

Abstract
Nonstop proliferation and vigorous neovascularization are two prominent characteristics of cancer. Antiangiogenic therapy has emerged as an important modality in treatment of solid tumors. Our previous work demonstrated that microparticles derived from apoptotic T-lymphocytes (LMPs) not only reduced the viabilities of high-proliferating cells, but also exhibited potent antiangiogenic effects through inhibition of the vascular endothelial growth factor (VEGF)/VEGF receptor 2 signalling pathway. In the present study, we extended these studies to explore the anticancer potential of LMPs using a murine model of Lewis lung carcinoma (LLC). Results show that intratumoral injection of LMPs (2.5 mg/kg) decreased tumor size by more than 50% relative to control. Tumor microvessel density and VEGF-A levels were also markedly reduced upon LMPs treatment. To elucidate the underlying mechanisms of LMPs-mediated antitumor activity, LLC cells were utilized in in vitro experiments. LMPs suppressed VEGF-A protein levels in LLC cells and led to inhibition of LLC cell viability and proliferation. In addition, knockdown of the low-density lipoprotein receptor (LDLR) expression reduced the uptake of LMPs into LLC cells and attenuated the inhibitory effects of LMPs on cell growth and VEGF-A expression. Our findings demonstrate that LMPs exert antiangiogenic and proapoptotic effects that lead to inhibition of lung carcinoma by reducing VEGF-A levels and LDLR mediates the anti-VEGF effect of LMPs through translocating LMPs into LLC cells. These results suggest that LMPs are promising antiangiogenic therapeutic agent and represent a new therapeutic strategy for treating lung carcinomas.
Key words: lewis lung carcinoma, lymphocyte-derived microparticles, VEGF-A, low density lipoprotein receptor, angiogenesis, cell viability
Introduction
Cellular microparticles (MPs) are small membrane vesicles released from the plasma membrane of virtually all cell types upon activation or apoptosis. During their formation, lipid rafts and their accompanying cholesterol and proteins accumulate where the membrane buds. MPs constitute a heterogeneous population of submicron elements differing in cellular origin, number, size, antigenic composition and functional properties.1 They have been identified as vectors of the intercellular exchange of biologic information, such as induction of endothelial modifications, angiogenesis or differentiation.2 Consistent with these properties, we have demonstrated that Lymphocyte-derived microparticles (LMPs) inhibit endothelial cell proliferation, migration and angiogenesis by antagonizing the vascular endothelial growth factor (VEGF)/VEGF receptor 2 (VEGFR2) pathway.3
Cancer is a disease manifested by uncontrolled cell growth that presents over 100 distinct clinical pathologies. All malignant solid tumors depend on angiogenesis for growth to a clinically relevant size, and for invasion and metastasis.4 Tumors activate the angiogenic switch by increasing expression of proangiogenic factors such as VEGF—a key mediator of tumor angiogenesis and endothelial cell survival, proliferation and motility.5,6 Overexpression of VEGF has been associated with poor prognostic outcome for patients with cancer.7 Thus, VEGF or tumor angiogenesis offer uniquely attractive therapeutic targets that are shared by most, and perhaps all types of human cancers.5,8–10
Mammalian cells obtain cholesterol necessary for membrane or steroid hormone synthesis by de novo synthesis or uptake via the low-density lipoprotein receptor (LDLR).11 Although cells rely predominantly on the LDLR pathway for their cholesterol needs,12 cancer cells in particular require large amounts of cholesterol for cell membrane synthesis when cell replication is rapid and uncontrolled.13 Moreover, numerous studies have suggested an inverse relationship between cholesterol levels and cancer risk.14–17 Several lines of evidence have also supported a link between LDLR and carcinomas. First, in numerous carcinoma cell lines, LDLR activity is increased during the growth phase and decreased in quiescent cells.13 Second, some malignant cell lines exhibit higher LDLR activity than nonneoplastic cells.18–21 Third, tumor growth is suppressed in LDLR-deficient mice.22 However, to date, no studies have investigated the role of LDLR in the uptake of membrane microparticles, including LMPs, into cancer cells.
Here, we sought to examine the effect of LMPs on tumorigenesis in the mouse Lewis lung carcinoma (LLC) model and in LLC cells. Two reasons facilitated this approach: the LLC model is largely dependent on angiogenesis and its tumors are known to grow very rapidly in syngenic mice, while lung cancer—the most common cancer worldwide—is one of the few with an increasing incidence and for which effective treatments are limited.23 The current findings reveal that LMPs significantly diminish LLC tumor growth by inducing microvascular degeneration and inhibiting VEGF-A expression; LDLR plays an important role in this process by facilitating the uptake of LMPs into cancer cells.
Results
LMPs inhibit tumor growth in LLC-bearing mice.
Our first objective was to investigate the antitumor efficacy of LMPs in the C57BL/6 Lewis lung carcinoma (LLC) model. In the first experiment, we investigated the effect of LMPs on tumorigenesis. We injected subcutaneously into the flanks of C57BL/6 mice with 0.5 × 106 LLC cells combined with PBS or LMPs and measured tumor weights 10 days later. Compared with control (LLC cells in PBS), tumor size was reduced by an average of 60% in mice treated with a combination of LLC cells and 50 µg LMPs (p < 0.001) (Fig. 1A and B). To further ensure this phenotype, 0.5 × 106 LLC cells were inoculated subcutaneously for 7 days (tumors reached approximately 30 mm2), and 4 consecutive intratumoral injections with LMPs (2.5 mg/kg) were performed. Similarly, tumor weight was decreased by 50% in LLC-bearing mice treated with LMPs compare to control group receiving PBS; (p < 0.05 vs. control) (Fig. 1C and D).
Figure 1.

Inhibitory effects of LMPs on tumor growth in LLC-bearing mice. (A and B) C57BL/6 female mice were sacrificed after 10 days s.c. inoculation with 0.5 × 106 LLC cells in PBS or 50 µg LMPs. Primary tumors were collected and weighed. Representative images (A) and quantifications (B) are presented. (C) After s.c. inoculation with 0.5 × 106 LLC cells for 7 days, C57BL/6 mice were intratumorly injected with 50 µL PBS (control) or 2.5 mg/kg LMPs every 2 days for 4 consecutive injections. Tumors were collected on day 14 and weighed. Representative images of the tumors are shown in (C) and quantification of tumor weight in (D). In each case, values are depicted as mean ± SEM of 8 (B) or 5 (D) mice per group. *p < 0.05, ***p < 0.001 vs. control.
LMPs decrease microvessel density and VEGF-A expression in LLC tumors.
Having recently demonstrated that LMPs potently inhibit angiogenesis3—an essential process for solid tumor growth and progression,4 we next evaluated the in vivo effect of LMPs on tumor microvasculature. Results show that microvessel density in established tumors, as revealed by TRITC-lectin staining, was approximately 40% lower in LMPs-treated tumors compared with control (Fig. 2A and B, p < 0.05). Tumor levels of the major proangiogenic factor, VEGF-A, were also significantly reduced upon LMPs treatment (Fig. 2C, p < 0.01).
Figure 2.

Effect of LMPs on LLC tumor angiogenesis in vivo. (A) Representative images of microvessels in the tumors developed from the mice received LLC and LLC + LMPs injection subcutaneously, stained with TRITC-lectin. (B) Microvessel density was quantified in three different fields (400x). (C) VEGF-A levels were measured via ELISA from fresh frozen LLC tumor lysates and normalized to protein concentrations. Values are presented as percentage of control (set to 100%). *p < 0.05, **p < 0.01 vs. control.
LMPs reduce LLC cell viability by inhibiting proliferation and inducing apoptosis.
Because high growth rate is a prominent feature of cancer cells, we investigated the effects of LMPs on LLC cell integrity and putative in vitro mechanisms. LMPs dose-dependently reduced LLC cell viability and proliferation as assessed by MTT assay and [3H]-thymidine DNA incorporation assay, respectively (Fig. 3A and B, p < 0.001). To determine whether these anti-proliferation effects were associated with increased apoptosis, two techniques were used. In the annexin-V binding assay, apoptotic cells, as characterized by annexin V positivity, increased from approximately 7% in control to 16% in LMPstreated cells, the induction was small but significant (Fig. 3C, p < 0.001). These results were confirmed by a DNA fragmentation assay, which revealed the characteristic pattern of DNA laddering in total DNA from LMPs-treated LLC cells; DNA laddering was absent in control cells (Fig. 3D).
Figure 3.
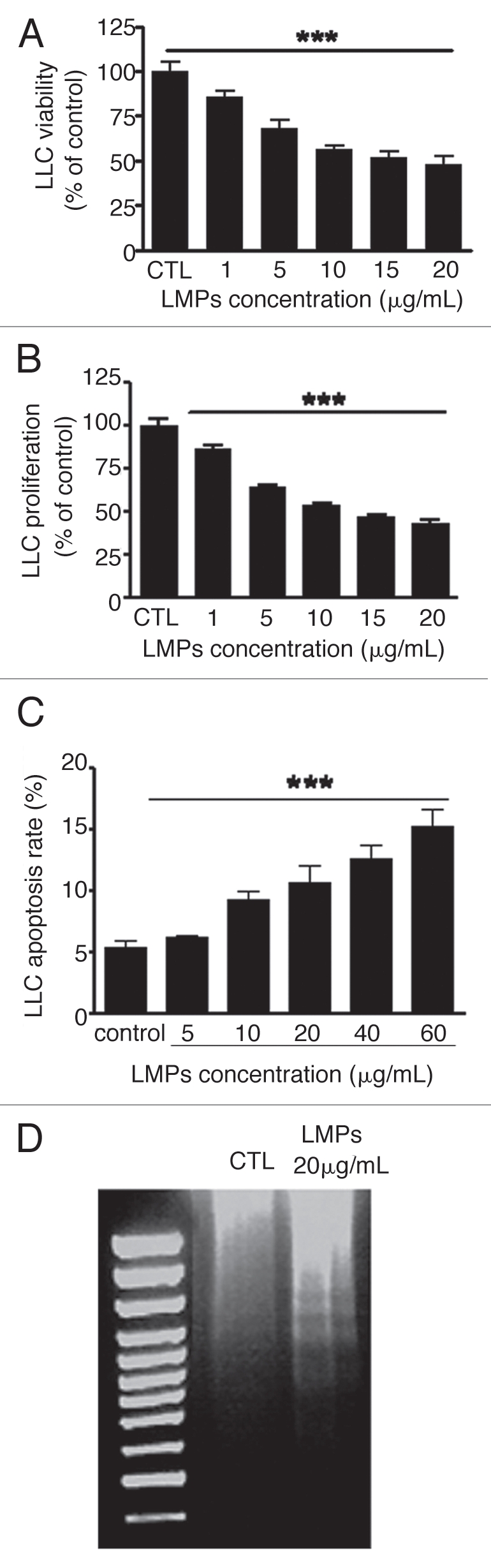
Effects of LMPs on LLC cell viability, proliferation and apoptosis. LLC cell viability (A) and proliferation (B) was measured after 24-h incubation with indicated concentrations of LMPs. (C) Apoptosis was determined by flow cytometry after LMPs treatment (24 h) and expressed as the percentage of apoptotic cells relative to the total number of cells per condition. (D) Representative image of DNA fragmentation assay performed in LLCs with or without 20 ug/mL LMPs. Values are depicted as means ± SEM of 3–5 independent experiments performed in duplicate (C) or triplicate (A and B). The control group was set to 100%. ***p < 0.001 vs. control.
LMPs reduce VE GF expression and VE GF-induced migration in LLC cells.
To assess whether LMPs interfere with VEGF-A expression in vitro, VEGF-A protein concentrations were measured via ELISA in LLC cells treated for 24 h with 20 µg/mL LMPs. LMPs significantly reduced VEGF-A levels in both culture medium (p < 0.01) and cell lysates (p < 0.05) (Fig. 4A). The effect of LMPs on VEGF-induced LLC cell migration was also examined; data show that cell migration substantially decreased by more than 60% after LMPs treatment (10 µg/mL, 72 h) (Fig. 4B and C, p < 0.001). As shown in Figure 4B, there was almost no cell migrating off the coverslip without additional VEGF, which suggested that LLC cell migration is VEGF-dependent.
Figure 4.
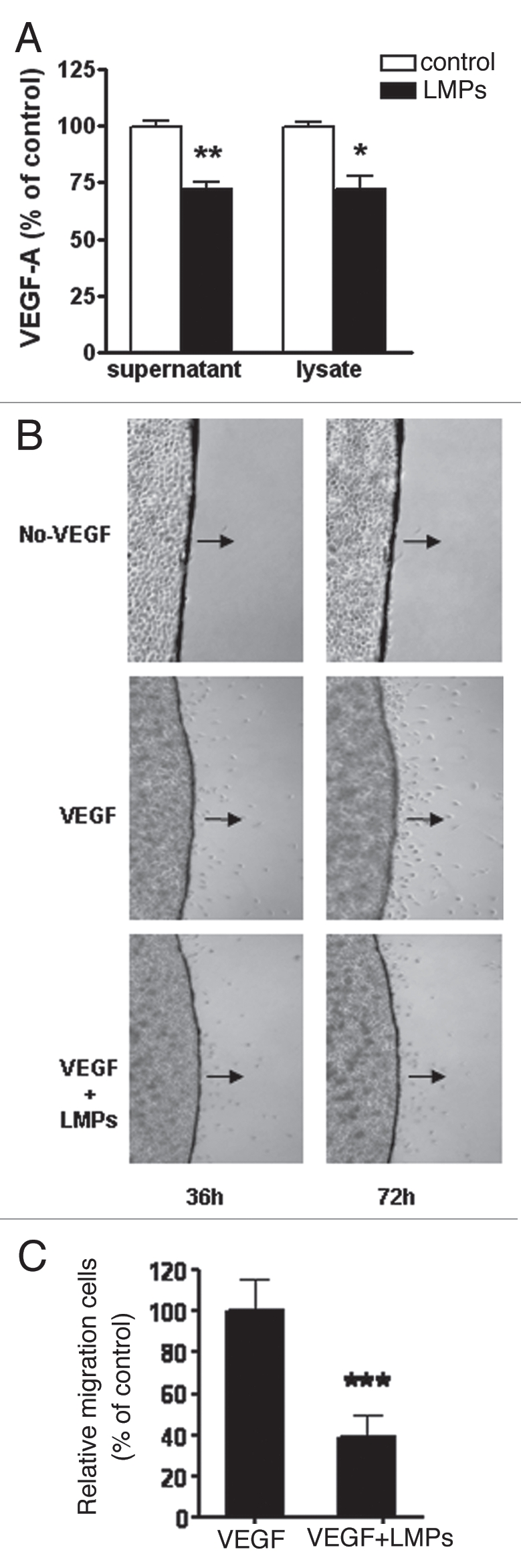
Effect of LMPs on VEGF-A expression and VEGF-induced cell migration. (A) After LMPs treatment (20 µg/mL, 24 h), VEGF-A levels in LLC culture medium and cell lysates were measured by ELISA and normalized to protein concentrations. The values in the control group were set to equal 100%. Values are means ± SEM of 3–5 individual experiments, each performed in triplicate. *p < 0.05, **p < 0.01 vs. control. (B) Representative images of VEGF-induced LLC cell migration in the presence or absence of LMPs (10 µg/mL). Photographs were taken 36 h and 72 h after LMPs treatment. Black arrows indicate direction of cell migration. Images (4x) are from three independent experiments performed in duplicate. (C) Cell migration at 72 h was quantified by MTT assay and is presented as the relative cell migration rate compared to VEGF alone. *p < 0.05.
Knockdown of LDLR expression decreases LMPs uptake into LLC cells.
Studies using Jurkat T-cell-derived MPs have shown that LMPs are transferred to macrophages by endocytosis.24 In order to elucidate whether similar mechanisms were operative in our experiments, we generated DiI-labelled LMPs by incubating apoptotic CEM T cells with the fluorescent lipophilic membrane dye DiI. Following exposure of LLC cells to DiI-LMPs (10 µg/mL, 1 h) distinct DiI staining was detectable by spectrofluorometery and increased in a time-dependent fashion (Fig. 5A and B). Fluorescent microscopy revealed the presence of DiI-labeled microparticles along the cell membrane and in intracellular compartments. To exclude the possibility that DiI was being released from stained LMPs, LLC cells were incubated with microparticle-free supernatant obtained from the last DiI-LMPs wash. Microparticle-free supernatant did not yield DiI-positive LLC cells (data not shown). Insights into the mechanisms of LMPs uptake into LLC cells were further explored via LDLR knockdown experiments. LDLR is a key receptor involved in the endocytosis of cholesterol and other ligands.12 We tested the inhibitory efficiency of siRNA-LDLR by western blot; transfection of LLC cells with 40 nM siRNA-LDLR decreased LDLR protein expression by 60% (Fig. 5C and D). This concentration was used in subsequent experiments. siRNA-LDLR transfected LLC cells were then incubated with DiI-LMPs for 24 h followed by preparation of membrane and cytosolic fractions for spectrofluorometer readings. As shown in Figure 5E and F, downregulation of LDLR expression significantly reduced DiI-LMPs uptake and distribution in both membrane and cytosolic compartments (p < 0.05).
Figure 5.

Influence of LDLR on the uptake of LMPs into LLC cells. (A) Representative fluorescent images of DiI-LMPs uptake into LLC cells. Photos were taken after LLC cells were incubated with 20 µg/mL DiI-LMPs at different time points (10x and 40x magnification in upper and lower parts, respectively). (B) Uptake of DiI-LMPs into LLC cells was evaluated by spectrofluorometry and presented as mean fluorescent intensity (MFI). (C) Efficiency of LDLR downregulation via siRNA in LLC cells. LDLR protein expression was determined by western blot in LLC cells transfected with or without siRNA-LDLR and (D) presented as percentage of control (scramble siRNA). (E and F) Incubation of DiI-LMPs with LLC cells transfected with siRNA-LDLR for 24 h. Fluorescence intensity of DiI-LMPs was assessed in intact cells (E) and in membrane or cytosolic fractions (F) and expressed as a percentage of control (scramble siRNA). *p < 0.05, *p < 0.01 vs. control.
LDLR mediates the antitumor effects of LMPs in LLC cells.
Results thus far implicated LDLR in the uptake of LMPs. To test whether LDLR also mediated the tumor inhibitory effects of LMPs, VEGF-A protein expression was analyzed by western blot in siRNA-LDLR transfected LLC cells. Treatment with LMPs suppressed VEGF-A protein expression (p < 0.001); this effect was partially, yet significantly, attenuated in siRNA-LDLR transfected LLC cells (Fig. 6A and B, p < 0.05). In addition, knockdown of LDLR expression abrogated the inhibitory effects of LMPs on cell viabilty (Fig. 6C, p < 0.05). These data were confirmed following blockade of LDLR expression using an anti-LDLR antibody; preincubation of LLC cells with 15 µg/mL anti-LDLR antibody significantly prevented the effect of LMPs on cell growth (Fig. 6D, p < 0.05 vs. LMPs).
Figure 6.

Interfering LMPs effect by LDLR. (A) LLC cells were transfected with siRNA-LDLR subsequent to treatment with LMPs (20 µg/mL). VEGF-A protein expression was determined by western blot analysis and normalized to β-actin. Values are presented relative to control (scramble siRNA without LMPs treatment). (B) Data are presented as means ± SEM. #p < 0.05 vs. scramble siRNA + LMPs, ***p < 0.001 vs. scramble siRNA. (C) Cell viability was assessed in LLC cells transfected with siRNA-LDLR and treated with 20 µg/mL LMPs for 24 h. Data are presented as percentage of control (scramble siRNA). #p < 0.05 vs. control + LMPs. (D) Cell viability was assessed in LLC cells preincubated with 15 µg/mL LDLR antibody followed by LMPs treatment (20 µg/mL, 24 h). Data are presented as percentage of control. #p < 0.05 vs. LMPs, ***p < 0.001 vs. CTL.
Discussion
Although MPs were first described as “cell dust” or “inert cell debris”,25 in recent years, their biological importance has emerged. Mounting evidence suggests that MPs are potent biological agents implicated in exchanging biological signals, interacting with target cells and inducing both beneficial and detrimental responses. This is due to their ability to regulate genes involved in inflammation, oxidative stress and vascular function (reviewed in ref. 2). While we and others have demonstrated an important role for LMPs in regulating angiogenesis, little is known regarding the effects of LMPs on tumorigenesis (Fig. 1). The present study investigated the antitumor efficacy of LMPs in the LLC tumor model.
Malignant growth of tumor cells depends on deregulation of transcription factor activity to maintain the transformed state. Tumors appear to activate the angiogenic switch by changing the balance of angiogenesis inducers and countervailing inhibitors.5 In the clinic, agents that block VEGF signalling have shown promising results against various types of tumors.26 Clinical trials have also revealed a significant correlation between VEGF expression and lung cancer prognosis.27–29 In our study, LMPs significantly inhibited LLC tumor growth and microvessel density and limited local production of VEGF-A (Fig. 2). Mechanistic aspects of these in vivo findings were delineated through experiments using LLC cells. Here, LMPs decreased LLC cell viability and proliferation (Fig. 3A and B) with associated increases in apoptosis (Fig. 3C and D). These anti-proliferative effects were also observed in other cancer cell lines including Hela, human breast cancer (MCF-7, M4A4) and neuroblastoma (data not shown).
In neoplasia, increased production of VEGF is responsible for maintaining vascular integrity and regulating cell proliferation and migration of neoplastic cells.6,30,31 Our data support this premise and show that LMPs inhibited tumor activity in a VEGF-A-dependent manner. Indeed, LMPs significantly limited VEGF-A expression in both LLC cell culture medium and intracellular components (Fig. 4A) and reduced VEGF-induced cell migration (Fig. 4B and C). Moreover, we also observed the presence of VEGF receptor 2 on LLC cells (data not shown), which suggests that VEGF has an autocrine function in which LLC act as both VEGF producers and target cells. As expected, blocking VEGF-A activity by anti-VEGF antibody strongly decreased cell proliferation, however, co-treatment with LMPs and antibody of VEGF did not result in a synergistic reduction of cell growth (unpublished data). Taken together, these data support the notion that LMPs suppress LLC tumor growth in vivo by targeting tumor angiogenesis and cell growth by interfering with the VEGF-A pathway.
Although there is a large body of evidence suggesting that MPs can be transferred between cells, the mechanism of MPs transfer differs between various acceptor cell types,32 while the specific mechanisms by which MPs are taken up by lung carcinoma cells remains to be established. Phosphatidylserine on apoptotic cells is recognized by the phosphatidylserine receptor (PSR) and allows the recognition and removal of apoptotic cells by macrophages.33–36 Recent studies using MPs released from Jurkat T-cells have demonstrated that LMPs are transferred to macrophages by endocytosis.24,37 In our study, LMPs showed annexin V positivity, thus indicating the presence of phosphatidylserine on their surface. However, because the PSR antibody (mAb 217) did not inhibit LMPs uptake into LLC cells (data not shown), PSR was not implicated in this process. Taking into account the plasma membrane components of LMPs and the physiological function of LDLR—which is highly expressed in lung cancer cells,20 we hypothesized that LDLR was involved in the uptake of LMPs into LLC cells. This hypothesis was corroborated by the following observations: (1) LMPs were first identified in the plasma membranes of LLC cells, which seemed to be the primary site of action. With time, LMPs accumulated in the cytoplasm (Fig. 5A); (2) Downregulation of LDLR expression in LLC cells significantly dampened the transfer of LMPs to both membrane and cytoplasm components (Fig. 5 E and F); (3) Modulation of LDLR levels in LLC cells altered the effect of LMPs on VEGF-A expression (Fig. 6A and B); (4) Downregulation of LDLR expression or blockade of LDLR activity with an antibody attenuated the inhibitory effect of LMPs on cell viability (Fig. 6C and D).
In conclusion, we demonstrate that the anti-neoplastic effect of LMPs may extend beyond angiostasis. LMPs inhibited tumor cell migration and proliferation and reduced tumor cell production of VEGF. In addition, the antitumor effects of LMPs in lung carcinoma cells are, at least in part, dependent on LDLR activity. Spontaneous T-cell infiltration into human cancers is increasingly recognized as a favourable prognostic sign,38–40 although the concentration of microparticles derived from T cells in LLC tumor tissues is far below the effective doses (unpublished data), we hypothesize that LMPs transfer into tumor cells may suppress proangiogenic and progrowth response under pathophysiological conditions. In these settings, enhanced generation of LMPs may counterbalance endogenous and exogenous VEGF signals, but it does not exclude the possibility that LMPs modulate other proangiogenic factors such as keratinocyte-derived chemokine, basic fibroblast growth factor (bFGF), etc. Although our study implicates LDLR in the uptake of LMPs, the components of LMPs involved in triggering this process, as well as the specific receptors and subsequent events mediating LMPs antitumor effects remain to be elucidated.
Materials and Methods
Antibodies and reagents.
VEGF-A rabbit polyclonal antibody, LDLR antibody (sc-11824), mouse LDLR silencing RNA (siRNA; sc-35803) and scramble siRNA (Santa Cruz Biotechnology, CA); Actinomycin D, 3-(4,5-dimethyl thiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) and TRITC-lectin (Sigma Aldrich); [3H]-thymidine (Amersham, Mississauga, Ontario, Canada); optimal cutting temperature (OCT) medium (Sakura Finetek, Torrance, CA); vybrant apoptosis assay kit, fluorescent microbeads (1 µm) and 1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine (DiI; Molecular Probes, Eugene, OR, USA); mouse VEGF-A ELISA (RayBiotech, Inc., Norcross, GA).
Cell culture and production of LMPs.
Mouse LLC cells (LLC1, CRL-1642) were purchased from ATCC (Manassas, USA) and cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco BRL, Long Island, NY) supplemented with 10% FBS, 100 units/mL penicillin and 100 µg/mL streptomycin. LMPs were generated and characterized as described previously.3 Briefly, CEM T cells were treated with 0.5 µg/ml actinomycin D for 24 h, and a supernatant was obtained by centrifugation at 750 g for 15 min and then at 1,500 g for 5 min to remove cells and large debris. MPs from the supernatant were washed after three centrifugation steps (50 min at 12,000 g) and recovered in saline or basic cell culture media. Washing medium from the last supernatant was used as control. LMPs were characterized with annexin V staining by fluorescence-activated cell sorting (FACS) analysis. The concentrations of LMPs were determined using the Bio-Rad protein assay. For fluorescent DiI labelled LMPs (DiI-LMPs), DiI was added to CEM T cells 24 hours (h) before actinomycin D treatment.
Tumor growth and treatment in LL C-bearing mice.
Six-week old female C57BL/6 mice were purchased from Charles River (St-Constant, Quebec, Canada) and used according to a protocol approved by the Animal Care Committee of the Research Center of CHU Sainte-Justine. Mice were housed in a room maintained at 25 ± 1°C with 55% relative humidity and given food and water ad libitum. After 3 days, mice were anesthetized and used in 2 independent experiments. In the first experiment, mice were randomly selected to receive subcutaneous (s.c.) injections of 0.5 × 106 LLC cells (in 50 µL PBS) (control group) or 0.5 × 106 LLC cells plus 50 µg LMPs (treatment group) on the right flank. After 10 days, mice were sacrificed and primary tumors collected and weighed. In a second experiment, mice were s.c. inoculated with 0.5 · 106 LLC cells on the right flank. One week later, mice were randomly grouped to receive intratumoral injection: control group—50 µL PBS; treatment group—2.5 mg/kg LMPs (in 50 µL PBS) every 2 days for 4 consecutive injections. Mice were sacrificed 15 days post-LLC inoculation; locally growing tumors were separated from skin and muscles, weighed and snap frozen in liquid nitrogen. During and after each treatment, mice were observed ≥3 times weekly for signs of toxicity (lethargy, bloating, ruffling of fur).
Analysis of tumor vasculature.
Tumors were fixed with paraformaldehyde, embedded in OCT medium and sectioned (∼15 µm thick) with a cryostat (Microm International, HM500 O). Sections were fixed in cold (−20°C) methanol for 10 minutes. After 2 washes (1% PBS-Triton), sections were blocked (1% PBS-Triton/3% BSA) and incubated overnight with the microvascular specific marker—TRITC-conjugated lectin (1:100 in 0.1% PBS-Triton/3% BSA/4oC). Sections were then washed with 0.1% PBS-Triton (3 times), counterstained with DAPI and visualized by epifluorescent microscopy. Microvessel density was defined as the number of TRITC-lectin microvessels per 200X field. Vessel counts were determined using Image-Pro Plus 4.5 software;41,42 average counts from 3 different fields was used for statistical evaluation.
Quantification of VE GF-A in tumor and LLC cell lysates.
Quantification of VEGF-A in tumor supernatants, LLC culturing medium and LCC cell lysates was achieved using commercial VEGF-A ELISA kits according to the manufacturer's instructions. Tumor supernatants were obtained by mincing fresh frozen tumor tissue followed by centrifugation at 400 g (10 min). LLC culture media and cell lysates were obtained from LLC cells treated with 20 µg/mL LMPs for 24 h. VEGF-A levels were normalized to protein concentrations.
Cell viability and proliferation assays.
Cells at approximately 60% confluence were incubated for 24 h with vehicle or the indicated concentrations of LMPs. Cell viability was estimated by mitochondrial-dependent reduction of MTT as described previously. 3 LLC cell proliferation was evaluated by [3H]-thymidine incorporation assay. Briefly, 4 × 104 LLC cells were plated into 24-well plates, serum starved (24 h) and thereafter cultured in complete medium containing different concentrations of LMPs for an additional 24 h.
Cell migration assay.
Cell migration was determined using a coverslip border migration assay.3 Briefly, 0.5 × 106 LLC cells were seeded onto 12 mm coverslips in a 24-well plate. Cells were serum starved for 4 h and proliferation was inhibited by adding 10 µg/mL mitomycin C for 30 min. Next, coverslips were carefully removed, washed with fresh media and transferred into a 12-well plate containing 10 ng/mL VEGF in the presence or absence of 10 µg/mL LMPs. Images were captured after 36 h and 72 h using an Axiovert 200M inverted microscope (Zeiss). After 72 h, coverslips were removed and the proportion of migrated cells quantified by MTT assay.
Apoptosis and DNA fragmentation analysis.
LLC cells were treated with indicated concentrations of LMPs for 24 h, followed by incubation with reagents from the Vybrant Apoptosis Assay Kit. Apoptosis was determined by flow cytometry according to the manufacturer's protocol and expressed as the percentage of apoptotic cells relative to the total number of cells per condition. For the DNA fragmentation assay, LLC cells were seeded at 60% confluence, incubated with 20 µg/mL LMPs and harvested after 24 h. DNA was isolated as described.43 DNA fragmentation was assessed by resolving 20 µg DNA per sample. Electrophoresis was performed on a 1.6% agarose gel containing ethidium bromide and visualized with a UV illuminator.
Downregulation of LDLR with siRNA.
LLC cells were grown to 50% confluence and transfected using Lipofectamine 2,000 with scrambled siRNA or sequence-specific siRNA targeting LDLR (Santa Cruz Biotechnology); 40 nM siRNA-LDLR showed 60% knock-down efficiency, therefore this concentration was used in subsequent DiI-LMPs uptake experiments.
Uptake of DiI-labelled LMPs (DiI-LMPs) into LLC cells.
LLC cells (60 × 103) were seeded onto 12-well plates with coverslips. The following day, cells were incubated with 20 µg/mL DiI-LMPs for indicated time periods. After treatment, one set of cells was fixed and nuclei were counterstained with DAPI (1:3,000, 5 min). Bound or ingested cells were detected by red fluorescence and observed using a Nikon eclipse E800 epifluorescent microscope with Nikon digital camera DXM 1200. Photos were taken at 10x and 40x magnification. A second set of cells was collected for spectrofluorometer readings (SPECTRAmaxGEMINI XS, Molecular Devices) and the mean fluorescent intensity (MFI) was determined. To investigate whether LDLR mediated the uptake of LMPs, 20 µg/mL DiI-LMPs were added to LLCs transfected with siRNA-LDLR for 24 h. DiI fluorescence intensity was then measured in intact cells and in membrane and cytosolic fractions. Cytosolic and membrane fractions were prepared from a protein extraction kit according to the manufacturer's instructions (ZmTech Scientific Company, Montreal, Canada).
Western blot analysis.
Protein extraction from cells and western blots was performed as previously described3,44 using 40 µg total protein and anti-VEGF-A (1:200), anti-LDLR antibody (1:200) or anti-β-actin (1:50,000) antibodies. Proteins were visualized using the ECL western blotting detection system (Perkin Elmer) and quantified via densitometry (Image-Pro Plus software, ver. 4.1; Media Cybernetics, Silver Spring, MD).
Statistical analysis.
All experiments were repeated at least three times and values are presented as mean ± SEM. Statistical analysis was performed using Prism software (GraphPad Software, San Diego, CA). Data were analyzed by one-way ANOVA followed by post-hoc Bonferroni tests for comparison among means. Student's t-test was used to assess the differences between control and treatment groups. Statistical significance was set at p < 0.05.
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (MOP#86631).
Abbreviations
- LLC
Lewis lung carcinoma
- LMPs
lymphocyte-derived microparticles
- VEGF-A
vascular endothelial growth factor-A
- VEGFR2
vascular endothelial growth factor receptor 2
- LDLR
low density lipoprotein receptor
- MPs
plasma membrane microparticles
- MTT
3-(4,5-dimethyl thiazol-2yl)-2,5-diphenyl tetrazolium bromide
- OCT
optimal cutting temperature
- DiI
1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine
- DiI-LMPs
fluorescent DiI labelled LMPs
- PSR
phosphatidylserine receptor
- siRNA
silencing RNA
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/12533
References
- 1.Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: two sides of the coin. Physiology (Bethesda) 2005;20:22–27. doi: 10.1152/physiol.00029.2004. [DOI] [PubMed] [Google Scholar]
- 2.Meziani F, Tesse A, Andriantsitohaina R. Microparticles are vectors of paradoxical information in vascular cells including the endothelium: role in health and diseases. Pharmacol Rep. 2008;60:75–84. [PubMed] [Google Scholar]
- 3.Yang C, Mwaikambo BR, Zhu T, Gagnon C, Lafleur J, Seshadri S, et al. Lymphocytic microparticles inhibit angiogenesis by stimulating oxidative stress and negatively regulating VEGF-induced pathways. Am J Physiol. 2008;294:467–476. doi: 10.1152/ajpregu.00432.2007. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Retrospective: Judah Folkman (1933–2008) Science. 2008;319:1055. doi: 10.1126/science.1156080. [DOI] [PubMed] [Google Scholar]
- 7.Fontanini G, Faviana P, Lucchi M, Boldrini L, Mussi A, Camacci T, et al. A high vascular count and overexpression of vascular endothelial growth factor are associated with unfavourable prognosis in operated small cell lung carcinoma. Br J Cancer. 2002;86:558–563. doi: 10.1038/sj.bjc.6600130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folkman J. Angiogenesis and angiogenesis inhibition: an overview. Exs. 1997;79:1–8. doi: 10.1007/978-3-0348-9006-9_1. [DOI] [PubMed] [Google Scholar]
- 9.Bouck N, Stellmach V, Hsu SC. How tumors become angiogenic. Adv Cancer Res. 1996;69:135–174. doi: 10.1016/s0065-230x(08)60862-3. [DOI] [PubMed] [Google Scholar]
- 10.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 11.Auwerx JH, Chait A, Wolfbauer G, Deeb SS. Involvement of second messengers in regulation of the low-density lipoprotein receptor gene. Mol Cell Biol. 1989;9:2298–2302. doi: 10.1128/mcb.9.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein JL, Brown MS. Progress in understanding the LDL receptor and HMG-CoA reductase, two membrane proteins that regulate the plasma cholesterol. J Lipid Res. 1984;25:1450–1461. [PubMed] [Google Scholar]
- 13.Gal D, MacDonald PC, Porter JC, Smith JW, Simpson ER. Effect of cell density and confluency on cholesterol metabolism in cancer cells in monolayer culture. Cancer Res. 1981;41:473–477. [PubMed] [Google Scholar]
- 14.Williams RR, Sorlie PD, Feinleib M, McNamara PM, Kannel WB, Dawber TR. Cancer incidence by levels of cholesterol. Jama. 1981;245:247–252. [PubMed] [Google Scholar]
- 15.Kagan A, McGee DL, Yano K, Rhoads GG, Nomura A. Serum cholesterol and mortality in a Japanese-American population: the Honolulu Heart program. Am J Epidemiol. 1981;114:11–20. doi: 10.1093/oxfordjournals.aje.a113157. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Palmieri MR, Sorlie PD, Costas R, Jr, Havlik RJ. An apparent inverse relationship between serum cholesterol and cancer mortality in Puerto Rico. Am J Epidemiol. 1981;114:29–40. doi: 10.1093/oxfordjournals.aje.a113171. [DOI] [PubMed] [Google Scholar]
- 17.Steenland K, Nowlin S, Palu S. Cancer incidence in the National Health and Nutrition Survey I. Follow-up data: diabetes, cholesterol, pulse and physical activity. Cancer Epidemiol Biomarkers Prev. 1995;4:807–811. [PubMed] [Google Scholar]
- 18.Ho YK, Smith RG, Brown MS, Goldstein JL. Low-density lipoprotein (LDL) receptor activity in human acute myelogenous leukemia cells. Blood. 1978;52:1099–1114. [PubMed] [Google Scholar]
- 19.Vitols S, Gahrton G, Ost A, Peterson C. Elevated low density lipoprotein receptor activity in leukemic cells with monocytic differentiation. Blood. 1984;63:1186–1193. [PubMed] [Google Scholar]
- 20.Vitols S, Peterson C, Larsson O, Holm P, Aberg B. Elevated uptake of low density lipoproteins by human lung cancer tissue in vivo. Cancer Res. 1992;52:6244–6247. [PubMed] [Google Scholar]
- 21.Gueddari N, Favre G, Hachem H, Marek E, Le Gaillard F, Soula G. Evidence for upregulated low density lipoprotein receptor in human lung adenocarcinoma cell line A549. Biochimie. 1993;75:811–819. doi: 10.1016/0300-9084(93)90132-c. [DOI] [PubMed] [Google Scholar]
- 22.Trieu VN, Uckun FM. Low density lipoprotein (LDL)-mediated suppression of Lewis lung carcinoma in hypercholesterolemic LDL receptor-deficient mice. Biochem Biophys Res Commun. 1999;255:377–381. doi: 10.1006/bbrc.1999.0184. [DOI] [PubMed] [Google Scholar]
- 23.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 24.Distler JH, Huber LC, Hueber AJ, Reich CF, 3rd, Gay S, Distler O, Pisetsky DS. The release of microparticles by apoptotic cells and their effects on macrophages. Apoptosis. 2005;10:731–741. doi: 10.1007/s10495-005-2941-5. [DOI] [PubMed] [Google Scholar]
- 25.Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. 1967;13:269–288. doi: 10.1111/j.1365-2141.1967.tb08741.x. [DOI] [PubMed] [Google Scholar]
- 26.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 27.Decaussin M, Sartelet H, Robert C, Moro D, Claraz C, Brambilla C, Brambilla E. Expression of vascular endothelial growth factor (VEGF) and its two receptors (VEGF-R1-Flt1 and VEGF-R2-Flk1/KDR) in non-small cell lung carcinomas (NSCLCs): correlation with angiogenesis and survival. J Pathol. 1999;188:369–377. doi: 10.1002/(SICI)1096-9896(199908)188:4<369::AID-PATH381>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 28.Niki T, Iba S, Tokunou M, Yamada T, Matsuno Y, Hirohashi S. Expression of vascular endothelial growth factors A–D and their relationships to lymph node status in lung adenocarcinoma. Clin Cancer Res. 2000;6:2431–2439. [PubMed] [Google Scholar]
- 29.Kajita T, Ohta Y, Kimura K, Tamura M, Tanaka Y, Tsunezuka Y, et al. The expression of vascular endothelial growth factor C and its receptors in non-small cell lung cancer. Br J Cancer 20. 2001;85:255–260. doi: 10.1054/bjoc.2001.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Restucci B, Borzacchiello G, Maiolino P, Martano M, Paciello O, Papparella S. Expression of vascular endothelial growth factor receptor Flk-1 in canine mammary tumours. J Comp Pathol. 2004;130:99–104. doi: 10.1016/j.jcpa.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Bonnesen B, Pappot H, Holmstav J, Skov BG. Vascular endothelial growth factor A and vascular endothelial growth factor receptor 2 expression in non-small cell lung cancer patients: relation to prognosis. Lung Cancer. 2009;66:314–318. doi: 10.1016/j.lungcan.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Koppler B, Cohen C, Schlondorff D, Mack M. Differential mechanisms of microparticle transfer toB cells and monocytes: anti-inflammatory propertiesof microparticles. Eur J Immunol. 2006;36:648–660. doi: 10.1002/eji.200535435. [DOI] [PubMed] [Google Scholar]
- 33.Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka Y, Schroit AJ. Insertion of fluorescent phosphatidylserine into the plasma membrane of red blood cells. Recognition by autologous macrophages. J Biol Chem. 1983;258:11335–11343. [PubMed] [Google Scholar]
- 35.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–216. [PubMed] [Google Scholar]
- 36.Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem. 2001;276:1071–1077. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- 37.Huber LC, Jungel A, Distler JH, Moritz F, Gay RE, Michel BA, et al. The role of membrane lipids in the induction of macrophage apoptosis by microparticles. Apoptosis. 2007;12:363–374. doi: 10.1007/s10495-006-0622-7. [DOI] [PubMed] [Google Scholar]
- 38.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piersma SJ, Jordanova ES, van Poelgeest MI, Kwappenberg KM, van der Hulst JM, Drijfhout JW, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007;67:354–361. doi: 10.1158/0008-5472.CAN-06-3388. [DOI] [PubMed] [Google Scholar]
- 40.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 41.Luo X, Slater JM, Gridley DS. Enhancement of radiation effects by pXLG-mEndo in a lung carcinoma model. Int J Radiat Oncol Biol Phys. 2005;63:553–564. doi: 10.1016/j.ijrobp.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 42.Brault S, Gobeil F, Jr, Fortier A, Honore JC, Joyal JS, Sapieha PS, et al. Lysophosphatidic acid induces endothelial cell death by modulating the redox environment. Am J Physiol Regul Integr Comp Physiol. 2007;292:1174–1183. doi: 10.1152/ajpregu.00619.2006. [DOI] [PubMed] [Google Scholar]
- 43.Bossolasco M, Veillette F, Bertrand R, Mes-Masson AM. Human TDE1, a TDE1/TMS family member, inhibits apoptosis in vitro and stimulates in vivo tumorigenesis. Oncogene. 2006;25:4549–4558. doi: 10.1038/sj.onc.1209488. [DOI] [PubMed] [Google Scholar]
- 44.Sennlaub F, Valamanesh F, Vazquez-Tello A, El-Asrar AM, Checchin D, Brault S, et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003;108:198–204. doi: 10.1161/01.CIR.0000080735.93327.00. [DOI] [PubMed] [Google Scholar]