

Abstract
Cortical intermediate progenitors (IPs) comprise a secondary neuronal progenitor pool that arises from radial glia (RG). IPs are essential for generating the correct number of cortical neurons, but the factors that regulate the expansion and differentiation of IPs in the embryonic cortex are essentially unknown. In this study, we show that the Wnt–β-catenin pathway (canonical Wnt pathway) regulates IP differentiation into neurons. Upregulation of Wnt–β-catenin signaling by overexpression of Wnt3a in the neocortex induced early differentiation of IPs into neurons and the accumulation of these newly born neurons at the subventricular zone/intermediate zone border. Long-term overexpression of Wnt3a led to cortical dysplasia associated with the formation of large neuronal heterotopias. Conversely, downregulation of Wnt–β-catenin signaling with Dkk1 during mid and late stages of neurogenesis inhibited neuronal production. Consistent with previous reports, we show that Wnt–β-catenin signaling also promotes RG self-renewal. Thus, our findings show differential effects of the Wnt–β-catenin pathway on distinct groups of cortical neuronal progenitors: RG self-renewal and IP differentiation. Moreover, our findings suggest that dysregulation of Wnt signaling can lead to developmental defects similar to human cortical malformation disorders.
Introduction
Projection neurons of the neocortex are generated from two progenitor pools, radial glia (RG) and RG-derived intermediate progenitors (IPs) (Molyneaux et al., 2007). Analyses of the behaviors of RG and IPs through the course of neurogenesis suggest that their patterns of behavior are coordinated. During early neurogenesis, the RG pool produces neurons via asymmetric divisions that replace the radial glial cell and produce one neuron. As neurogenesis proceeds, the RG instead produce an IP along with one replacement RG. IPs dramatically amplify the neurogenic pool before terminal neurogenic divisions (Noctor et al., 2007, 2008). Thus, as neurogenesis continues, the RG pool is primarily static, whereas the IP pool expands so that IPs take over the bulk of neuron production.
The Wnt–β-catenin pathway is believed to be a key regulator of this developmental progression. Wnts regulate many developmental processes, including cell proliferation, specification, differentiation, and migration (Logan and Nusse, 2004; Clevers, 2006). In the canonical signaling pathway, Wnt ligands bind to Frizzled–LRP5/6 receptor complexes at the plasma membrane promoting gene expression through β-catenin and lymphoid enhancer-binding factor (LEF)/T-cell factor (Tcf) transcription factor activity (Logan and Nusse, 2004; Clevers, 2006). Loss-of-function studies in LRP6 and β-catenin decrease cortical neuronal production (Woodhead et al., 2006; Zhou et al., 2006). This phenotype is supported by gain-of-function studies that suggest that the pathway promotes self-renewal of neuroepithelial progenitors during early neurogenesis (Chenn and Walsh, 2002; Machon et al., 2007; Wrobel et al., 2007). For instance, expression of a dominant-active form of β-catenin promotes RG self-renewal and inhibition of IP production (Wrobel et al., 2007). However, studies conducted during mid neurogenesis have yielded inconsistent results. Focal ablation of β-catenin promotes RG cell cycle exit and neuronal differentiation (Woodhead et al., 2006). This is consistent with Wnt signaling maintaining the progenitor state in early neurogenesis. Other studies contradict this result, demonstrating a role for the Wnt–β-catenin pathway in promoting neuronal differentiation through N-myc and the neurogenic transcription factor Ngn1/2 (Hirabayashi et al., 2004; Israsena et al., 2004; Kuwahara et al., 2010). Together, these findings implicate multiple roles for the Wnt–β-catenin pathway but also reveal that additional studies are necessary to reach a clearer understanding of the functions of the pathway in neurogenesis. It is our hypothesis that the Wnt–β-catenin pathway regulates both proliferation and differentiation in vivo in a context-dependent manner, likely separated by either progenitor state or developmental time. Importantly, some previous experiments suggest that the Wnt–β-catenin pathway specifically regulates IPs (Viti et al., 2003; Hirabayashi et al., 2004; Kuwahara et al., 2010), but there has been little direct consideration of this question.
We set out to determine whether the Wnt–β-catenin pathway regulates RG and IPs independently. We used secreted factors of the pathway, Wnt3a ligand or Wnt inhibitor Dkk1, to avoid the caveats of transcription factor manipulation, including activation of signaling in cells not normally responsive to Wnt ligands and the cell adhesion consequences of β-catenin. Our study uncovered a novel role for the Wnt–β-catenin pathway in promoting IP differentiation and the formation of neuronal heterotopias.
Materials and Methods
Animals.
In utero electroporations were performed on CD1 wild-type (Charles River Laboratories) and BAT–gal transgenic heterozygous (Maretto et al., 2003) mice. RNA in situ hybridization and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) reaction were performed on tissue obtained from CD1 and BAT–gal mice, respectively. All animal protocols were approved by the University of California, San Francisco Institutional Animal Care and Use Committee.
In utero electroporation, expression plasmids, and bromodeoxyuridine labeling.
Electroporations were performed as described previously by Li et al. (2008). Electroporations were conducted on embryonic day 13.5 (E13.5) or E15.5 embryos, allowed to continue gestation in utero, and harvested at E14.5, E16.5, E18.5, or postnatal day 2 (P2). DNA solution (100–300 nl of 2–3 μg/μl plasmid/10%Trypan Blue/10 m Tris-HCl, pH 8.0) was injected in the left lateral ventricles of cerebral cortices. DNA solution was allowed to diffuse throughout the ventricle before exposing ventricles to electric field at 33 V for E13.5 and 37 V for E15.5. The following plasmids were used: M38 TOP-destabilized enhanced green fluorescence protein (TOP–dGFP) (Addgene plasmid 17114; R. Moon, University of Washington, Seattle, WA); pCIG (GFP) (Megason and McMahon, 2002); pCIG2mouseWnt3a (Wnt3a), subcloned with pCIG2 (Hand et al., 2005) and mouse Wnt3a cDNA cloned from Ambion cDNA library; pCIGchickenWnt3a (Wnt3a) (Galli et al., 2004); pCIGLEF1–VP16, subcloned with pCIG and LEF1–VP16 (LEF1 fused to the herpes simplex virus VP16 transactivation domain; for info, contact G.L. at grant.li@ucsf.edu); pCIGmouseDkk1 and pCIGΔaa29-48humanβ-catenin (provided by L.W. Burrus, San Francisco State University, San Francisco, CA) [Δaa29–48humanβ-catenin (Tetsu and McCormick, 1999)]; and pCImRFP (provided by J. L. R. Rubenstein, University of California, San Francisco). Water-solubilized bromodeoxyuridine (BrdU) was injected into the peritoneal cavity to a final dose of 50 μg BrdU/g bodyweight.
Immunohistochemistry and X-gal reaction.
Mouse brains were collected, fixed, cryoprotected, and coronally cryosectioned at 20 μm using standard methods. Sections were immunolabeled and stained with X-gal using standard methods. The following primary antibodies were used: chicken anti-GFP (1:800; Aves Labs), mouse anti-BrdU (B44, 1:50; BD Biosciences), mouse anti-MAP2 (2a and 2b, 1:500; BD Biosciences Pharmingen), mouse anti-neuronal nuclei (NeuN) (1:200, Millipore Bioscience Research Reagents), rabbit anti-active Caspase3 (1:500; BD Pharmingen), rabbit anti-CDP/Cux1 (M-222, 1:200; Santa Cruz Biotechnology), rabbit anti-Tbr2 (1:500; Millipore Bioscience Research Reagents), rabbit anti-Pax6 (1:200; Covance), rat anti-Ctip2 (25B6, 1:300 to 1:800; Abcam), and rat anti-Ki67 (TEC-3, 1:300; Dako). Primary antibodies were detected with goat secondary antibodies conjugated to Alexa fluorophores (Invitrogen). β-Galactosidase enzymatic activity was detected with X-gal substrate (Invitrogen).
In situ hybridization.
Mouse brain sections were prepared as described for immunohistochemistry. RNA in situ hybridization was performed as described by Li et al. (2008). Axin2 digoxigenin-labeled RNA probe was generated from pYX–Asc–mouseAxin2 (catalog #EMM1002-7496341; Open Biosystems).
Image analysis, quantification, and statistical analysis.
Images were captured with QI imaging CCD camera and QCapture Pro software. For each separate experiment, three or more embryos were used for qualitative analyses (n ≥ 3), and five or more embryos were used for quantitative analyses (n ≥ 5) except for dGFP+, Tbr2+ cell counts, in which three embryos were used (n = 3). Embryos from two or more litters were used for each separate experiment. For cell counts at E14.5 and E16.5, cells within a 100-μm-wide neocortical column were counted. For cell counts at P2, cells within a 300-μm-wide neocortical column were counted. Length and thickness were quantified with the NIH ImageJ measuring tool and by taking the average of three different measurements of adjacent regions per sample. Normalized data [unelectroporated (unEp)-normalized] were normalized by dividing measurements or counts of the electroporated neocortex by measurements or counts of the unelectroporated neocortex of the same rostrocaudal level of the same brain. Quantified results were expressed as the mean ± SEM for n given samples. Two-tailed Student's t test with unequal variance was used to analyze data except for Dkk1 electroporations in which one-tailed tests were used. A value of p < 0.05 was considered statistically significant.
Results
Ectopic Wnt3a causes cortical dysplasia and neuronal heterotopias
We analyzed the effect of excess Wnt–β-catenin signaling by expressing Wnt3a in the neocortex at E13.5, a time point when RG symmetric self-renewal has already slowed to bypass the early strong effects on this process, and analyzed the results at P2 when neurogenesis and migration have primarily subsided. We chose to ectopically express Wnt3a for several reasons. (1) Loss-of-function mutation of Wnt3a leads to dramatic disruption of neocortical development (Lee et al., 2000). (2) Wnt3a, Wnt2b, and Wnt8b are thought to generate the medial-high to lateral-low gradient of Wnt–β-catenin activity that has been implicated in the regulation of patterning and neurogenesis in the dorsal cortex. (3) Wnt3a has been shown to activate the Wnt–β-catenin pathway in many biological contexts. (4) Other Wnts with specific expression in the neocortex (Wnt5b, Wnt7a, and Wnt7b) have not been correlated with roles in neurogenesis. Moreover, Wnt7a and Wnt7b have instead been associated with dendritic complexity and endothelial/vasculature regulation (Hall et al., 2000; Rosso et al., 2005; Daneman et al., 2009). (5) Expression of a secreted ligand of the Wnt pathway incorporates regulation from the extracellular space to the nucleus and should elicit effects on cells normally responsive to Wnt ligands, unlike expression of transcription factors of the Wnt pathway.
We used in utero electroporation of plasmids to ectopically express GFP, as a control, or Wnt3a in the developing mouse neocortex. We first tested whether electroporated Wnt3a upregulated the Wnt–β-catenin pathway in BAT–gal transgenic and wild-type embryos. The BAT–gal transgene is a Wnt–β-catenin pathway reporter allele with β-galactosidase under the control of LEF/Tcf-responsive elements (Maretto et al., 2003). We detected electroporated regions by immunolabeling for GFP and the expression of the BAT–gal transgene by X-gal colorimetric reaction with β-galactosidase in coronal sections of mouse brains. Wnt3a, but not GFP, strongly upregulated the expression of the BAT–gal transgene (Fig. 1A–D). Moreover, it is clear that Wnt3a elicits a non-cell-autonomous effect. Increased β-galactosidase activity was seen beyond electroporated regions (solid lines indicate the electroporated region in Fig. 1C,D; dashed lines indicate non-cell-autonomous activity in D). To confirm that Wnt3a upregulates endogenous signaling as well, we analyzed Axin2 mRNA expression, a direct target of the Wnt–β-catenin pathway, by RNA in situ hybridization. Wnt3a electroporation at E13.5, but not GFP, increased and laterally extended Axin2 mRNA expression by E14.5 (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). Thus, ectopic expression of Wnt3a in the neocortex upregulates the Wnt–β-catenin pathway within 24 h of electroporation.
Figure 1.
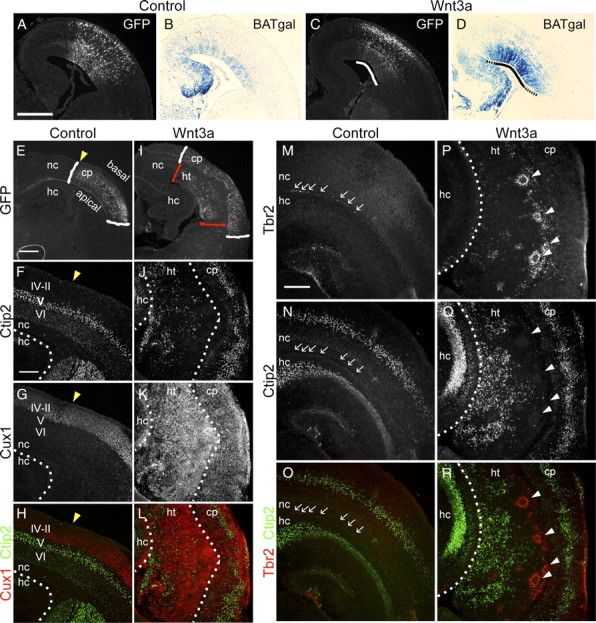
Ectopic Wnt3a causes cortical dysplasia and neuronal heterotopias. A–D, The capacity of ectopic Wnt3a to induce Wnt–β-catenin signaling was tested by electroporating E13.5 BAT–gal transgenic embryos with control or Wnt3a expression plasmids. Electroporated embryos were analyzed at E16.5 for GFP expression of electroporation plasmids and β-galactosidase expression of the BAT–gal transgene. A, C, Immunolabeling for GFP. B, D, X-gal staining for β-galactosidase activity. Black solid line in D demarcates the region similar to the electroporated region in C (white solid line). Dashed lines in D demarcate X-gal staining beyond the electroporated region. E–R, Characterization of P2 brains electroporated at E13.5 by immunolabeling for GFP, Ctip2, and Cux1 layer neuron markers and Tbr2 IP marker. E, I, White brackets indicate region of GFP localization in the cortical plate (cp), and red brackets indicate localization of GFP in a heterotopic structure apical to the cortical plate. Yellow arrowheads in E–H indicate the medial boundary of GFP electroporation. F–H, J–L, Immunolabeling for layer VI–V and layer IV–II neurons with Ctip2 (F, H, J, L) and Cux1 (G, H, K, L) antibodies, respectively. M–R, Analysis of IP distribution with Tbr2 immunolabeling. Arrows in M–O indicate IPs in control sample, and white arrowheads in P–R indicate IPs in rosette formation in the experimental sample. Scale bars: A, E, 500 μm; F, M, 200 μm. hc, Hippocampus; ht, heterotopia; nc, neocortex.
To test the ultimate effects of Wnt3a overexpression, we analyzed electroporated brains at P2, 8 d after electroporation. The GFP-expressing cells in control brains were distributed throughout the cortical plate, whereas many GFP+ cells in Wnt3a-electroporated brains were clustered in a mass adjacent to the ventricle [Fig. 1E,I, cortical plate (cp) GFP, cp/white brackets; ectopic mass GFP, heterotopia (ht)/red brackets]. To investigate the nature of these cell clusters, we analyzed the expression of the neuronal markers NeuN, Ctip2, and Cux1 by immunolabeling. NeuN is a pan-neuronal marker, whereas Ctip2 and Cux1 labels specific layers of the cortical plate. Ctip2 is expressed moderately by layer VI and strongly by layer V early-born/deep layer neurons (Fig. 1F,H), whereas Cux1 is expressed strongly by layer IV–II late-born/upper layer neurons (Fig. 1G,H). Labeling with NeuN (supplemental Fig. S2B,D, available at www.jneurosci.org as supplemental material), Ctip2, and Cux1 (Fig. 1J–L) showed that Wnt3a-induced cell masses are predominantly made up of neuronal cells. We further analyzed the composition of these heterotopias with the MAP2 antibody, a dendritic marker, and found that these large heterotopias are densely composed of disorganized dendrites (supplemental Fig. S2F,H, available at www.jneurosci.org as supplemental material).
To determine how long-term induction of Wnt signaling affects the organization of IPs remaining in the cortex by P2, we analyzed the expression of Tbr2 by immunolabeling. In control brains, there were few remaining Tbr2+ cells found in the subventricular zone (SVZ) (Fig. 1M–O, arrows). However, in Wnt3a-electroporated brains, there were many more Tbr2+ cells in disorganized clumps or organized in rosettes in an expanded SVZ adjacent to the heterotopic neuronal mass (Fig. 1P–R, rosettes, arrowheads). Thus, long-term upregulation of the Wnt–β-catenin pathway expands the population and dramatically alters the organization of these cells. These factors are consistent with characteristics that would lead to production of excess neurons. Furthermore, disorganization and rosette reorganization of IPs could cause newly born neurons to migrate incorrectly, which could lead to the disorganized cortical plate and periventricular heterotopia seen at P2.
The formation of an extra-neocortical neuronal mass larger than the normal epithelium suggests that Wnt3a ectopic expression induces excess progenitor proliferation but does not ultimately hinder neuronal differentiation. These results confirm a function for the Wnt–β-catenin pathway in promoting progenitor proliferation as described previously (Chenn and Walsh, 2002; Logan and Nusse, 2004; Woodhead et al., 2006; Zhou et al., 2006; Wrobel et al., 2007). Moreover, it potentially implicates dysregulation of Wnt–β-catenin signaling pathway in the occurrence of neuronal migration disorders, as suggested previously (Zhou et al., 2006; Freese et al., 2010).
Wnt3a promotes expansion of RG and differentiation of IPs
To begin to determine the mechanism of Wnt3a effects that lead to the dramatic changes seen at P2, we examined the distribution of neuronal progenitors 3 d after electroporation, at E16.5. We immunolabeled for Pax6 to label RG and Tbr2 to label IPs. Because the thickness and density of cells of Pax6+ ventricular zone (VZ) appeared greater after Wnt3a electroporation compared with control (Fig. 2 A,C, yellow brackets), we measured the thickness of the VZ, counted Pax6+ VZ cells, and determined that both the thickness and cell number of the VZ are significantly greater in Wnt3a-electroporated brains than in controls (VZ thickness: control, n = 6, average ± SEM of 1.05 ± 0.016 unEp-normalized VZ thickness; Wnt3a, n = 6, average ± SEM of 1.28 ± 0.035 unEp-normalized VZ thickness, pvalue = 0.0005; Wnt3a/control ratio = 1.22; Pax6+ VZ cells: control, n = 6, average ± SEM of 1.08 ± 0.030 unEp-normalized cell number/100 μm column; Wnt3a, n = 6, average ± SEM of 1.28 ± 0.067 unEp-normalized cell number/100 μm column, pvalue = 0.03; Wnt3a/control ratio = 1.18) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). This is consistent with Wnt3a inducing RG to undergo self-renewing divisions, an established function of the Wnt–β-catenin pathway.
Figure 2.
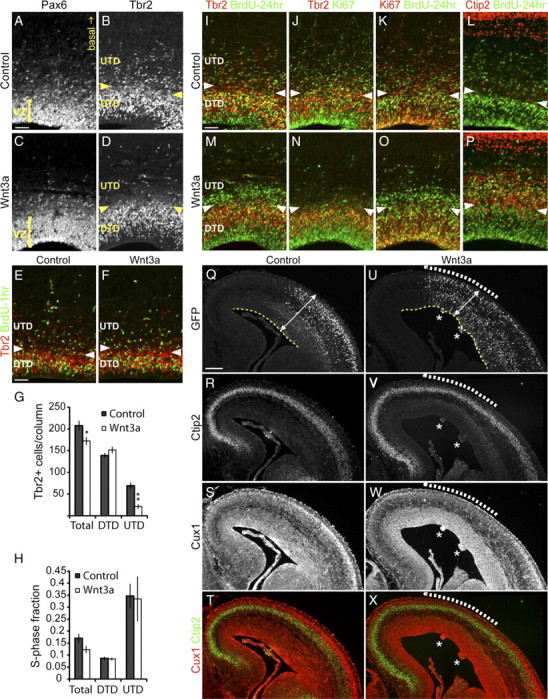
Wnt3a promotes expansion of RG and differentiation of IPs. Brains electroporated at E13.5 were analyzed at E16.5 for the distribution of progenitors and neurons by immunolabeling. A, C, Analysis of RG distribution with Pax6 immunolabeling. Yellow bracket indicates the thickness of the ventricular zone. In B, D–F, and I–P, the boundary between the UTD and DTD subpopulations of Tbr2+ IPs is demarcated by the pairs of yellow or white arrowheads. B, D, Examination of IP distribution with Tbr2 immunolabeling. E–H, Analysis of the numbers and S-phase fraction of IPs. BrdU was injected 1 h before harvest for S-phase fraction analysis. E, F, Representative samples used for quantification of Tbr2+ cells numbers in G and the S-phase fraction of Tbr2+ cell populations in H with Tbr2 and BrdU immunolabeling. I–P, Lineage tracing of progenitors in S-phase with BrdU injected at E15.5, 24 h before harvest and analysis at E16.5. I, M, Coimmunolabeling for BrdU and Tbr2 to determine the localization of IPs born at E15.5. J,K and N,O, Triple immunolabeling for BrdU, Ki67, and Tbr2 to analyze the cell cycle state of BrdU-labeled cells. L, P, Coimmunolabeling for BrdU and Ctip2 to determine the differentiation state of BrdU-labeled cells. Q–X, Triple immunolabeling for GFP, Ctip2, and Cux1 to determine the effect of Wnt3a on neuron production. Q, U, GFP immunolabeling shows the extent of plasmid expression. White dashed lines in U–X indicate the region in which Ctip2 and Cux1 expression are most strongly affected. Q, U, Analysis of neocortex morphology. White double-ended arrows and yellow dashed lines indicate the thickness and trace the length of the neocortex, respectively. Nodules positive for ectopic Ctip2 is indicated with a white asterisk in U–X. Scale bars: A, E, I, 50 μm; Q, 200 μm. Error bars represent SEM. In G, *p < 0.05 and **p < 0.001.
Next we analyzed the distribution of Tbr2+ cells. We demarcated the expression pattern of Tbr2 into two domains: the “deep Tbr2 domain” (DTD), consisting of Tbr2+ cells within the VZ and SVZ (Tbr2 domain below the arrowheads in Fig. 2B), and the “upper Tbr2 domain” (UTD), consisting of the more sparsely distributed Tbr2+ cells in the intermediate zone (IZ) (Tbr2 domain above the arrowheads in Fig. 2B). Strikingly, Wnt3a electroporation led to a dramatic depletion of UTD IPs compared with control (Fig. 2B,D). There are several potential cellular mechanisms for this change: Wnt3a may (1) block production of IPs from RG, (2) decrease proliferation of IPs, (3) block transition of IPs from the DTD to UTD, (4) induce early differentiation of IPs into neurons, and (5) force IPs to become a non-neuronal Tbr2-negative (Tbr2−) cell population.
Normally, an increase in RG proliferation and population would be expected to lead to increased production of IPs. However, we observed a clear decrease of IPs in the UTD of experimental samples (Fig. 2B,D). It seems likely that induction of RG proliferation by Wnt3a might also be associated with inhibiting transition of RG into the IP state, thus reducing the total IP population. Previous work by Wrobel et al. (2007) has shown that genetically inducing RG proliferation with a dominant-active allele of β-catenin (ΔEx3β-catenin) inhibits the production of IPs. To test whether this is the explanation for loss of IPs after Wnt3a electroporation, we quantified the number of Tbr2+ cells in the total and Tbr2 subdomains (DTD and UTD) [Fig. 2E,F (quantification: control, n = 7; Wnt3a, n = 9) and G]. Because IPs should be generated and seen first in the DTD (at the VZ–SVZ border), the numbers of cells in the DTD should be reduced if there is a block in IP production from RG. Indeed, the total number of Tbr2+ cells is significantly decreased in Wnt3a-electroporated brains compared with controls (total: control, average ± SEM of 207 ± 11.6 cells/100 μm column; Wnt3a, average ± SEM of 172 ± 9.7 cells/100 μm column, pvalue = 0.037) (Fig. 2G), but the number of Tbr2+ cells in the DTD is unchanged (DTD: control, average ± SEM of 139 ± 5.6 cells/100 μm column; Wnt3a, average ± SEM of 151 ± 7.8 cells/100 μm column, pvalue = 0.219) (Fig. 2G). Instead, there is a dramatic decrease in UTD IPs after Wnt3a electroporation compared with control (UTD: control, average ± SEM of 69 ± 7.6 cells/100 μm column; Wnt3a, average ± SEM of 21 ± 5.6 cells/100 μm column, pvalue = 0.0003) (Fig. 2G). Thus, the decrease in total Tbr2+ cells is more likely attributable to the depletion of UTD Tbr2+ cells rather than a failure to generate DTD Tbr2+ cells from RG.
The loss of Tbr2+ cells in the UTD could be attributable to a failure of IPs to continue to proliferate in this zone. To assess this, we calculated the mitotic fraction of Tbr2+ cells in the DTD and UTD by acute BrdU labeling (50 μg BrdU/g body weight 1 h before harvest at E16.5). We quantified the total, DTD, and UTD fraction of Tbr2+ cells that are BrdU+ [Fig. 2E,F (quantification: control, n = 7; Wnt3a, n = 6) and H]. The S-phase fraction of DTD Tbr2+ cells in Wnt3a-electroporated brains is similar to controls (DTD: control, average ± SEM of 0.086 ± 0.0077/100 μm column; Wnt3a, average ± SEM of 0.084 ± 0.0062/100 μm column, pvalue = 0.828) (Fig. 2H). Interestingly, although the number of UTD Tbr2+ cells is dramatically decreased, the S-phase fraction of UTD and total Tbr2+ cells after Wnt3a electroporation is also similar to controls (UTD: control, average ± SEM of 0.347 ± 0.052/100 μm column; Wnt3a, average ± SEM of 0.335 ± 0.094/100 μm column, pvalue = 0.913; total: control, average ± SEM of 0.170 ± 0.019/100 μm column; Wnt3a, average ± SEM of 0.123 ± 0.015/100 μm column, pvalue = 0.080) (Fig. 2H). Thus, although there is a dramatic loss of UTD IPs, the level of proliferation of IPs is maintained.
To test whether UTD IPs are absent because they have stopped expressing Tbr2, we traced the lineage of IPs by pulsing with BrdU 24 h before harvesting embryos. This method labeled progenitors that are in the S-phase at E15.5 and allowed us to visualize their location at E16.5. If the absence of UTD IPs is not attributable to failed production of this population but is attributable to downregulation of Tbr2 expression, we would see an extra pool of BrdU+ cells that are Tbr2− in Wnt3a-electroporated regions but not in controls. Indeed, we observed such a large pool of BrdU+, Tbr2− cells in the apical-most region of the UTD of Wnt3a-electroporated brains but not in controls (Fig. 2I,M). To test whether the BrdU+, Tbr2− pool of cells are no longer dividing or have just lost Tbr2 expression, we immunolabeled with Ki67, an active-cell cycle marker. This confirmed that the BrdU+, Tbr2− cells found in the UTD are also Ki67− and, thus, are no longer proliferative (Fig. 2J,K,N,O).
The disappearance of UTD IPs and the concomitant appearance of BrdU+, Tbr2− postmitotic cells in the UTD suggest that ectopic Wnt3a induces UTD IPs to become postmitotic. To confirm that the BrdU+, Tbr2− cell population are differentiated neurons, we immunolabeled with Ctip2 antibody. At E16.5, Ctip2 is only faintly expressed in some cells in progenitor domains but relatively more strongly expressed by postmigratory, differentiated neurons localized in the cortical plate. We found that electroporation of Wnt3a but not GFP lead to increased expression of Ctip2 in progenitor domains. Moreover, a large number of the UTD-accumulated BrdU+, Tbr2− cells also express Ctip2 (Fig. 2L,P). Thus, upregulation of the Wnt–β-catenin pathway with Wnt3a induces UTD IPs to differentiate into neurons.
The IP pool is critical for generating the correct number of cortical plate neurons. Intermediate progenitors amplify the neuronal output of RG by providing one or two extra steps of self-renewing divisions before producing a pair of neurons on its last symmetric division. We therefore hypothesized that premature differentiation of IPs and accumulation of the resulting neurons in the UTD in Wnt3a-electroporated regions would lead to a reduction in the number of neurons reaching the cortical plate during this period. To address this hypothesis, we examined Ctip2 and Cux1 expression by immunolabeling at E16.5, 3 d after electroporation. Within the cortical plate, the expression patterns of both markers were reduced in Wnt3a-electroporated regions compared with controls (Fig. 2Q–X, white dashed lines). The population of late-born/upper layers neurons labeled by Cux1 was more strongly affected, being either absent or thinner in Wnt3a-eletroporated regions relative to the unaffected regions and controls (Fig. 2S,T,W,X, white dashed lines). To quantify these observations, we measured the thickness of both Ctip2 and Cux1 cortical plate layers and determined that these layers in Wnt3a-electroporated brains are significantly thinner than controls (Ctip2 layer thickness: control, n = 6, average ± SEM of 1.07 ± 0.031 unEp-normalized thickness; Wnt3a, n = 6, average ± SEM of 0.66 ± 0.050 unEp-normalized thickness, pvalue = 0.00009; Wnt3a/control ratio = 0.61; Cux1 layer thickness: control, average ± SEM of 0.96 ± 0.037 unEp-normalized thickness; Wnt3a, average ± SEM of 0.39 ± 0.004 unEp-normalized thickness, pvalue = 0.00009; Wnt3a/control ratio = 0.41) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). To rule out a significant contribution of cell death to this effect, we immunolabeled for active Caspase3, a marker of cell death, and observed no significant increase of apoptosis in Wnt3a-electroporated brains compared with controls (supplemental Fig. S5A–D, available at www.jneurosci.org as supplemental material). These results suggest that premature differentiation of IPs and accumulation of these newly born cells in the UTD in Wnt3a-electroporated brains lead to reduction in the number of neurons reaching the cortical plate.
Newborn neocortical neurons use RG processes as a migratory scaffolding; therefore, the failure of newly born neurons to reach the cortical plate might also be attributed to loss of RG cell integrity. We analyzed the structure of the radial processes using Nestin antibody. Nestin is localized in the cell bodies, extensions, and end feet of RG cells, thus allowing examination of RG extensions and pial connection of their end feet. The general appearance of Nestin immunolabeling is similar between control and experimental (supplemental Fig. S6B,E, available at www.jneurosci.org as supplemental material). Moreover, the appearance of extensions in the cortical plate and end feet at the pial surface look unperturbed by Wnt3a overexpression (supplemental Fig. S6C,F, available at www.jneurosci.org as supplemental material). Thus, our analyses indicate that perturbation of RG morphology and pial connection is likely not a primary factor in the formation of Wnt3a-induced heterotopias.
Lateral expansion of the neocortical epithelium is achieved by self-renewal of RG. At E16.5, the neocortices electroporated with Wnt3a are generally longer than controls (Fig. 2Q,U, yellow dashed lines). In addition, in regions electroporated with Wnt3a, the cortical wall was frequently thinner than regions distant from the site of electroporation and in controls (Figs. 1A–D, solid line; 2Q,U, white double-ended arrows). We measured the length and thickness of neocortices and determined that Wnt3a-electroporated neocortices were significantly longer and thinner than controls (neocortex length: control, n = 6, average ± SEM of 0.98 ± 0.036 unEp-normalized length; Wnt3a, n = 6, average ± SEM of 1.38 ± 0.063 unEp-normalized length, pvalue = 0.0006, Wnt3a/control ratio = 1.41; neocortex thickness: control, n = 6, average ± SEM of 1.02 ± 0.016 unEp-normalized thickness; Wnt3a, n = 6, average ± SEM of 0.79 ± 0.060 unEp-normalized thickness, pvalue = 0.011, Wnt3a/control ratio = 0.78) (supplemental Fig. S7A,B, available at www.jneurosci.org as supplemental material). This morphology is consistent with the abnormal expansion of the RG pool observed at E16.5 (Fig. 2A,C) in Wnt3a-electroporated brains and may ultimately contribute to the production of excess neurons observed at P2 once this increased cohort of RG produces neurons.
Intermediate progenitors are targets of Wnt signaling in vivo
If the Wnt–β-catenin pathway is physiologically important for IP differentiation, then endogenous Wnt–β-catenin signaling must be active in IPs. To test whether IPs are responding to endogenous Wnts, we electroporated TOP–dGFP Wnt–β-catenin pathway reporter plasmid into E13.5 brains and analyzed expression of dGFP at E14.5. First, to test the relative expression of dGFP in expressing cells, we coelectroporated TOP–dGFP with a control plasmid ubiquitously expressing red fluorescent protein (RFP) (pCImRFP). Because RFP is under the control of a constitutive promoter, the level of RFP expression serves as a visual measure of the relative electroporation efficiency. We observed cells strongly expressing RFP that were either weakly or strongly expressing dGFP (Fig. 3A1,A4). Similarly, we observed cells weakly expressing RFP that were either weakly or strongly expressing dGFP (Fig. 3A2,A3). This expression heterogeneity suggests that the Wnt reporter expression differences that we observe are not solely attributable to differences in plasmid number but rather probably readout of endogenous levels of Wnt signaling. Thus, as others have also concluded (Woodhead et al., 2006), the TOP–dGFP plasmid introduced into the neocortex by electroporation can be used to identify the presence of endogenous Wnt–β-catenin signaling.
Figure 3.
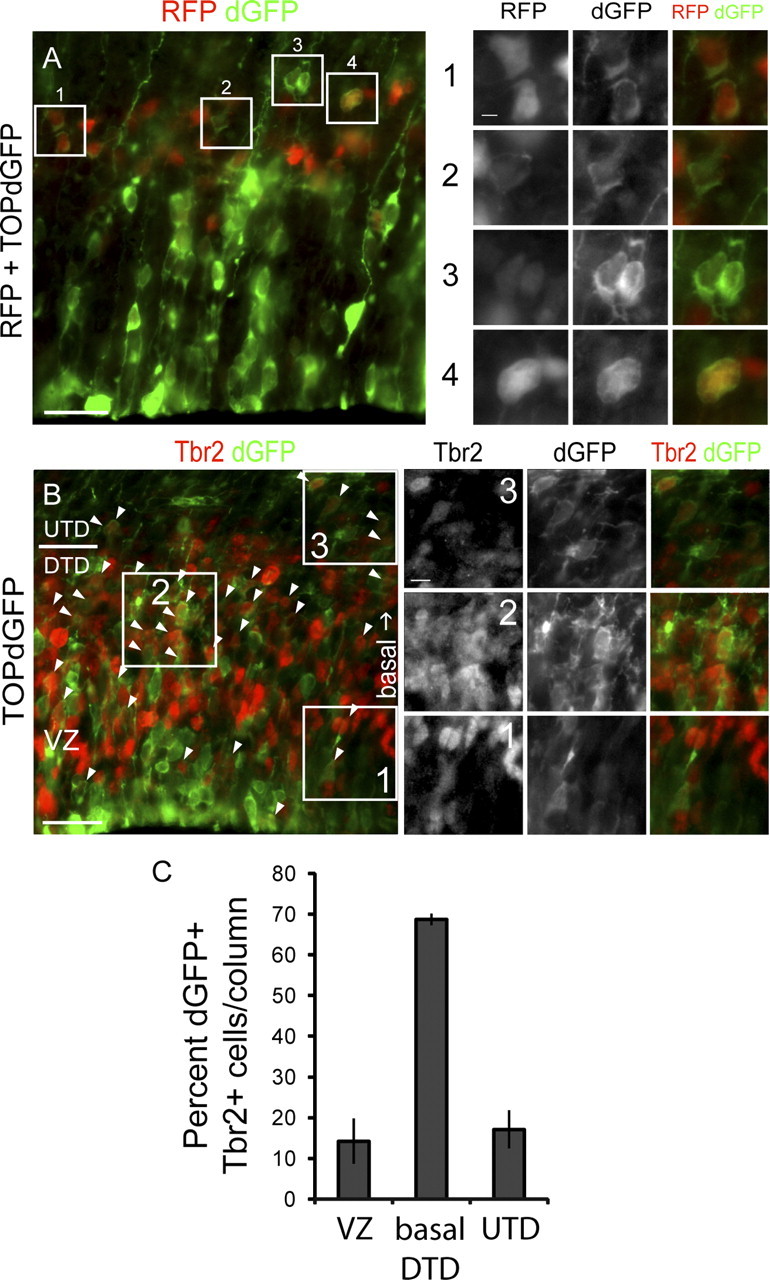
Intermediate progenitors can be targets of Wnt signaling in vivo. Analysis of Wnt–β-catenin activity in IPs. Embryos were electroporated with the TOP–dGFP Wnt–β-catenin signaling reporter at E13.5 and analyzed at E14.5 for dGFP expression to visualize Wnt–β-catenin signaling activity. A, Coelectroporation of pCImRFP and TOP–dGFP plasmids to determine whether dGFP expression is affected by electroporation efficiency. Electroporation efficiency is measured by the fluorescence level of RFP, which is expressed under a ubiquitous promoter. A1, Cells expressing strong RFP and weak dGFP. A2, A cell expressing weak RFP and dGFP. A3, Cells expressing weak RFP and strong dGFP. A4, Cells expressing strong RFP and dGFP. B, Immunolabeling for Tbr2 in brains electroporated with TOP–dGFP alone to determine whether Wnt–β-catenin signaling is active in IPs. Arrowheads indicate cells coexpressing Tbr2 and dGFP. Examples of Tbr2+, dGFP+ cells in the ventricular zone (B1), basal DTD (B2), and UTD (B3). C, Quantification of the numbers of dGFP+, Tbr2+ cells within the VZ, basal DTD, and emerging UTD. Scale bars: A, B, 25 μm; A1, 3.125 μm; B3, 6.25 μm.
We next analyzed the colocalization of Tbr2 and dGFP to determine whether IPs are included among the cells responsive to endogenous Wnt–β-catenin signaling. Many Tbr2-expressing cells were dGFP+ (Fig. 3B, arrows). In the VZ, few Tbr2+ cells were also dGFP+ (Fig. 3B1), whereas in the most basal area of the DTD, many more are doubly expressing Tbr2 and dGFP (Fig. 3B2). By E14.5, some Tbr2+ cells have emerged from the DTD and migrated into the UTD (Fig. 3B). These UTD Tbr2+ cells also express dGFP (Fig. 3B3). We quantified the number of dGFP+, Tbr2+ cells that are localized within these domains and verified that IPs within the basal DTD are most actively responding to endogenous Wnt–β-catenin signaling at E14.5 (dGFP+, Tbr2+ cells, n = 3; VZ, average ± SEM of 14 ± 5.56% cells/100 μm column; basal DTD, average ± SEM of 69 ± 1.42% cells/100 μm column; UTD, average ± SEM of 17 ± 4.71% cells/100 μm column) (Fig. 3C). These observations indicate that Tbr2-expressing IPs respond to endogenous Wnt–β-catenin signaling.
Wnt3a promotes differentiation of neurons in progenitor domains and disorganization of RG and IP distribution
The BrdU+, Tbr2−, Ctip2+ postmitotic cells that accumulated in the UTD in Wnt3a-electroporated regions are likely immature neurons born from the missing UTD IP population. To determine whether these cells will mature into the neurons that later comprise the heterotopia seen at P2, we looked for ectopic expression of neuronal markers at E18.5 (5 d after electroporation). Normally, Ctip2 expression is only strong in layers VI and V of the cortical plate at E18.5 (Fig. 4B,D). However, in Wnt3a-electroporated regions, there was ectopic expression of Ctip2 in progenitor domains (Fig. 4B,D,F,H, ht). Cux1 is typically expressed by some cells in progenitor domains but mostly by differentiated neurons in layer IV–II of the cortical plate at E18.5 (Fig. 4C,D). However, in Wnt3a-electroporated regions, Cux1 expression was dramatically expanded in the progenitor/IZ domains and decreased in the cortical plate (Fig. 4C,D,G,H). Together with our previous findings, our results suggest that ectopic Wnt3a induces premature differentiation of UTD IPs and accumulation of these cells in progenitor domains. This hypothesis is consistent with the decreased thickness of the cortical plate and increased thickness of the heterotopic region (Figs. 1E,I, cortical plate (cp)/white brackets, heterotopia (ht)/red brackets; 4A–H, cp/white double-ended arrows, ht/yellow double-ended arrows).
Figure 4.

Wnt3a promotes differentiation of neurons in progenitor domains. Characterization of E18.5 brains electroporated at E13.5 by triple immunolabeling for GFP and neuronal markers Ctip2 and Cux1. A–H, White dashed lines demarcate the boundaries between the cortical plate (cp), heterotopia (ht), neocortex (nc), choroid plexus (p), and hippocampus (hc). White double-headed arrows indicate the thickness of the cortical plate, and yellow double-headed arrows indicate the thickness of the heterotopia. A, E, Immunolabeling for GFP show distribution of GFP in the cortical plate and heterotopia. Immunolabeling for Ctip2 (B, D, F, H) and Cux1 (C, D, G, H) show expression of layer neurons in the cortical plate and heterotopia. Scale bars: A, 200 μm. IV–II, Layer IV-II; V, layer V; VI, layer VI.
To further understand the progression of heterotopia formation, we analyzed the distribution of RG and IPs at E18.5, 5 d after Wnt3a electroporation. As before, we used Pax6 to label RG and Tbr2 to label IPs. Using these immunomarkers, we noted scattering of progenitors and reorganization of progenitors into rosettes (Fig. 5A–D, scattered cells/arrows, rosettes/arrowheads, Pax6; 5I–L, Tbr2). We further tested whether RG and IPs in Wnt3a-electroporated brains remain active in the cell cycle by coimmunolabeling with Ki67. We observed that a large proportion of RG and IPs are Ki67+ and, thus, are actively in the cell cycle (Fig. 5E–H, Pax6; M–P, Tbr2). Thus, Wnt3a overexpression leads to premature differentiation of IPs to neurons and disorganization of progenitor distribution without completely depleting the RG and IP pool.
Figure 5.
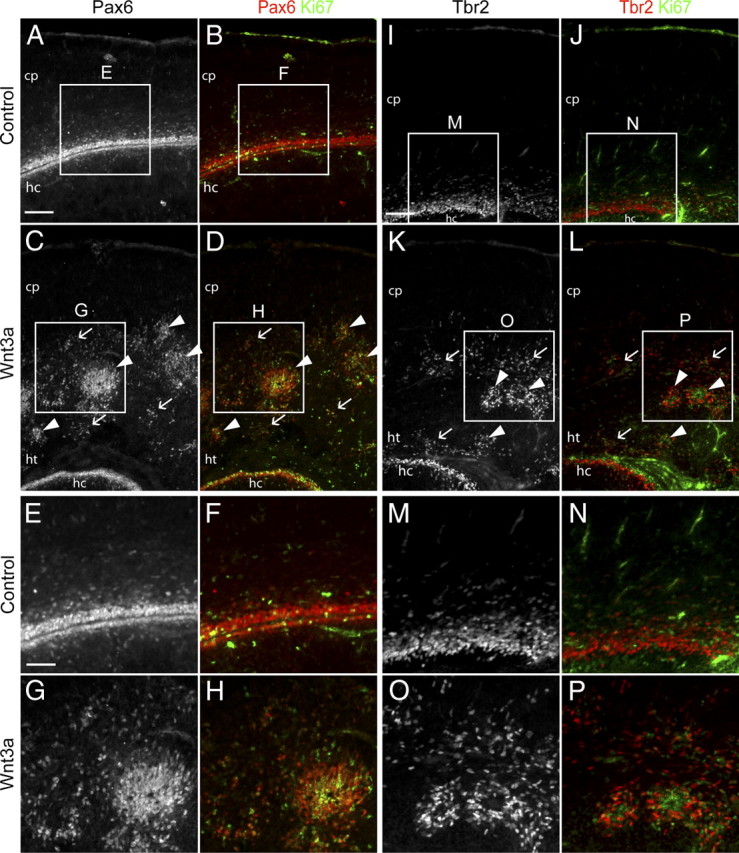
Wnt3a promotes disorganization of RG and IP distribution. Characterization of E18.5 brains electroporated at E13.5 by immunolabeling for progenitor markers. A–P, Immunolabeling for Pax6, Tbr2, and the active cell cycle marker Ki67 to show the distribution and cell cycle state of progenitors. C, D, K, L, Arrows indicate examples of scattered progenitors, and arrowheads indicate examples of progenitors in rosette formations. Higher-magnification pictures in E–H and M–P correspond to boxed areas in A–D and I–L and illustrate colocalization of Ki67 with Pax6 and Tbr2. cp, Cortical plate; ht, heterotopias; hc, hippocampus. Scale bars: A, 200 μm; E, 100 μm.
Dominant activation of Wnt signaling drives ectopic neuronal differentiation
To determine whether cell-autonomous activation of Wnt signaling leads to similar effects as ectopic Wnt3a expression, we tested whether a strong dominant-active form of LEF1 could elicit the same effects. The LEF/Tcf family of transcription factors transduce the transcriptional output of the Wnt–β-catenin pathway. We used a dominant-active form of LEF1 fused to the transactivation domain of the VP16 protein from the herpes simplex virus (LEF1–VP16). Consistent with the effects of Wnt3a electroporation, LEF1–VP16 promoted the differentiation of Tbr2+ IPs in the UTD 3 d after electroporation (Fig. 6A–P). The cohort of LEF1–VP16-electroporated cells did not express Tbr2 (Fig. 6B–D,F–H, arrowheads) or Ki67 but strongly expressed Ctip2 (Fig. 6I–P, arrows). We saw similar effects after expression of a dominant-active form of β-catenin (Δ29–48βcat) (supplemental Fig. S8A–H, arrows, available at www.jneurosci.org as supplemental material).
Figure 6.
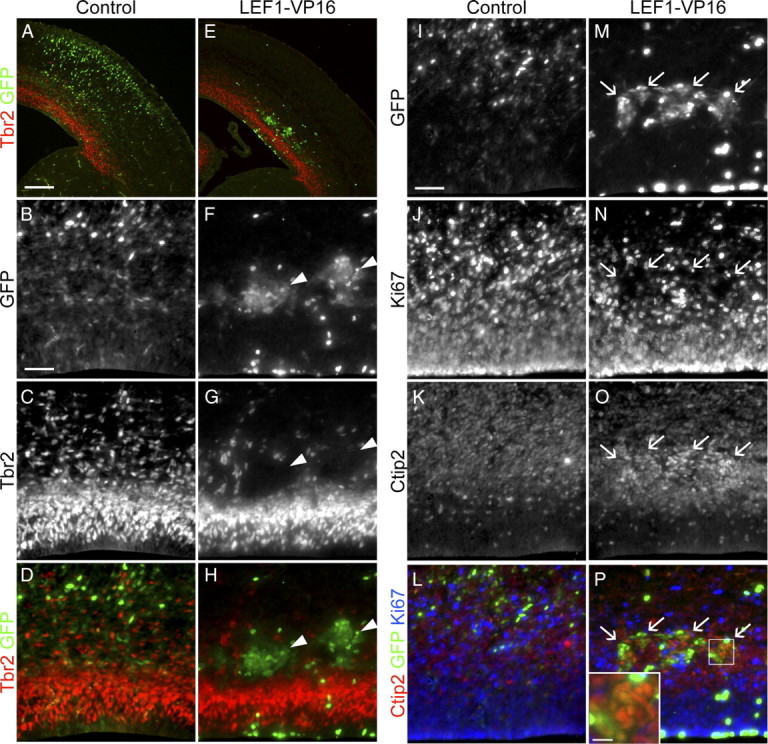
Dominant activation of Wnt signaling drives ectopic neuronal differentiation. Embryos electroporated at E13.5 with a dominant-active form of LEF1 (LEF1–VP16) were analyzed at E16.5 to test the effect of dominant activation of the Wnt–β-catenin pathway on the distribution and molecular identity of IPs. A, E, Immunolabeling for GFP and Tbr2 IP marker to determine the localization of electroporated cells. B–D, F–H, Higher-magnification images of samples A and E. Arrowheads in F–H indicate the positions of cell cohorts electroporated with LEF1–VP16 expressing GFP but not Tbr2. I–P, Triple immunolabeling for GFP, the active cell cycle marker Ki67, and the neuronal marker Ctip2. Arrows in M–P indicate cell cohorts electroporated with LEF1–VP16 coexpressing GFP and Ctip2 but not Ki67. Inset in P is a higher magnification of the boxed area in P for illustrating the colocalization of GFP and Ctip2. Scale bars: A, 200 μm; B, I, 50 μm; inset in P, 12.5 μm.
Dkk1 inhibits the production of neurons during mid and late neurogenesis
To determine whether Wnt signaling is required for the normal production of neurons in the neocortex, we ectopically expressed Dkk1, a secreted antagonist of the Wnt–β-catenin pathway coreceptor LRP6, and analyzed the production of neocortical neurons. We used Ctip2 to label early-born/deep layer neurons and Cux1 to label late-born/upper layer neurons. We then counted the numbers of neurons of layer V and layer IV–II, neurons primarily born after the time point of electroporation at E13.5. Ectopic expression of Dkk1 at E13.5 (Fig. 7A–H) inhibited the production of layer V neurons but not the production of layer IV–II neurons [Ctip2: control, n = 6 average ± SEM of 92 ± 7.06 cells/300 μm column; Dkk1, n = 5, average ± SEM of 69 ± 7.37 cells/300 μm column, pvalue = 0.027 (Fig. 7I); Cux1: control, n = 6, average ± SEM of 792 ± 23.45 cells/300 μm column; Dkk1, n = 5, average ± SEM of 744 ± 48.55 cells/300 μm column, pvalue = 0.186 (Fig. 7J)]. On one hand, this result suggests that Dkk1 might selectively regulate deep layer neuron production. However, we wondered whether instead the effects of Dkk1 were merely transient but general so that, if electroporated later, it would inhibit superficial neuron generation instead. To test this, we expressed Dkk1 at E15.5 (Fig. 7K–R) and found that it specifically inhibited production of layer IV–II [Ctip2: control, n = 6, average ± SEM of 108 ± 5.20 cells/300 μm column; Dkk1, n = 6, average ± SEM of 107 ± 3.80 cells/300 μm column, pvalue = 0.487 (Fig. 7S); Cux1: control, n = 6, average ± SEM of 595 ± 35.09 cells/300 μm column; Dkk1, n = 6, average ± SEM of 514 ± 21.86 cells/300 μm column, pvalue = 0.043 (Fig. 7T)]. These results suggest that inhibition of Wnt signaling by Dkk1 generally inhibits neurogenesis without specifically regulating different layer specificities and are consistent with the phenotype of the Wnt3a electroporation experiments: the agonist Wnt3a generates extra neurons, whereas the antagonist Dkk1 inhibits the production of neurons. Moreover, because IPs generate the majority of neurons after E15.5, our results further imply a role for the Wnt–β-catenin pathway in the regulation of IP behavior.
Figure 7.
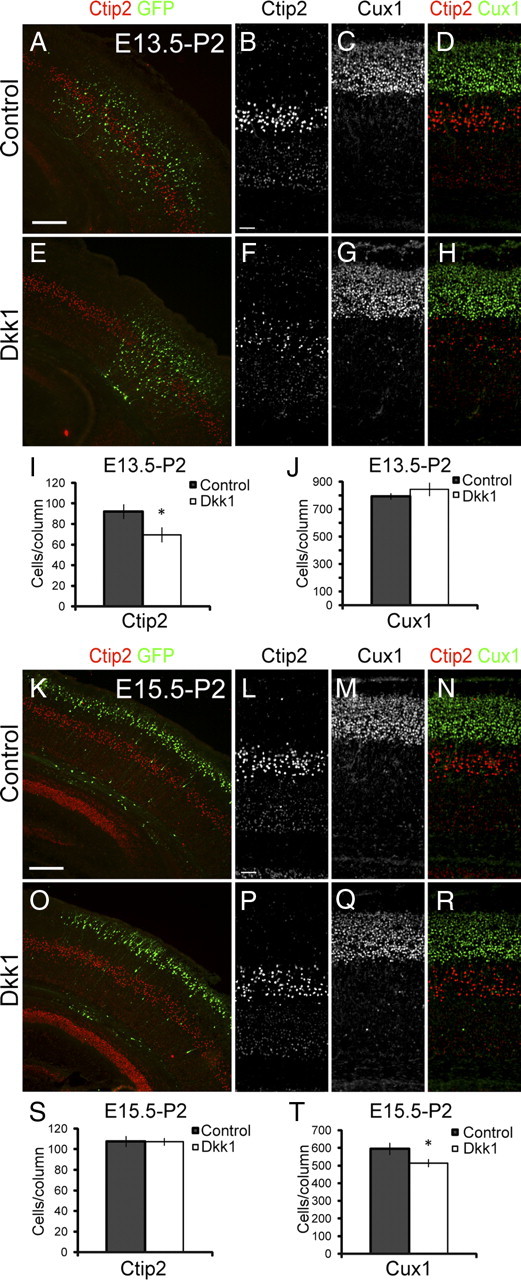
Dkk1 inhibits the production of neurons during mid and late neurogenesis. A–J, Brains electroporated at E13.5 with Dkk1 Wnt–β-catenin pathway inhibitor were analyzed at P2 to test the effect of Dkk1 starting at mid neurogenesis. A, E, Immunolabeling for Ctip2 and GFP shows distribution of electroporated cells. B–D, F–H, Higher magnification of samples A and E immunolabeled with Ctip2 and Cux1. I, Quantification of the numbers of Ctip2+ layer V neurons. J, Quantification of the numbers of Cux1+ layer IV–II neurons. K–T, Brains electroporated at E15.5 with Dkk1 were analyzed at P2 to test the effect of Dkk1 on late neurogenesis. K, O, Immunolabeling for Ctip2 and GFP shows distribution of electroporated cells. L–N, P–R, Higher magnification of samples K and O immunolabeled with Ctip2 and Cux1. S, Quantification of the numbers of Ctip2+ layer V neurons. T, Quantification of the numbers of Cux1+ layer IV–II neurons. Scale bars: A, K, 200 μm; B, L, 50 μm. Error bars represent SEM. *p < 0.05.
Discussion
The IP stem cell pool is critical for the generation of the correct numbers and subtypes of neurons during mammalian neurogenesis. In this study, we show that the Wnt–β-catenin pathway is active in neocortical IPs and required for production of projection neurons during mid and late stages of neurogenesis. We further show that the Wnt–β-catenin pathway plays a pivotal role in regulating IP differentiation and that dysregulation of the pathway can lead to cortical dysplasia and the formation of neuronal heterotopias.
The Wnt–β-catenin pathway regulates IP differentiation
Our data show that induction of the Wnt–β-catenin pathway with excess ligand leads to early differentiation of IPs into neurons. These neurons fail to migrate to the cortical plate and accumulate within the UTD (Fig. 8A). Together, these findings indicate that the Wnt–β-catenin pathway regulates the timing or progression of IP differentiation. These findings have not been described previously by other in vivo studies, perhaps because other studies relied solely on expression of intracellular signaling molecules that may have additional cell-autonomous consequences. Like our results, previous in vitro studies did show that Wnt–β-catenin signaling induces neuronal differentiation in neural precursor cell cultures (Hirabayashi et al., 2004). Moreover, the promoters of the proneuronal basic helix–loop–helix transcription factors, neurogenin1 (Ngn1) and neurogenin2 (Ngn2), contain LEF/Tcf elements and can be activated by β-catenin in vitro (Hirabayashi et al., 2004; Israsena et al., 2004), and loss-of-function mutation of β-catenin leads to loss of Ngn2 expression (Backman et al., 2005). In accord with these previous findings, our data further implicates the Wnt–β-catenin pathway in specifically regulating the timing or progression of differentiation of IPs into projection neurons.
Figure 8.

The Wnt–β-catenin pathway regulates RG self-renewal and IP differentiation. A, Schematic comparing the effect of ectopic Wnt3a to the normal distribution of IPs and newly born IP-derived neurons. The current understanding of IP behavior indicates that IPs differentiate as they migrate from the ventricular zone toward the cortical plate. Our results indicate that excess Wnt3a advances the maturation process of IPs, causing IPs to ectopically differentiate into neurons in the UTD. The resulting neurons fail to migrate to the cortical plate and accumulate in the UTD to form a neuronal heterotopia. B, Our results also show that the Wnt–β-catenin pathway (WβP) plays a dual role in neocortical neurogenesis, promoting RG self-renewal and IP differentiation.
Superficially, there appears to be a contradiction between the ample evidence that the Wnt–β-catenin pathway promotes symmetric progenitor proliferation during early and mid neurogenesis (Chenn and Walsh, 2002, 2003; Zechner et al., 2003; Woodhead et al., 2006; Wrobel et al., 2007) and the studies showing that, beginning at mid neurogenesis, the Wnt–β-catenin pathway promotes differentiation of neuronal progenitors (Hirabayashi et al., 2004; Israsena et al., 2004; Hirabayashi and Gotoh, 2005; Guillemot, 2007). Retroviral infection of RG with either S33Yβcat or Wnt7a led to enrichment of infected cells in the SVZ (Viti et al., 2003; Kuwahara et al., 2010). These affected cells could be arrested in the SVZ as a result of early differentiation of IPs, similar to what we have observed with our results. Thus, our results along with other studies suggest that the Wnt–β-catenin pathway regulates both self-renewal and differentiation but in different pools of progenitors: self-renewal in RG and differentiation in IPs (Fig. 8B).
Complementing our findings using excess Wnt ligand, we found that downregulation of endogenous Wnt–β-catenin signaling with Dkk1 inhibits neuronal production during mid and late stages of neurogenesis. This implies a required role for the Wnt–β-catenin pathway in the regulation of IP behavior, because IPs generate a large proportion of neurons during mid neurogenesis and may function as the only neuron-generating progenitor pool during late neurogenesis (Noctor et al., 2007, 2008). Downregulation of endogenous Wnt–β-catenin signaling with Dkk1 likely reduces the number of IPs that differentiate over a given time period, leading to less neurons produced over the course of neurogenesis.
The differential effects of both Wnt3a and LEF1–VP16 on the UTD subpopulation of IPs suggest that the DTD and UTD IP subpopulations have some distinctions. A dramatic loss of UTD IPs that coincided with the formation of ectopic neurons in the UTD was observed in Wnt3a-electroporated brains, but the size and proliferation level of DTD IPs were unaffected. Thus, it appears that there is specificity in the response of UTD IPs to Wnt–β-catenin signaling. We defined the subpopulations of Tbr2+ IPs into the DTD and UTD domains in this study based on the Wnt3a phenotype we observed because there are currently no known molecular markers that define the DTD and UTD IP subpopulations. However, based on our results, there must be some cellular distinctions between these subpopulations of IPs, although they may have a precursor–product relationship.
Excess Wnt signaling leads to cortical dysplasia and neuronal heterotopias
Long-term upregulation of the Wnt–β-catenin pathway with Wnt3a resulted in cortical dysplasia associated with the formation of large neuronal heterotopias. The heterotopias are composed of both early-born/deep layer and late-born/upper layer projection neurons and contained disorganized axonal (data not shown) and dendritic processes (supplemental Fig. S2F,H, available at www.jneurosci.org as supplemental material). These features are similar to those found in cortical malformations of patients (Fox and Walsh, 1999; Lu and Sheen, 2005; Lian and Sheen, 2006). Thus, our findings indicate possible roles for the Wnt–β-catenin pathway in cortical malformation disorders.
We observed several factors that likely contributed to these findings in the Wnt3a-electroporated brains. Ectopic differentiation of IPs in the UTD is most likely the direct factor for the formation of ectopic neurons in progenitor domains. We observed the stepwise expansion of the pool of ectopic neurons in progenitor domains from E16.5 to E18.5 to P2. Second, the abnormal horizontal expansion of RG, observed at E16.5, would eventually lead to extra IPs and neurons. To this end, a dramatically larger population of Tbr2+ IPs is present at P2 in Wnt3a-electroporated brains compared with controls. Third, we observed in some samples Ctip2+ nodules protruding at the ventricular surface of the neocortex of Wnt3a-electroporated brains at E16.5 (Fig. 2U–X, asterisks). These are likely RG that have directly differentiated into neurons within the VZ. Last, disorganization and reorganization of RG and IPs into rosettes, which we observed at E18.5 and P2, could lead to disruption of migration of daughter neurons to the cortical plate.
Other studies demonstrating that Wnt–β-catenin signaling induces differentiation of neocortex-derived neural precursor cell cultures (Hirabayashi et al., 2004) and regulate the expression of Ngn1/2 proneuronal genes (Hirabayashi et al., 2004; Israsena et al., 2004; Backman et al., 2005) support our conclusion that the primary consequence of Wnt–β-catenin pathway activation is the regulation of neocortical IP cell differentiation. However, the dramatic effect we see on the position of neurons raises the possibility that there is a direct role for canonical Wnt signaling on cell motility, as shown in other contexts (Moon et al., 1993; Silhankova and Korswagen, 2007). Upregulation of Wnt–β-catenin signaling might directly inhibit IP or newborn neuron migration. It is also possible that the expression of Wnt3a also has effects on the noncanonical Wnt signaling pathway, which can also have direct migratory consequences. Future experiments that can differentiate between direct and indirect effects on migration or the neural differentiation program will be necessary to clarify this matter.
In conclusion, we have defined new roles for the Wnt signaling pathway in controlling the terminal differentiation of IPs and shown that dysregulation of this signaling axis can lead to cortical malformations similar to those seen in some patients. This will make it attractive in the future to consider the Wnt signaling pathway as a target for mutations in the inherited disorders of cortical migration.
Footnotes
This work was supported by National Institutes of Health Grants K02 MH074958 and R01 MH066084, the National Multiple Sclerosis Society, and a generous gift endowment from the family of Glenn W. Johnson Jr.
References
- Backman M, Machon O, Mygland L, van den Bout CJ, Zhong W, Taketo MM, Krauss S. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol. 2005;279:155–168. doi: 10.1016/j.ydbio.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JW, Walsh CA. Periventricular heterotopia and the genetics of neuronal migration in the cerebral cortex. Am J Hum Genet. 1999;65:19–24. doi: 10.1086/302474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese JL, Pino D, Pleasure SJ. Wnt signaling in development and disease. Neurobiol Dis. 2010;38:148–153. doi: 10.1016/j.nbd.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli LM, Willert K, Nusse R, Yablonka-Reuveni Z, Nohno T, Denetclaw W, Burrus LW. A proliferative role for Wnt-3a in chick somites. Dev Biol. 2004;269:489–504. doi: 10.1016/j.ydbio.2004.01.041. [DOI] [PubMed] [Google Scholar]
- Guillemot F. Cell fate specification in the mammalian telencephalon. Prog Neurobiol. 2007;83:37–52. doi: 10.1016/j.pneurobio.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Hall AC, Lucas FR, Salinas PC. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT–7a signaling. Cell. 2000;100:525–535. doi: 10.1016/s0092-8674(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron. 2005;48:45–62. doi: 10.1016/j.neuron.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y. Stage-dependent fate determination of neural precursor cells in mouse forebrain. Neurosci Res. 2005;51:331–336. doi: 10.1016/j.neures.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development. 2004;131:2791–2801. doi: 10.1242/dev.01165. [DOI] [PubMed] [Google Scholar]
- Israsena N, Hu M, Fu W, Kan L, Kessler JA. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev Biol. 2004;268:220–231. doi: 10.1016/j.ydbio.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Kuwahara A, Hirabayashi Y, Knoepfler PS, Taketo MM, Sakai J, Kodama T, Gotoh Y. Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development. 2010;137:1035–1044. doi: 10.1242/dev.046417. [DOI] [PubMed] [Google Scholar]
- Lee SM, Tole S, Grove E, McMahon AP. A local Wnt–3a signal is required for development of the mammalian hippocampus. Development. 2000;127:457–467. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- Li G, Adesnik H, Li J, Long J, Nicoll RA, Rubenstein JL, Pleasure SJ. Regional distribution of cortical interneurons and development of inhibitory tone are regulated by Cxcl12/Cxcr4 signaling. J Neurosci. 2008;28:1085–1098. doi: 10.1523/JNEUROSCI.4602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian G, Sheen V. Cerebral developmental disorders. Curr Opin Pediatr. 2006;18:614–620. doi: 10.1097/MOP.0b013e328010542d. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Lu J, Sheen V. Periventricular heterotopia. Epilepsy Behav. 2005;7:143–149. doi: 10.1016/j.yebeh.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Machon O, Backman M, Machonova O, Kozmik Z, Vacik T, Andersen L, Krauss S. A dynamic gradient of Wnt signaling controls initiation of neurogenesis in the mammalian cortex and cellular specification in the hippocampus. Dev Biol. 2007;311:223–237. doi: 10.1016/j.ydbio.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–2098. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Moon RT, DeMarais A, Olson DJ. Responses to Wnt signals in vertebrate embryos may involve changes in cell adhesion and cell movement. J Cell Sci Suppl. 1993;17:183–188. doi: 10.1242/jcs.1993.supplement_17.26. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Kriegstein AR. Contribution of intermediate progenitor cells to cortical histogenesis. Arch Neurol. 2007;64:639–642. doi: 10.1001/archneur.64.5.639. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Kriegstein AR. Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J Comp Neurol. 2008;508:28–44. doi: 10.1002/cne.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2005;8:34–42. doi: 10.1038/nn1374. [DOI] [PubMed] [Google Scholar]
- Silhankova M, Korswagen HC. Migration of neuronal cells along the anterior-posterior body axis of C. elegans: Wnts are in control. Curr Opin Genet Dev. 2007;17:320–325. doi: 10.1016/j.gde.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Viti J, Gulacsi A, Lillien L. Wnt regulation of progenitor maturation in the cortex depends on Shh or fibroblast growth factor 2. J Neurosci. 2003;23:5919–5927. doi: 10.1523/JNEUROSCI.23-13-05919.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead GJ, Mutch CA, Olson EC, Chenn A. Cell-autonomous β-catenin signaling regulates cortical precursor proliferation. J Neurosci. 2006;26:12620–12630. doi: 10.1523/JNEUROSCI.3180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel CN, Mutch CA, Swaminathan S, Taketo MM, Chenn A. Persistent expression of stabilized beta-catenin delays maturation of radial glial cells into intermediate progenitors. Dev Biol. 2007;309:285–297. doi: 10.1016/j.ydbio.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner D, Fujita Y, Hülsken J, Müller T, Walther I, Taketo MM, Crenshaw EB, 3rd, Birchmeier W, Birchmeier C. beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol. 2003;258:406–418. doi: 10.1016/s0012-1606(03)00123-4. [DOI] [PubMed] [Google Scholar]
- Zhou CJ, Borello U, Rubenstein JL, Pleasure SJ. Neuronal production and precursor proliferation defects in the neocortex of mice with loss of function in the canonical Wnt signaling pathway. Neuroscience. 2006;142:1119–1131. doi: 10.1016/j.neuroscience.2006.07.007. [DOI] [PubMed] [Google Scholar]