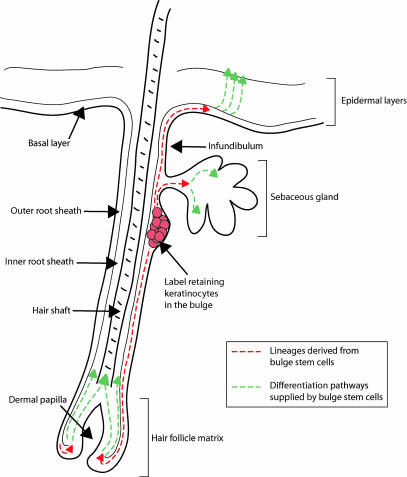
Diagram of the hair follicle and cell lineages supplied by epidermal stem cells. A compartment of multipotent stem cells is located in the bulge, which lies in the outer root sheath (ORS) just below the sebaceous gland. Contiguous with the basal layer of the epidermis, the ORS forms the external sheath of the hair follicle. The interior or the inner root sheath (IRS) forms the channel for the hair; as the hair shaft nears the skin surface, the IRS degenerates, liberating its attachments to the hair. The hair shaft and IRS are derived from the matrix, the transiently amplifying cells of the hair follicle. The matrix surrounds the dermal papilla, a cluster of specialized mesenchymal cells in the hair bulb. The multipotent stem cells found in the bulge are thought to contribute to the lineages of the hair follicle, sebaceous gland, and the epidermis (see red dashed lines). Transiently amplifying progeny of bulge stem cells in each of these regions differentiates as shown (see green dashed lines).
Two studies confirmed the relevance of the label retention of bulge keratinocytes by dissecting rat (16) and human (17) hair follicles and evaluating different regions of the follicles for clonogenicity in vitro. In rat whisker follicles, of ≈740 total colony-forming cells per follicle, 95% were found in the bulge, and the remaining 5% were found in the matrix region. In human scalp follicles, the highest clonogenicity was found in a region directly below the bulge; these cells were found to have tremendous proliferative capacity, with theoretical output of as many as 1.7 × 1038 progeny from a single cell (17). In a creative confirmation that LRCs have stem cell properties, LRCs cultured from epidermis after a long chase were found to be more clonogenic than pulse-labeled cells from a similarly aged animal (13). Thus, the most clonogenic cells and the cells with highest label-retaining capacity in the mammalian haired epidermis occur in the hair follicle, in or near the bulge region.
Hair cycling, the repeated regeneration of hair follicles to produce new hair shafts over the lifetime of the organism, is a useful model by which to study stem cell properties. According to the “bulge activation hypothesis,” bulge stem cells are stimulated to divide and produce a new germinative hair matrix only after receiving signals from specialized hair follicle mesenchymal cells (12). In support of this theory, bulge cells have been shown to divide before regrowth of the follicle (18).
Functional Characteristics of Skin Epithelial Stem Cells
Locating the putative epidermal stem cells represents a major advance in the field, allowing scientists to move forward with respect to the biochemical and functional characteristics of this important population of cells. Other stem cell fields, such as the hematopoietic system, are replete with cell surface markers that identify nearly every cell type, starting with stem cells and extending through the most differentiated forms of the progeny types. Specific markers of epidermal stem cells, however, are not yet known. Although these cells can be identified either in vivo by label retention or in vitro by clonogenicity, neither method of identification presently allows easy isolation of stem cells for analysis. Therefore, there is a strong need for specific epidermal stem cell markers.
One class of candidate stem cell markers is the integrin family of transmembrane receptors, whose members are responsible for the attachment of the basal layer of the epidermis to its underlying substratum, the basement membrane (reviewed in ref. 19). Basement membrane is rich in extracellular matrix (ECM) (20) proteins, many of which constitute the ligands for integrin αβ heterodimers. When cultured human keratinocytes were isolated by fluorescence-activated cell sorting (FACS) on the basis of their surface integrin β1 levels, cells with the highest fluorescence displayed a moderately increased colony-forming efficiency in vitro (21). In this study, colony-forming efficiency correlated with the speed of cell adherence to integrin ligands, including type IV collagen (α2β1) and ECM proteins secreted by keratinocytes (21). In a different study, human keratinocytes sorted for the hemidesmosomal integrin α6, which partners with β4 to robustly attach to basement membrane component laminin 5, were shown to have higher proliferative potential than those sorted for the focal adhesion integrin β1, which partners promiscuously with α2 (type IV collagen), α3 (laminin 5), α5 (fibronectin), and α9 (tenascin) in keratinocytes (22).
Integrin expression levels can change when cells are transferred from living skin to culture conditions, potentially introducing caveats to the extrapolation of in vitro integrin expression data to in vivo stem cells. In this case, however, the colony-forming efficiency of human keratinocytes obtained directly from skin also correlated with rapid adhesion to type IV collagen, a feature characteristic of cells with elevated α2 integrin (23). Additionally, immunohistochemical analysis of intact human skin from different regions seems to display heterogeneity in β1 integrin expression levels; patches of increased expression have been postulated to contain stem cells (23). Early studies suggested a role for β1 integrin signaling in the prevention of terminal differentiation (20). Mice conditionally lacking integrin β1 in skin epithelial cells exhibit severe defects in basement membrane assembly and organization, underscoring a role for these integrins not only in attachment to but also assembly of extracellular matrix (refs. 24 and 25; see also ref. 26). Wound healing is impaired in these mice; although β1 null keratinocytes proliferate adequately in vivo after wounding, they migrate ineffectively, resulting in delayed reepithelialization (27). Embryonic stem cells lacking β1 integrin show reduced ability to differentiate into keratinocytes, a defect that can be partially rescued by dermal derived growth factors (28). Whether the skin stem cell compartment depends on β1 integrin has been more difficult to judge given the severity in phenotype of the β1-null skin.
Recently, two studies investigating the transcriptional profiles of hematopoietic, neural, and embryonic stem cells have found integrins to be up-regulated in these stem cells as compared with their transiently amplifying progeny (29, 30). Integrin α6 was present in both lists, and β1 was present in one of the two. Because stem cells are restricted to the basal layer of either hair follicle bulge or interfollicular epidermis, molecules instrumental in cell-substratum adhesion are conceptually interesting stem cell markers. It is possible that stem cells require strong adherence to the basement membrane to maintain their stem cell characteristics or their position in the stem cell niche. Despite the intrigue, most if not all proliferating cells use integrins in adhesion. Thus, the usefulness of integrins as stem cell markers is limited by the uncertainty of interpretation of their levels of expression relative to transit-amplifying stem cell progeny.
The transferrin receptor is another surface marker shown to differ in its expression between stem cells and proliferating progeny. In this case, reduced surface expression of the transferrin receptor has been associated with human keratinocyte stem cells. Sorting of primary skin cells on the basis of integrin α6 and transferrin receptor found that LRCs were enriched in α6-high, transferrin receptor-low cells, whereas cells actively dividing were enriched in the α6-high, transferrin receptor-high population (31).
Even in the absence of cell surface markers useful for isolation of stem cells, skin biologists have made advances in understanding some of the molecules important in conversion from stem cell to transit-amplifying cell. One such example is the protooncogene c-myc, a transcriptional regulator of proliferation in a large variety of cell types, including skin keratinocytes (32, 33). Interestingly, overexpression of c-myc in transgenic mouse skin results in what appears to be depletion of the multipotent skin stem cells of the bulge, as judged by a reduction in LRCs and impaired wound healing (34, 35). Surprisingly, increasing c-myc expression also seems to cause a cell fate change from hair follicle progenitor cells to sebum-producing cells, suggesting that c-myc levels may influence not only the decision of stem cell daughters to become transit-amplifying cells, but also the decision of which lineage to adopt.
Another factor associated with stem cells and/or their conversion to transit-amplifying cells is the transcription factor p63, a homologue of p53. p63 is known to be expressed in epithelial stem cells of the corneal limbus in vivo and in the holoclone-generating skin keratinocytes in vitro (36). Mice harboring a targeted deletion at the p63 gene locus have a profound defect in epidermal development (37, 38). In early development, when the skin epithelium is a single layer, the p63-null skin appears normal. As the process of stratification proceeds, however, p63-null skin becomes progressively denuded of epithelium, leaving only a few remaining cells that express suprabasal and not basal markers (37).
These studies suggest that p63 and c-myc are both important regulators of skin keratinocyte function. A major issue still unresolved for both these factors is the extent to which they govern stem cell maintenance versus the production of transit-amplifying progeny.
The Multipotency of Skin Epithelial Stem Cells: What Is the Relationship Between Interfollicular and Bulge Stem Cells?
Substantial evidence supports the idea that stem cells in the interfollicular epidermis are less potent than bulge stem cells, leading to speculation that they are progeny, perhaps unipotent progeny, of multipotent bulge cells. In contrast to bulge cells, interfollicular stem cells do not have a clearly defined niche. There are more slow-cycling stem cells in the bulge than in the interfollicular epidermis (12), and the longevity of label retention in bulge cells is longer than that in interfollicular cells (39, 40). Cells cultured from the bulge also have a higher clonogenic potential than interfollicular cells (17). Curiously, LRCs of the bulge require repeated treatment with 12-O-tetradecanoylphorbol-13-acetate (PMA) to induce proliferation, whereas interfollicular LRCs are more easily induced to divide (39). Finally, superficial burns that destroy the interfollicular epidermis but leave intact the hair follicles do not require skin grafting, whereas deeper burns in which the hair follicles are destroyed cannot regrow epithelium except from the edges (41).
The multipotency of bulge cells is demonstrated by the fact that these special cells can give rise to all lineages of skin epithelia, including interfollicular epidermis (Fig. 1B). Cells isolated from dissected bulge regions are capable of differentiating into stratified epidermis when grown to confluence in culture and transplanted onto athymic mice (17). The label-retaining property of slow-cycling stem cells has been exploited to evaluate bulge cell contribution to multiple epidermal lineages. Cells that retain label after an 8-week chase, which reside exclusively in the bulge by the technique used in this study, contribute to all of the layers of the hair follicle (40). Pulse labeling with two different nucleotides, timed precisely (based on cell cycle length) to label cells of the infundibulum region of the hair follicle, demonstrated an efflux of hair follicle cells out into interfollicular epidermis in both normal and wounded states (40). In another assay, the multipotency of bulge stem cells was demonstrated by transplantation of β-galactosidase-expressing transgenic whisker bulge cells into the bulge region of follicles from an unlabeled recipient mouse (42). Over time, β-galactosidase-expressing cells from transplanted bulges populated all of the epithelial compartments of the resulting chimeric follicles, including the sebaceous gland and the infundibular region above the bulge that is thought to be most similar to interfollicular epidermis.
Although the evidence supporting multipotency of the bulge cells is compelling, the characteristics of stem cells outside this niche are less certain. Are interfollicular epidermal cells unipotent or multipotent? To what extent, if any, do they differ from bulge stem cells? The answers to these questions await more extensive knowledge about the molecular characteristics of bulge cells. Recently, however, the Wnt signaling pathway has been linked to the ability of skin epithelial cells to acquire and/or maintain features of multipotent stem cells (43-45). At the heart of this pathway is β-catenin, a multifunctional protein that is stabilized when cells receive a Wnt signal. β-catenin is required in some way to activate members of a DNA-binding protein family referred to as the Lef/Tcf family (reviewed in ref. 46). In skin, Wnt signals are received by multipotent embryonic skin epithelial cells before their commitment to form a hair follicle (47). When specialized skin mesenchymal cells inhibit a second signaling pathway, the bone morphogenetic protein (BMP) pathway, the multipotent epithelial cells express Lef1 and become committed to forming a hair follicle (48). Both the Wnt and BMP signaling pathways appear to be functionally important to making a hair follicle, as judged by the fact that mice with disrupted function of Lef1 (49-51), Noggin (52), or β-catenin (53, 54) are all severely impaired in their ability to form hair follicles.
Normally, only bulge stem cells are thought to retain multipotency in postnatal skin. However, when β-catenin is constitutively stabilized in transgenic mouse skin, the adult interfollicular epidermis behaves as embryonic skin, seemingly able to choose between an epidermal and hair follicle fate (44). Interestingly, when the specialized mesenchymal cells (dermal papilla cells) of the hair follicle are exposed to Wnt signaling, they too appear to retain their hair follicle-inducing power (55). Therefore, Wnt signals may be able to act both on the epithelium to induce stem cell-like properties and on the mesenchyme to maintain its stem cell recruiting properties.
The degree of Lef1/Tcf activity may be critical in determining the outcome of stem cell lineage determination. When Lef1 is overexpressed in the skin and oral epithelium, occasional hairs and teeth are seen in inappropriate places (43). When a form of Lef1 that is unable to associate with β-catenin is overexpressed, hair follicle cells adopt a sebaceous cell fate in inappropriate places, and epidermal cysts form in place of some secondary hair follicles (50, 51). Similarly, when β-catenin is conditionally targeted for removal in skin, epidermal cysts are observed in place of hair follicles (53). In the future, it will be interesting to explore the levels and effects of other proteins that are now known to influence the status of Lef1/Tcf activity (reviewed in ref. 56). In this regard, the recent parallel findings of Kielman et al. (57) showing that adenomatous polyposis coli (APC) influences stem cell lineage determination in embryonic stem cells by controlling the dosage of β-catenin signaling are fascinating. In addition, there are now a number of reports that suggest a more global role for β-catenin and its partners in stem cells and fate specification (see refs. 58-60).
Clinical Applications of Skin Epithelial Stem Cells: Grafting of Cultured Keratinocytes
Basic research into stem cell biology is partly oriented toward the eventual possibility of harvesting stem cells from a patient, modifying or expanding them, and reimplanting them to treat disease. Skin keratinocytes have proved useful for this already, because of their accessibility and ability to be cultured. The most prominent clinical use of cultured keratinocytes is in creating confluent epithelial sheets that can be gently removed from the culture dish and applied to reconstitute the epithelial portion of burns, chronic wounds, and ulcers. The advantage of this method is the use of the patient's own skin, which represents the optimal long-term repopulation strategy. Today, the most commonly used skin grafting technique employs a different approach, the use of split-thickness grafts taken from unaffected skin. This method is effective, but it is limited by the available surface area of unaffected skin and creates some degree of additional injury. The use of cultured keratinocytes allows a much greater surface to be covered and requires a smaller area of unaffected skin from which to harvest the keratinocytes for culture. At present, the use of cultured keratinocytes is limited by the length of time needed to grow the epithelial sheets in vitro, during which time the patient is susceptible to infection. The epithelial sheets are also extremely fragile and do not adhere well to some burn surfaces. Under development are skin substitutes that could function as dermal equivalents to hold the expanding keratinocytes, improve adhesiveness to the burn wound, and form a temporary wound cover to reduce infection rates. If subconfluent keratinocyte cultures could be implanted into the dermal equivalent, then this graft could be applied very early after the burn injury, with the epithelial cover maturing while the dermal equivalent functions as a temporary dressing, obviating the need for two surgeries (61).
Grafting of cultured skin epithelial stem cells has other potential applications besides replacing burned skin; in particular, it is exciting to consider the possibility of using the cultured keratinocytes as delivery instruments for gene therapy. Two groups have devised methods to use cultured human keratinocytes to correct inborn metabolic skin diseases (62, 63). Keratinocytes were harvested from patients with recessive dystrophic epidermolysis bullosa, and the genetic defect was corrected either by genomic integration of the correct sequence using a bacteriophage integrase or by transgene expression using a lentivirus. The repaired keratinocytes were expanded in culture and grafted onto nude mice to produce healthy epithelia in which the defect was corrected. Although neither study included grafting back onto the original human being suffering from skin disease, these efforts represent a major advance toward the possibility of manipulating stem cells to treat human disease.
Conclusions
The search for the biochemical regulators of skin epithelial stem cell self-renewal and production of daughter cells that will populate one or several lineages of the epidermis is ongoing. The field has advanced significantly over the past three decades, especially with respect to the presence and location of discrete stem cell compartments. Some initial work has implicated the integrins as cell surface markers that, although not specific, may be useful to enrich populations of cells for stem cells to allow characterization. C-myc and p63 have also been identified as regulators of stem cell fate. In addition, recent years have experienced a flurry of reports that implicate Wnt signaling, β-catenin, and Lef1/Tcf transcriptional regulation in stem cell maintenance and/or lineage determination. Despite these advances, scientists are not yet able to reliably isolate stem cells from skin epithelium to exhaustively study their transcriptional and functional characteristics. Eventually, when techniques exist to find the answers to these questions, doctors may be able to not only use skin epithelial stem cells to grow new skin to treat burns, but also to treat genetic diseases of skin and possibly even nonskin origin. We don't yet know whether it might be possible in the future to genetically engineer keratinocytes to inducibly secrete peptide hormones, such as insulin as a treatment for diabetes, or growth hormone as a treatment for growth hormone deficiency (64). Nor do we know whether keratinocyte stem cells possess sufficient plasticity to be differentiated into nonkeratinocyte cell types to correct defects of other tissues. The studies of the past three decades indicate that the future of skin stem cell research holds great promise.
Acknowledgments
We thank our scientific colleagues and past and present Fuchs laboratory members, who have advanced our understanding of epithelial stem cells. E.F. is an Investigator of the Howard Hughes Medical Institute. The research leading to this article was supported by National Institutes of Health Grant R01-AR31737.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Regenerative Medicine,” held October 18-22, 2002, at the Arnold and Mabel Beckman Center of the National Academies of Science and Engineering in Irvine, CA.
Abbreviation: LRC, label-retaining cell.
References
- 1.Fuchs, E. & Cleveland, D. W. (1998) Science 279, 514-519. [DOI] [PubMed] [Google Scholar]
- 2.Kalinin, A. E., Kajava, A. V. & Steinert, P. M. (2002) BioEssays 24, 789-800. [DOI] [PubMed] [Google Scholar]
- 3.Gandarillas, A. (2000) Exp. Gerontol. 35, 53-62. [DOI] [PubMed] [Google Scholar]
- 4.Potten, C. S. (1975) Br. J. Dermatol. 93, 649-658. [DOI] [PubMed] [Google Scholar]
- 5.Rheinwald, J. G. & Green, H. (1975) Cell 6, 331-343. [DOI] [PubMed] [Google Scholar]
- 6.Rheinwald, J. G. & Green, H. (1977) Nature 265, 421-424. [DOI] [PubMed] [Google Scholar]
- 7.Barrandon, Y. & Green, H. (1987) Proc. Natl. Acad. Sci. USA 84, 2302-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potten, C. S., Schofield, R. & Lajtha, L. G. (1979) Biochim. Biophys. Acta 560, 281-299. [DOI] [PubMed] [Google Scholar]
- 9.Mackenzie, I. C. (1997) J. Invest. Dermatol. 109, 377-383. [DOI] [PubMed] [Google Scholar]
- 10.Bickenbach, J. R. & Mackenzie, I. C. (1984) J. Invest. Dermatol. 82, 618-622. [DOI] [PubMed] [Google Scholar]
- 11.Potten, C. S., Kovacs, L. & Hamilton, E. (1974) Cell Tissue Kinet. 7, 271-283. [DOI] [PubMed] [Google Scholar]
- 12.Cotsarelis, G., Sun, T. T. & Lavker, R. M. (1990) Cell 61, 1329-1337. [DOI] [PubMed] [Google Scholar]
- 13.Morris, R. J. & Potten, C. S. (1994) Cell Proliferation 27, 279-289. [DOI] [PubMed] [Google Scholar]
- 14.Hardy, M. H. (1992) Trends Genet. 8, 55-61. [DOI] [PubMed] [Google Scholar]
- 15.Alonso, L. & Fuchs, E. (2003) Genes Dev. 17, 1189-1200. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi, K., Rochat, A. & Barrandon, Y. (1993) Proc. Natl. Acad. Sci. USA 90, 7391-7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rochat, A., Kobayashi, K. & Barrandon, Y. (1994) Cell 76, 1063-1073. [DOI] [PubMed] [Google Scholar]
- 18.Wilson, C., Cotsarelis, G., Wei, Z. G., Fryer, E., Margolis-Fryer, J., Ostead, M., Tokarek, R., Sun, T. T. & Lavker, R. M. (1994) Differentiation (Berlin) 55, 127-136. [DOI] [PubMed] [Google Scholar]
- 19.Watt, F. M. (2002) EMBO J. 21, 3919-3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams, J. C. & Watt, F. M. (1989) Nature 340, 307-309. [DOI] [PubMed] [Google Scholar]
- 21.Jones, P. H. & Watt, F. M. (1993) Cell 73, 713-724. [DOI] [PubMed] [Google Scholar]
- 22.Kaur, P. & Li, A. (2000) J. Invest. Dermatol. 114, 413-420. [DOI] [PubMed] [Google Scholar]
- 23.Jones, P. H., Harper, S. & Watt, F. M. (1995) Cell 80, 83-93. [DOI] [PubMed] [Google Scholar]
- 24.Brakebusch, C., Grose, R., Quondamatteo, F., Ramirez, A., Jorcano, J. L., Pirro, A., Svensson, M., Herken, R., Sasaki, T., Timpl, R., et al. (2000) EMBO J. 19, 3990-4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raghavan, S., Bauer, C., Mundschau, G., Li, Q. & Fuchs, E. (2000) J. Cell Biol. 150, 1149-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giancotti, F. G. & Ruoslahti, E. (1999) Science 285, 1028-1032. [DOI] [PubMed] [Google Scholar]
- 27.Grose, R., Hutter, C., Bloch, W., Thorey, I., Watt, F. M., Fassler, R., Brakebusch, C. & Werner, S. (2002) Development (Cambridge, U.K.) 129, 2303-2315. [DOI] [PubMed] [Google Scholar]
- 28.Bagutti, C., Hutter, C., Chiquet-Ehrismann, R., Fassler, R. & Watt, F. M. (2001) Dev. Biol. 231, 321-333. [DOI] [PubMed] [Google Scholar]
- 29.Ivanova, N. B., Dimos, J. T., Schaniel, C., Hackney, J. A., Moore, K. A. & Lemischka, I. R. (2002) Science 298, 601-604. [DOI] [PubMed] [Google Scholar]
- 30.Ramalho-Santos, M., Yoon, S., Matsuzaki, Y., Mulligan, R. C. & Melton, D. A. (2002) Science 298, 597-600. [DOI] [PubMed] [Google Scholar]
- 31.Tani, H., Morris, R. J. & Kaur, P. (2000) Proc. Natl. Acad. Sci. USA 97, 10960-10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gandarillas, A. & Watt, F. M. (1997) Genes Dev. 11, 2869-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandarillas, A. & Watt, F. M. (1995) Oncogene 11, 1403-1407. [PubMed] [Google Scholar]
- 34.Frye, M., Gardner, C., Li, E. R., Arnold, I. & Watt, F. M. (2003) Development (Cambridge, U.K.) 130, 2793-2808. [DOI] [PubMed] [Google Scholar]
- 35.Waikel, R. L., Kawachi, Y., Waikel, P. A., Wang, X. J. & Roop, D. R. (2001) Nat. Genet. 28, 165-168. [DOI] [PubMed] [Google Scholar]
- 36.Pellegrini, G., Dellambra, E., Golisano, O., Martinelli, E., Fantozzi, I., Bondanza, S., Ponzin, D., McKeon, F. & De Luca, M. (2001) Proc. Natl. Acad. Sci. USA 98, 3156-3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang, A., Schweitzer, R., Sun, D., Kaghad, M., Walker, N., Bronson, R. T., Tabin, C., Sharpe, A., Caput, D., Crum, C. & McKeon, F. (1999) Nature 398, 714-718. [DOI] [PubMed] [Google Scholar]
- 38.Mills, A. A., Zheng, B., Wang, X. J., Vogel, H., Roop, D. R. & Bradley, A. (1999) Nature 398, 708-713. [DOI] [PubMed] [Google Scholar]
- 39.Morris, R. J. & Potten, C. S. (1999) J. Invest. Dermatol. 112, 470-475. [DOI] [PubMed] [Google Scholar]
- 40.Taylor, G., Lehrer, M. S., Jensen, P. J., Sun, T. T. & Lavker, R. M. (2000) Cell 102, 451-461. [DOI] [PubMed] [Google Scholar]
- 41.Green, H. (1991) Sci. Am. 265, 96-102. [DOI] [PubMed] [Google Scholar]
- 42.Oshima, H., Rochat, A., Kedzia, C., Kobayashi, K. & Barrandon, Y. (2001) Cell 104, 233-245. [DOI] [PubMed] [Google Scholar]
- 43.Zhou, P., Byrne, C., Jacobs, J. & Fuchs, E. (1995) Genes Dev. 9, 700-713. [DOI] [PubMed] [Google Scholar]
- 44.Gat, U., DasGupta, R., Degenstein, L. & Fuchs, E. (1998) Cell 95, 605-614. [DOI] [PubMed] [Google Scholar]
- 45.Andl, T., Reddy, S. T., Gaddapara, T. & Millar, S. E. (2002) Dev. Cell 2, 643-653. [DOI] [PubMed] [Google Scholar]
- 46.Nusse, R. (1999) Trends Genet. 15, 1-3. [DOI] [PubMed] [Google Scholar]
- 47.DasGupta, R. & Fuchs, E. (1999) Development (Cambridge, U.K.) 126, 4557-4568. [DOI] [PubMed] [Google Scholar]
- 48.Jamora, C., DasGupta, R., Kocieniewski, P. & Fuchs, E. (2003) Nature 422, 317-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Genderen, C., Okamura, R. M., Farinas, I., Quo, R. G., Parslow, T. G., Bruhn, L. & Grosschedl, R. (1994) Genes Dev. 8, 2691-2703. [DOI] [PubMed] [Google Scholar]
- 50.Merrill, B. J., Gat, U., DasGupta, R. & Fuchs, E. (2001) Genes Dev. 15, 1688-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niemann, C., Owens, D. M., Hulsken, J., Birchmeier, W. & Watt, F. M. (2002) Development (Cambridge, U.K.) 129, 95-109. [DOI] [PubMed] [Google Scholar]
- 52.Botchkarev, V. A., Botchkareva, N. V., Roth, W., Nakamura, M., Chen, L. H., Herzog, W., Lindner, G., McMahon, J. A., Peters, C., Lauster, R., et al. (1999) Nat. Cell Biol. 1, 158-164. [DOI] [PubMed] [Google Scholar]
- 53.Huelsken, J., Vogel, R., Erdmann, B., Cotsarelis, G. & Birchmeier, W. (2001) Cell 105, 533-545. [DOI] [PubMed] [Google Scholar]
- 54.DasGupta, R., Rhee, H. & Fuchs, E. (2002) J. Cell Biol. 158, 331-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kishimoto, J., Burgeson, R. E. & Morgan, B. A. (2000) Genes Dev. 14, 1181-1185. [PMC free article] [PubMed] [Google Scholar]
- 56.Brantjes, H., Barker, N., van Es, J. & Clevers, H. (2002) Biol. Chem. 383, 255-261. [DOI] [PubMed] [Google Scholar]
- 57.Kielman, M. F., Rindapaa, M., Gaspar, C., van Poppel, N., Breukel, C., van Leeuwen, S., Taketo, M. M., Roberts, S., Smits, R. & Fodde, R. (2002) Nat. Genet. 32, 594-605. [DOI] [PubMed] [Google Scholar]
- 58.Korinek, V., Barker, N., Moerer, P., van Donselaar, E., Huls, G., Peters, P. J. & Clevers, H. (1998) Nat. Genet. 19, 379-383. [DOI] [PubMed] [Google Scholar]
- 59.Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. (2001) Nature 414, 105-111. [DOI] [PubMed] [Google Scholar]
- 60.Wong, M. H., Huelsken, J., Birchmeier, W. & Gordon, J. I. (2002) J. Biol. Chem. 277, 15843-15850. [DOI] [PubMed] [Google Scholar]
- 61.Harris, P. A., Leigh, I. M. & Navsaria, H. A. (1998) Burns 24, 591-593. [DOI] [PubMed] [Google Scholar]
- 62.Ortiz-Urda, S., Thyagarajan, B., Keene, D. R., Lin, Q., Fang, M., Calos, M. P. & Khavari, P. A. (2002) Nat. Med. 8, 1166-1170. [DOI] [PubMed] [Google Scholar]
- 63.Chen, M., Kasahara, N., Keene, D. R., Chan, L., Hoeffler, W. K., Finlay, D., Barcova, M., Cannon, P. M., Mazurek, C. & Woodley, D. T. (2002) Nat. Genet. 32, 670-675. [DOI] [PubMed] [Google Scholar]
- 64.Wang, X., Zinkel, S., Polonsky, K. & Fuchs, E. (1997) Proc. Natl. Acad. Sci. USA 94, 219-226. [DOI] [PMC free article] [PubMed] [Google Scholar]