

Abstract
Historically, ErbB3 has been overlooked within the ErbB receptor family due to its perceived lack of tyrosine kinase activity. We have previously demonstrated that in pancreatic cancer ErbB3 is the preferred dimerization partner of EGFR, ErbB3 protein expression level directly correlates with the anti-proliferative effect of erlotinib (an EGFR-specific tyrosine kinase inhibitor), and transient knockdown of ErbB3 expression results in acquired resistance to EGFR-targeted therapy. In this study, we develop a stable isogenic model of ErbB3 expression in an attempt to decipher ErbB3's true contribution to pancreatic cancer tumorigenesis and to examine how this receptor affects cellular sensitivity to EGFR-targeted therapy. Analysis of the EGFR-ErbB3 heterodimer demonstrates that ligand-induced PI3K-AKT signaling is limited to ErbB3-expressing cells and that this signaling cascade can be partially abrogated by inhibiting EGFR function with erlotinib. Using our model of exogenous ErbB3 expression we showed a direct relationship between ErbB3 protein levels and increased pancreatic cancer cell proliferation in vitro. In vivo, ErbB3+PANC-1 xenografts had a significantly larger tumor volume than PANC-1 control xenografts (ErbB3-PANC-1) and displayed increased sensitivity to EGFR-targeted therapy. In pancreatic cancer, ErbB3 appears to be critically involved in EGFR signaling as evidenced by its profound effect on cellular proliferation and its ability to influence response to EGFR-targeted therapy.
Key words: ErbB3, EGFR, pancreatic cancer, EGFR-targeted therapy, tumorigenesis
Introduction
ErbB signaling is a key component to the growth, differentiation and survival of numerous cell types within the human body, and dysregulation of this signaling is implemented in the pathogenesis of various epithelial tumors.1–3 ErbB3 (HER3), along with the Epidermal Growth Factor Receptor (EGFR, HER1), ErbB2 (HER2) and ErbB4 (HER4), comprise the members of the ErbB family of receptor tyrosine kinases. Ligands for this family include those which preferentially bind to EGFR [epidermal growth factor (EGF), transforming growth factor α, amphiregulin, epiregulin, betacellulin, heparin-binding EGF and epigen] and neuregulin (NRG) which binds to ErbB3 and ErbB4.1,4,5 Upon ligand binding, these receptors undergo conformational change resulting in the ability to either homo- or heterodimerize leading to tyrosine phosphorylation and initiation of intracellular signaling.
ErbB3 has relatively weak tyrosine kinase activity and only exists as an obligate heterodimerization partner for the other members of the ErbB family.6 After binding neuregulin, ErbB3 actively dimerizes with EGFR or ErbB2, which in return, phosphorylates ErbB3 and instigates downstream signaling. Once thought to play a secondary role in cellular survival and proliferation due to its weak tyrosine kinase activity, it is now known that the intracellular domain of ErbB3 contains six binding sites for the p85 regulatory subunit of phosphoinositide 3-kinase (PI3K) and ErbB3 is now thought of as a potent platform for PI3K signaling.7–9 Ligand-induced phosphorylation of ErbB3 in NIH3T3 cells results in strong association of the receptor with p85/PI3K and subsequent cellular proliferation.10 This critical role of ErbB3 in PI3K/AKT-induced cellular proliferation has been demonstrated in lung and colon cancer using antibodies against ErbB311 and neuregulin,12 as well as, ErbB3 small interfering RNA.13
EGFR is overexpressed in many epithelial tumors including pancreatic adenocarcinoma, yet EGFR targeted therapy in patients with advanced pancreatic cancer has been associated with only modest results.14 We have previously demonstrated that pancreatic cancer cells that are sensitive to anti-EGFR therapy can be rendered resistant via siRNA-induced ErbB3 inhibition, further suggesting a critical influence of ErbB3 in pancreatic cancer EGFR signaling.15 A similar function of ErbB3 on EGFR signaling has been described by others not only in pancreatic cancer,16 but also in lung17,18 and colon carcinomas.16 These findings can be explained by an analysis of the chemical cross-linking patterns of ErbB receptors in pancreatic cancer cells which has revealed that ErbB3 is the preferential heterodimerization partner of EGFR.15 In clinical studies, several groups have demonstrated that ErbB3 is overexpressed19–21 in pancreatic cancer and that high expression of ErbB3 correlates with advanced stage and decreased overall survival.19 Additionally, overexpression of NRG-1β, a potent ErbB3 ligand, directly correlates with worse pancreatic cancer clinical prognosis.20 Beyond these findings, the function of ErbB3 in pancreatic adenocarcinoma remains largely unknown.
Utilizing an ErbB3-deficient cell line, we developed a stable isogenic model of ErbB3 expression to further describe the EGFR-ErbB3 heterodimer and its signaling cascades and to decipher the influence of ErbB3 on pancreatic cancer tumorigenesis and cellular response to EGFR-targeted therapy. Analysis of the EGFR-ErbB3 heterodimer demonstrates that ligand-induced PI3K-AKT signaling is limited to ErbB3-expressing cells and that this signaling cascade is significantly diminished yet not completely abrogated by EGFR-targeted therapy. Furthermore, using our isogenic model we have demonstrated that ErbB3 directly affects pancreatic cell proliferation and sensitivity to anti-EGFR therapy both in vitro and in vivo.
Results
Generation of an isogenic model of ErbB3 expression in pancreatic adenocarcinoma.
To investigate the impact that ErbB3 expression has on the pancreatic cancer cell, we utilized the ErbB3-deficient PANC-1 cell line to generate an isogenic model with varying levels of stablyexpressed exogenous full-length ErbB3. After transfection, several clones with different levels of expression were isolated. One of these clones was tested for the ability of exogenous ErbB3 to functionally dimerize. For this, we stimulated the ErbB3 transfected PANC-1 cells with NRG-1β, an ErbB3 ligand. We then subjected these cells to Bis(Sulfosuccinimidyl)-suberate (BS3) which covalently binds receptor dimers and preserves their cross-linking. Subsequent immunoblotting with an ErbB3-specific antibody identified a 185 kiloDalton (kDa) band consistent with the single ErbB3 protein as well as a ∼360 kDa band that corresponds in size to an ErbB3-associated heterodimer (Fig. 1A). ErbB3 functionality within the transfected clone cells was verified as stimulation with NRG-1β resulted in phosphorylation of ErbB3 and an increase in the level of phospho-AKT (Fig. 1B).
Figure 1.

Analysis of the transfected ErbB3 + PANC-1 cell line, confirms that ErbB3 is expressed, can dimerize, and can be phosphorylated by NRG-1β resulting in AKT signaling. (A) western blot analysis of ErbB3−PANC-1 and ErbB3+PANC-1 stable transfectant cells after stimulation with NRG-1β demonstrates expression and phosphorylation of ErbB3 in the ErbB3+PANC-1 cells. Significant increase in AKT activation is observed in the ErbB3+PANC-1 cell line. (B) western blotting for ErbB3 expression after covalently binding receptor dimers with Bis(Sulfosuccinimidyl)suberate (BS3) confirms the presence of ErbB3 dimerization within the ErbB3+PANC-1 cell line. As controls, ErbB3-associated dimers are absent in PANC-1 and present in AsPC-1 cells.
ErbB3 expression affects EGFR activation and allows persistent AKT signaling in the presence of erlotinib.
To specifically analyze the interactions between ErbB3 and EGFR and to further understand their downstream signaling pathways, we stimulated the EGFR-ErbB3 heterodimer in serum-free condition with receptor-specific ligands in the presence and absence of EGFR inhibition (Fig. 2A). We selected an intermediate and a high expressing ErbB3+PANC-1 clone, as well as the ErbB3−PANC-1 clone to analyze. EGF and NRG-1β were used to stimulate EGFR and ErbB3, respectively and erlotinib was utilized as an EGFR inhibitor.
Figure 2.

(A) Effect of ErbB3 expression on receptor heterodimerization and intra-cellular signaling in PANC-1 cells in the presence of EGF and NRG-1β stimulation. With ErbB3 present, EGF stimulation results in phosphorylation of EGFR and NRG-1β stimulation results in phosphorylation of ErbB3 as expected, but interestingly, in each scenario, activation of the non-target receptor is seen suggesting heterodimerization. Furthermore, ErbB3 expression permits AKT signaling with NRG-1β stimulation that, although diminished, persists in the presence of EGFR inhibition. ERK1/2 signaling is not effected by ErbB3 expression. (B) Western blotting of ErbB3 and EGFR expression in each of the 3 PANC-1 cell lines.
EGF and NRG-1β induced strong phosphorylation of EGFR and ErbB3 in ErbB3+PANC-1 cells, respectively. Each ligand also induced phosphorylation of the non-targeted receptor, further supporting the notion of EGFR-ErbB3 heterodimerization and receptor cross-activation. This effect was most pronounced in the panel of PANC-1 cells with high expression of ErbB3. Analysis of intracellular signaling cascades linked to the EGFR-ErbB3 heterodimer revealed several interesting findings. First, ErbB3 expression did not appear to affect the phosphorylation pattern of ERK1/2 when stimulated with EGF or NRG-1β in the presence of erlotinib, suggesting that MAPK signaling is not involved in the anti-proliferative effects of EGFR-targeted therapy. As expected, induction of ErbB3 expression resulted in NRG-1β-associated downstream phosphorylation of AKT, an effect that is not seen in ErbB3-PANC-1 cells treated with NRG-1β. More intriguing is the fact that NRG-1β-dependent activation of AKT is only partially diminished in the presence of EGFR inhibition with erlotinib. These findings confirm that ErbB3 signaling occurs primarily through the PI3K/AKT pathway, and in the presence of NRG-1β ligand, ErbB3 expression permits persistent intracellular signaling through AKT which can only be partially inhibited with anti-EGFR therapy. Interestingly, increased expression of ErbB3 did not affect the expression level of EGFR in our ErbB3 transfected PANC-1 cells (Fig. 2B).
ErbB3 expression increases in vitro cell proliferation and erlotinib sensitivity.
Next, we analyzed the effect of ErbB3 on cellular proliferation. ErbB3+PANC-1 cells displayed increased proliferation when compared to ErbB3−PANC-1 control cells in serum-rich media and the rate of proliferation directly correlated with the level of ErbB3 expression (p < 0.01; Fig. 3A). After 72 hours, intermediate and high ErbB3 expressing cells displayed a rate of proliferation 250% and 350% that of ErbB3−PANC-1 cells, respectively (p < 0.01). Similar results were observed after 96 hours and 120 hours. ErbB3−PANC-1 cells and ErbB3+PANC-1 high expressing cells were also propagated in serum-stressed conditions (0.5% FBS). Although not as drastic an effect as seen in serum-rich media, cells with high expression of ErbB3 demonstrated a 60% increase in proliferation when compared to ErbB3−PANC-1 cells suggesting the observed effect was not due to cell culture conditions (p < 0.05; Fig. 3B).
Figure 3.

In PANC-1, increased ErbB3 expression directly correlates with increased cellular proliferation (p < 0.01) and sensitivity to EGFR targeted therapy (p < 0.01). (A) When allowed to propagate in 10% serum containing media, high-expressing ErbB3+PANC-1 cells and intermediate-expressing ErbB3+PANC-1 cells proliferated at rates 3 and 2.5 times that of ErbB3−PANC-1 cells, respectively, and these findings were observed after 3, 4 and 5 days of propagation. (B) The proliferative advantage of ErbB3 expression is also seen in serum-stressed conditions (0.5% FBS). (C) After 96 hours of erlotinib treatment, ErbB3+PANC-1 cells demonstrated significantly more growth inhibition relative to DMSO-treated controls than ErbB3-PANC-1 cells. Percent inhibition directly correlated with the level of ErbB3 expression (p < 0.05).
In vitro knockdown of ErbB3 expression using siRNA confers resistance to erlotinib,15 and here, we attempted to determine whether introduction of ErbB3 can confer sensitivity to anti-EGFR targeted therapy. In order to do this, we treated ErbB3−PANC-1 and ErbB3+PANC-1 cells with erlotinib. We have previously reported that PANC-1 cell proliferation is relatively resistant to erlotinib.22 This finding was further supported by the fact that ErbB3−PANC-1 cells displayed almost no growth inhibition (less than 5%) after 96 hours of erlotinib treatment. Proliferation of ErbB3+PANC-1 cells, on the other hand, was significantly inhibited by erlotinib and the degree of inhibition directly correlated with increasing levels of ErbB3 protein expression (p < 0.05; Fig. 3C).
AKT inhibition affects PANC-1 cell proliferation.
We have previously demonstrated that pancreatic cancer cell AKT and ERK1/2 signaling is affected by ligand stimulation of EGFR and ErbB3.15 In order to further investigate the role of ERK1/2 and AKT signaling in the PANC-1 cell line, we selectively inhibited each of these downstream pathways and analyzed the effect on cell proliferation. As expected, PD98059 (15 µmol/L) and LY294002 (25 µmol/L) completely inhibited ERK1/2 and AKT activation, respectively, in each of the three PANC-1 cell lines with different levels of ErbB3 expression (Fig. 4A). Inhibition of AKT significantly decreased cellular proliferation in all cell lines, (Fig. 4B), while ERK1/2 inhibition had little effect on cell proliferation. This experiment confirms that ErbB3 induced PI3K/AKT signaling is actively involved in and has a potent effect on PANC-1 cell proliferation.
Figure 4.

Inhibition of AKT signaling significantly diminishes PANC-1 cell proliferation. (A) western blot demonstrating that LY294002 (25 µmol/L) and PD98059 (15 µmol/L) successfully inhibits AKT and ERK1/2 signaling, respectively, in all 3 PANC-1 cell lines. (B) Dose effect of LY294002 and PD98059 on PANC-1 cell proliferation after 48 hours. LY294002 resulted in a significant decrease is proliferation (p < 0.05) relative to DMSO treated cells, while PD98059 has no inhibitory effect on proliferation of PANC-1 cells.
ErbB3 expressing PAN C-1 xenografts display increased tumor volume and relative sensitivity to erlotinib.
Our next step was to validate our in vitro findings in a murine pancreatic cancer model with variable ErbB3 expression. ErbB3−PANC-1 and ErbB3+PANC-1 murine subcutaneous xenografts were established. After 5 weeks of growth, ErbB3+PANC-1 xenografts grew significantly larger with a mean tumor volume of 479.6 ± 60.7 mm3 compared to 261.1 ± 35.0 mm3 in ErbB3−PANC-1 xenografts (n = 8, p < 0.01; Fig. 5A). Daily intra-peritoneal erlotinib treatment had no significant effect on the size of ErbB3−PANC-1 xenografts, but resulted in a 51% reduction in tumor volume of the ErbB3+PANC-1 xenografts (479.6 ± 60.7 mm3 vs. 246.6 ± 28.3 mm3; p < 0.01; Fig. 5B). In summary, ErbB3+PANC-1 xenografts displayed greater tumorigenesis, and at the same time, exhibited greater relative response to anti-EGFR therapy than ErbB3−PANC-1 xenografts, suggesting a dual role for ErbB3 in these tumors.
Figure 5.

In PANC-1 xenografts, increased ErbB3 expression directly correlates with increased cellular proliferation (p < 0.05) and sensitivity to EGFR targeted therapy (p < 0.05). (A) After 5 weeks, ErbB3+PANC-1 xenografts had a significantly larger mean tumor volume (479.6 ± 60.7 mm3 vs. 261.1 ± 35.0 mm3; p < 0.05). (B) When treated with erlotinib, ErbB3+PANC-1 xenografts demonstrated a significant greater decrease in the rate of proliferation than did ErbB3−PANC-1 xenografts relative to vehicle-treated control groups. Tumor growth in each cell line is plotted with vehicle treated controls to demonstrate that ErbB3+PANC-1 xenografts displayed increased tumor proliferation, and that when treated with erlotinib, ErbB3+PANC-1 xenografts were not significantly larger than ErbB3−PANC-1 treated tumors. Data represents the mean ± SEM for 8 xenografts. (C) Western blot xenograft analysis confirms ErbB3 expression and shows that erlotinib results in diminished phospho-ErbB3 and phospho-AKT signaling in ErbB3+PANC-1 tumors.
Immunoblot analysis of individual tumors from each of the four cohorts revealed differences between ErbB3−PANC-1 and ErbB3+PANC-1 xenografts following erlotinib treatment (Fig. 5C). Phosphorylation of ErbB3 in ErbB3+PANC-1 xenografts was blocked by treatment with erlotinib. Of the 4 erlotinib-treated ErbB3+PANC-1 xenografts analyzed, 3 xenografts displayed significant downregulation of AKT activation, an effect that was not observed in the ErbB3−PANC-1 control xenografts treated with erlotinib. Xenograft ERK1/2 and AKT phosphorylation status was somewhat different compared to our in vitro studies (Fig. 2A). Our ErbB3−PANC-1 tumors demonstrate activated AKT and ERK1/2 in the presence of erlotinib suggesting more complex signaling facilitated through autocrine and paracrine stimulation within the surrounding microenvironment. The persistent activation of these proteins in the face of erlotinib further supports our finding that ErbB3−PANC-1 xenograft tumor volume is unaffected by erlotinib. Yet, when ErbB3 is expressed by PANC-1 cells, erlotinib therapy results in an interruption of AKT signaling and a significant decrease in tumor volume (Fig. 5B and 5C). Erlotinib is a specific EGFR inhibitor and does not directly inhibit ErbB3 signaling, thus by binding to EGFR this molecule inhibits EGFR-induced ErbB3 activation and subsequent AKT signaling.
Analysis of EGFR phosphorylation at tyrosine 845, tyrosine 992, tyrosine 1068 and tyrosine 1173 revealed no consistent pattern of EGFR activation or inhibition (data not shown), suggesting that EGFR phosphorylation is not indicative of pancreatic cancer cell response to erlotinib in vivo. In Figure 5C, tumor 4 in the ErbB3+PANC-1 treatment group has minimal ErbB3 expression and strong AKT phosphorylation. Interestingly, this tumor demonstrated a 440% increase in tumor size during treatment and thereby, appears resistant to erlotinib. Despite this 4-fold increase, this tumor had a final volume of 134.8 mm3, not significantly different than the ErbB3−PANC-1 xenografts. The fact that this relatively small tumor has minimal ErbB3 expression and is resistant to erlotinib further supports our conclusion that ErbB3 expression directly affects cancer cell proliferation and response to EGFR-targeted therapy.
ErbB3 mRNA is highly abundant in human pancreatic adenocarcinoma specimens.
We have demonstrated that ErbB3 actively heterodimerizes with EGFR and is intricately involved in pancreatic tumor proliferation by signaling through the PI3K/AKT pathway. To evaluate the significance of this receptor in the context of human pancreatic adenocarcinoma, 39 pancreatic cancer surgical specimens were analyzed for expression of ErbB3 and EGFR mRNA. Laser-capture microdissection was employed to ensure that only ductal adenocarcinoma cells were evaluated. In comparison to endogenous control RPLPO, all specimens demonstrated detectable, quantifiable levels of ErbB3 and EGFR transcript at varying levels (Fig. 6). ErbB3 mRNA had a wide range of expression among our patients and demonstrated no significant trends toward under- or overexpression. Interestingly, ErbB3 and EGFR expression were directly correlated (p = 0.0001; Fig. 6). This finding further supports the existence of a functional EGFR-ErbB3 heterodimer in pancreatic cancer.
Figure 6.
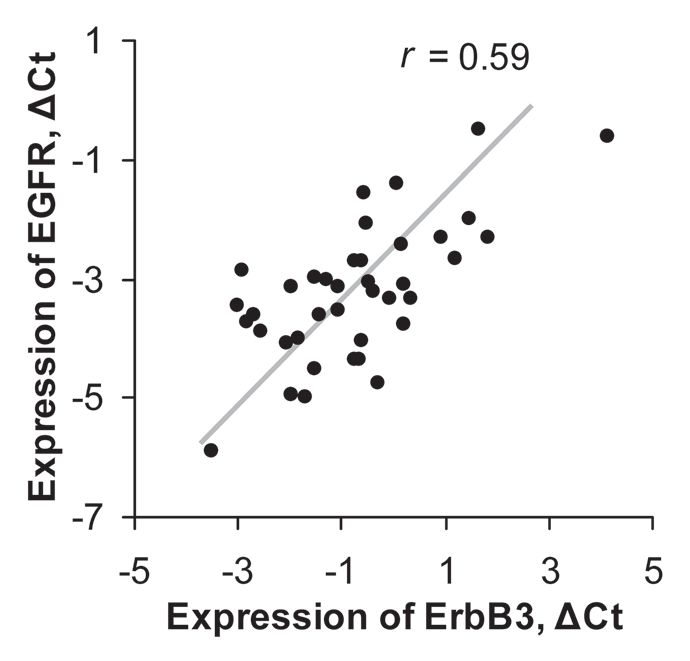
Expression levels of ErbB3 and EGFR mRNA in pancreatic cancer surgical specimens for 39 patients. ΔCt is calculated relative to expression of RPLPO.r is a Spearman correlation coefficient.
Discussion
Pancreatic cancer is the fourth most deadly carcinoma with an estimated 42,470 new cases and 35,240 attributed deaths in 2009.23 Nearly 95% of afflicted patients succumb to the disease process within 5 years; thus, long-term survival, even in patients with early stage disease remains extremely rare. Advancements in cancer treatment have had little effect on patient outcome as the 5-year overall survival from pancreatic cancer has increased from just 3% to 5% over the last 30 years.23 This dismal statistic, in combination with the known aggressive nature of this tumor, begs the point that novel therapeutic targets for pancreatic cancer are much needed.
ErbB3 is widely expressed in the murine fetal pancreas and appears to have a crucial role in the development of normal pancreas as ErbB3-/- knock-out experiments result in an embryonically lethal phenotype with embryos displaying drastically under-developed pancreata.24,25 These embryological findings suggest that ErbB3 is essential to the pancreatic ductal cell during organogenesis and that this receptor harbors the potential to drastically affect cell cycle progression possibly through receptor upregulation or differential ligand expression during organ development. Here, we have demonstrated that ErbB3 is expressed in all analyzed operative pancreatic adenocarcinoma specimens (39/39 = 100%), and it has been shown that pancreatic cancer specimens express significantly higher levels of ErbB3 mRNA transcript than normal controls.26 The vital role of this receptor in pancreatic organogenesis and its abundant expression in pancreatic cancer specimens suggests that the expression and activation of ErbB3 is involved in the malignant transformation of these cancer cells, and therefore, ErbB3 may be an important therapeutic target in pancreatic cancer.
In this study, we investigated the function of ErbB3 and identified a prominent role for this receptor in pancreatic adenocarcinoma proliferation and tumorigenesis. Traditionally, it was thought that ErbB3 played a moderator role in cellular proliferation acting as a simple scaffolding partner for the other signaling receptors, such as EGFR and ErbB2.5 In prostate cancer,27 lung cancer28 and melanoma,29 it has been speculated that the EGFR-ErbB3 heterodimer contributes to tumorigenesis, and here, utilizing pancreatic adenocarcinoma, we identified ErbB3 as the critical, rate-limiting component in EGFR-dependent cellular proliferation. We postulate that pancreatic cancer cellular proliferation is directly affected by the level of ErbB3 expression. While several groups have shown that silencing of ErbB3 expression diminishes basal cell proliferation,15,16,27 our study is the first attempt to demonstrate increased pancreatic cancer cell growth with the introduction of exogenous ErbB3. ErbB3 is closely associated with PI3K-AKT signaling8 and the potency of this receptor signaling cascade is only now being realized. Jones et al. recently demonstrated that three isoforms of PI3K displayed high affinity binding at multiple sites in ErbB3, identifying a connection between ErbB3 and intracellular signaling.30 Here, we demonstrate that ErbB3 and EGFR heterodimerize in the presence of their ligands resulting in activation of the ErbB3-PI3K-AKT axis and increased cellular proliferation, both in vitro and in vivo.
Erlotinib is the only anti-EGFR small molecule tyrosine kinase inhibitor that is FDA approved for treatment in advanced pancreatic cancer, and has no known activity against ErbB3. We demonstrated that, by introducing the ErbB3 receptor into a pancreatic cancer cell, we can confer sensitivity to erlotinib. This finding suggests that although EGFR is expressed on the cancer cell surface, significant cellular proliferation through this receptor does not proceed until ErbB3 is present. Only when present, can receptor dimerization and subsequent PI3K-induced cellular proliferation proceed, and these processes, as we have shown, are partially blocked by EGFR-targeted therapy. Breast cancer experiments demonstrate that ErbB3 expression is essential to proliferation in ErbB2-expressing cells31 and that ErbB3 activation results in sensitivity to ErbB2-targeted therapy. MCF7 breast cancer cells with stable expression of neuregulin demonstrate higher levels of ErbB3 phosphorylation, cellular proliferation and trastuzumab sensitivity than do MCF7 cells overexpressing ErbB2.32 These breast cancer data corroborate our findings in pancreatic cancer and stress that ErbB3 is a very active, critical component of efficient ErbB signaling and proliferation.
Our results portray an interesting two-faced image of ErbB3 in pancreatic cancer: on one hand it increases tumorigenesis; while on the other hand, ErbB3 expression improves the cell's response to anti-EGFR therapy. Based on our in vitro findings, it appears that the proliferative effect of ErbB3 can outweigh its beneficial effect in pancreatic cancer, especially when one considers ErbB3 overexpression and ligand abundance within the pancreatic cancer microenvironment. Recent reports have demonstrated that ErbB3 is critical to a cancer cell's escape from EGFR-targeted therapy by dimerization with other receptor tyrosine kinases, and this complex role of ErbB3 could be due to differential ligand expression.33,34 As mentioned above, stable expression of neuregulin in MCF7 cells with minimal levels of ErbB3 had a significant effect on cell proliferation further implementing neuregulin expression in tumorigenesis.32 The impact of autocrine and paracrine neuregulin expression should be investigated thoroughly in order to fully understand the impact of ErbB3 signaling on pancreatic cancer. Despite the effect of ligand expression, we have demonstrated that this receptor is highly expressed in pancreatic cancer cells and directly increases tumor cell proliferation, thereby possessing the two prerequisites of an ideal therapeutic target.
Although the clinical significance of ErbB3 expression in pancreatic adenocarcinoma remains to be determined, our findings suggest that pancreatic cancer patients could benefit from combining anti-EGFR therapy with specific anti-ErbB3 therapy and support testing this combination in a clinical trial.
Materials and Methods
Reagents.
Erlotinib (provided by OSI Pharmaceutical Inc., Melville, NY) was dissolved in DMSO to prepare a 20 mM stock solution. Epidermal Growth Factor (EGF; 1 mg/ml) and Heregulin-β1 (neuregulin-β1; 1 mg/ml) were purchased from Sigma (St. Louis, MO). LY294002, a specific inhibitor of PI3K and PD98059, a specific inhibitor of MAPK, were purchased from EMD Chemicals (Gibbstown, NJ).
Cell culture.
All cell lines were propagated in a humidified atmosphere containing 5% CO2 at 37°C. AsPC-1 and PANC-1 were purchased from the American Type Culture Collection (Rockville, MD) and propagated according to provider's recommendations.
Stable ErbB3 transfection.
PANC-1, an ErbB3 deficient cell line, was used to generate an isogenic model of ErbB3 expression. PANC-1 cells were co-transfected in a 10:1 ratio with a full length human ErbB3 cDNA in pCMV-SPORT6 vector (Thermo Fisher Scientific, Waltham, MA) and a pPuro vector (Clontech Laboratories, Inc., Mountain View, CA), which contains a puromycin resistance gene (ErbB3+PANC-1). pPuro vector only was used in control PANC-1 cells (ErbB3−PANC-1). All transfections were conducted using LipofectAMINE 2000 reagent (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer's protocols. Stable colonies of PANC-1 cells expressing various levels of ErbB3 were selected. For the duration of the experiments, all cells were propagated in the presence of puromycin (2 µg/mL). Clones maintained similar levels of ErbB3 expression after at least 30 passages. EGFR-ErbB3 heterodimer formation was analyzed using Bis(Sulfosuccinimidyl)suberate (BS)3 as previously described.15
Proliferation assay.
ErbB3+PANC-1 and ErbB3−PANC-1 were seeded in 96-well plates (1.5 × 103 cells/well), allowed to propagate overnight before treatment with erlotinib as indicated below. For analysis of ERK1/2 and PI3K inhibition, 1.0 × 103 cells were plated in 10% FBS in the presence of each inhibitor at the indicated concentrations and allowed to propagate for 72 hours. CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) was used to assess absolute proliferation in cell lines or relative proliferation between DMSO-treated control cells and erlotinib-treated cells.
Pancreatic cancer patient specimens and laser-capture microdissection (LCM).
After IRB-approved informed consent was obtained, tumor specimens were collected from pancreatic adenocarcinoma patients who underwent laparotomy with curative intent. Specimens were snap-frozen at the time of operation and stored at −80°C. Laser-capture microdissection (LCM) was performed as previously described.22 A pancreaticobiliary-trained pathologist (P.K.) evaluated each specimen and identified regions of adenocarcinoma cells prior to dissection. Approximately 3,000 tumor cells were captured from each specimen for analysis.
RNA extraction and reverse-transcriptase PCR (RT-PCR).
RNA from microdissected patient specimens was isolated with a RNAqueous Micro Kit (Ambion, Austin, TX) using the manufacturer's protocol. cDNA was synthesized in a thermal cycler with a High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). Quantitative real-time PCR was performed using TaqMan® Gene Expression EGFR, ERBB3 and RPLPO (housekeeping gene) Assays-on-Demand and TaqMan® Universal PCR Master Mix in an ABI Prism 7700 Detection System (Applied Biosystems). RT-PCR data is the average of triplicate experiments and represents expression value relative to RPLPO expression in the same specimen.
Western blotting.
Protein lysates were prepared from cell lines or pulverized frozen tumors and standard SDS-PAGE, western blotting and chemiluminescence was performed as previously described.15 To analyze ligand-induced ErbB signaling, upon reaching 80% confluence, cells were serum-starved overnight, treated with erlotinib (12 µM) for 4 hours, and then stimulated with either NRG-1β (0.22 µg/mL) or EGF (0.1 µg/mL) for 15 minutes before lysates were prepared. The following antibodies were obtained from Millipore (Temecula, CA): anti-pEGFR tyr845 and anti-pEGFR tyr1173; Sigma: anti-β-actin and anti-EGFR; Cell Signaling (Beverly, MA): anti-pEGFR tyr992, antip-EGFR tyr1068, anti-pErbB3 tyr1289, anti-ErbB3, anti-pAKT ser473, anti-AKT, anti-pp44/42 MAPK thr202/tyr204 and anti-MAPK. All antibodies were used as specified by the manufacturer. Unless otherwise specified, antibodies were diluted in 5% milk.
Animals.
Six week old female in-bred Fox Chase SCID mice were obtained from Charles River Laboratories (Hartford, CT). Animals were handled according to a protocol approved by The Institutional Animal Care and Use Committee (IACUC) at our university. Mice were allowed to acclimate to animal housing and xenografts were developed by subcutaneously injecting 1.5 × 107 cells (ErbB3+PANC-1 or ErbB3−PANC-1) to the murine flank bilaterally. Trice weekly, tumor volume was determined using digital caliper measurements and the formula:
[large diameter2 × small diameter]/2
After 14 days, all mice had measurable tumors and were sorted into equal volume treatment and control groups. Treatment group mice received 50 mg/kg erlotinib dissolved in vehicle (0.1 M NaCl, 0.05% Pluronic Acid in PBS) per treatment while control mice received vehicle only. All mice received 15 intraperitoneal injections over a 21 day period (cycle: 5 treatment days then 2 non-treatment days). After 21 days, mice were sacrificed and tumors were harvested and snap-frozen at −80°C.
Statistical analysis.
Correlations between ErbB3 and EGFR mRNA levels were nonparametrically compared by Spearman's rho employing SPSS software (Chicago, IL). All cell assay conditions were performed in six wells and repeated in triplicate independent runs and data is presented as mean ± SEM. In these assays, Student's t-test was used for comparison and the Mann-Whitney U-test was used to compare independent runs. Statistical significance was defined at p < 0.05.
Acknowledgements
Financial support was received from a NIH/NRSA T32 Training Grant (J.S.L.) and from the Robert E. Reed Gastrointestinal Oncology Research Foundation.
Abbreviations
- EGFR
epidermal growth factor receptor
- EGF
epidermal growth factor
- NRG
neuregulin (heregulin)
- PI3K
phosphoinositide 3-kinase
- ERK1/2
extracellular-signal related kinase1/2
- MAPK
mitogen-activated protein kinase
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/12532
References
- 1.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 2.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 4.Papageorgio C, Perry MC. Epidermal growth factor receptor-targeted therapy for pancreatic cancer. Cancer Invest. 2007;25:647–657. doi: 10.1080/07357900701522653. [DOI] [PubMed] [Google Scholar]
- 5.Sithanandam G, Anderson LM. The ERBB3 receptor in cancer and cancer gene therapy. Cancer Gene Ther. 2008;15:413–448. doi: 10.1038/cgt.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger MB, Mendrola JM, Lemmon MA. ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett. 2004;569:332–336. doi: 10.1016/j.febslet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Fedi P, Pierce JH, di Fiore PP, Kraus MH. Efficient coupling with phosphatidylinositol 3-kinase, but not phospholipase Cgamma or GTPase-activating protein, distinguishes ErbB-3 signaling from that of other ErbB/EGFR family members. Mol Cell Biol. 1994;14:492–500. doi: 10.1128/mcb.14.1.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HH, Sierke SL, Koland JG. Epidermal growth factor-dependent association of phosphatidylinositol 3-kinase with the erbB3 gene product. J Biol Chem. 1994;269:24747–24755. [PubMed] [Google Scholar]
- 9.Hellyer NJ, Cheng K, Koland JG. ErbB3 (HER3) interaction with the p85 regulatory subunit of phosphoinositide 3-kinase. Biochem J. 1998;333:757–763. doi: 10.1042/bj3330757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carraway KL, 3rd, Soltoff SP, Diamonti AJ, Cantley LC. Heregulin stimulates mitogenesis and phosphatidylinositol 3-kinase in mouse fibroblasts transfected with erbB2/neu and erbB3. J Biol Chem. 1995;270:7111–7116. doi: 10.1074/jbc.270.13.7111. [DOI] [PubMed] [Google Scholar]
- 11.Sithanandam G, Smith GT, Masuda A, Takahashi T, Anderson LM, Fornwald LW. Cell cycle activation in lung adenocarcinoma cells by the ErbB3/phosphatidylinositol 3-kinase/Akt pathway. Carcinogenesis. 2003;24:1581–1592. doi: 10.1093/carcin/bgg125. [DOI] [PubMed] [Google Scholar]
- 12.Venkateswarlu S, Dawson DM, St Clair P, Gupta A, Willson JK, Brattain MG. Autocrine heregulin generates growth factor independence and blocks apoptosis in colon cancer cells. Oncogene. 2002;21:78–86. doi: 10.1038/sj.onc.1205011. [DOI] [PubMed] [Google Scholar]
- 13.Sithanandam G, Fornwald LW, Fields J, Anderson LM. Inactivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and invasiveness of human lung adenocarcinoma cell line A549. Oncogene. 2005;24:1847–1859. doi: 10.1038/sj.onc.1208381. [DOI] [PubMed] [Google Scholar]
- 14.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 15.Frolov A, Schuller K, Tzeng CW, Cannon EE, Ku BC, Howard JH, et al. ErbB3 expression and dimerization with EGFR influence pancreatic cancer cell sensitivity to erlotinib. Cancer Biol Ther. 2007;6:548–554. doi: 10.4161/cbt.6.4.3849. [DOI] [PubMed] [Google Scholar]
- 16.Buck E, Eyzaguirre A, Haley JD, Gibson NW, Cagnoni P, Iwata KK. Inactivation of Akt by the epidermal growth factor receptor inhibitor erlotinib is mediated by HER-3 in pancreatic and colorectal tumor cell lines and contributes to erlotinib sensitivity. Mol Cancer Ther. 2006;5:2051–2059. doi: 10.1158/1535-7163.MCT-06-0007. [DOI] [PubMed] [Google Scholar]
- 17.Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–235. [PubMed] [Google Scholar]
- 18.Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 19.Friess H, Yamanaka Y, Kobrin MS, Do DA, Buchler MW, Korc M. Enhanced erbB-3 expression in human pancreatic cancer correlates with tumor progression. Clin Cancer Res. 1995;1:1413–1420. [PubMed] [Google Scholar]
- 20.Kolb A, Kleeff J, Arnold N, Giese NA, Giese T, Korc M, et al. Expression and differential signaling of heregulins in pancreatic cancer cells. Int J Cancer. 2007;120:514–523. doi: 10.1002/ijc.22360. [DOI] [PubMed] [Google Scholar]
- 21.Lemoine NR, Lobresco M, Leung H, Barton C, Hughes CM, Prigent SA, et al. The erbB-3 gene in human pancreatic cancer. J Pathol. 1992;168:269–273. doi: 10.1002/path.1711680305. [DOI] [PubMed] [Google Scholar]
- 22.Tzeng CW, Frolov A, Frolova N, Jhala NC, Howard JH, Vickers SM, et al. Pancreatic cancer epidermal growth factor receptor (EGFR) intron 1 polymorphism influences postoperative patient survival and in vitro erlotinib response. Ann Surg Oncol. 2007;14:2150–2158. doi: 10.1245/s10434-007-9409-5. [DOI] [PubMed] [Google Scholar]
- 23.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 24.Kritzik MR, Krahl T, Good A, Gu D, Lai C, Fox H, et al. Expression of ErbB receptors during pancreatic islet development and regrowth. J Endocrinol. 2000;165:67–77. doi: 10.1677/joe.0.1650067. [DOI] [PubMed] [Google Scholar]
- 25.Erickson SL, O'Shea KS, Ghaboosi N, Loverro L, Frantz G, Bauer M, et al. ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development. 1997;124:4999–5011. doi: 10.1242/dev.124.24.4999. [DOI] [PubMed] [Google Scholar]
- 26.Friess H, Wang L, Zhu Z, Gerber R, Schroder M, Fukuda A, et al. Growth factor receptors are differentially expressed in cancers of the papilla of vater and pancreas. Ann Surg. 1999;230:767–774. doi: 10.1097/00000658-199912000-00005. discussion 74–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soler M, Mancini F, Meca-Cortes O, Sanchez-Cid L, Rubio N, Lopez-Fernandez S, et al. HER3 is required for the maintenance of neuregulin-dependent and -independent attributes of malignant progression in prostate cancer cells. Int J Cancer. 2009;125:2565–2575. doi: 10.1002/ijc.24651. [DOI] [PubMed] [Google Scholar]
- 28.Engelman JA, Janne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci USA. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ueno Y, Sakurai H, Tsunoda S, Choo MK, Matsuo M, Koizumi K, et al. Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int J Cancer. 2008;123:340–347. doi: 10.1002/ijc.23465. [DOI] [PubMed] [Google Scholar]
- 30.Jones RB, Gordus A, Krall JA, MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 31.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuste L, Montero JC, Esparis-Ogando A, Pandiella A. Activation of ErbB2 by overexpression or by transmembrane neuregulin results in differential signaling and sensitivity to herceptin. Cancer Res. 2005;65:6801–6810. doi: 10.1158/0008-5472.CAN-04-4023. [DOI] [PubMed] [Google Scholar]
- 33.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 34.Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]