

Abstract
Neurite outgrowth assays are the most common phenotypic screen to assess chemical effects on neuronal cells. Current automated assays involve expensive equipment, lengthy sample preparation and handling, costly reagents and slow rates of data acquisition and analysis. We have developed a high throughput screen (HTS) for neurite outgrowth using a robust neuronal cell model coupled to fast and inexpensive visualization methods, reduced data volume and rapid data analysis. Neuroscreen-1 (NS-1) cell, a subclone of PC12, possessing rapid growth and enhanced sensitivity to NGF was used as a model neuron. This method reduces preparation time by using cells expressing GFP or native cells stained with HCS CellMask™ Red in a multiplexed 30 min fixation and staining step. A 2x2 camera binning process reduced both image data files and analysis times by 75% and 60% respectively, compared to current protocols. In addition, eliminating autofocus steps during montage generation reduced data collection time. Pharmacological profiles for stimulation and inhibition of neurite outgrowth by NGF and SU6656 were comparable to current standard method utilizing immunofluorescence detection of tubulin. Potentiation of NGF-induced neurite outgrowth by members of a 1,120-member Prestwick compound library as assayed using this method identified six molecules, including etoposide, isoflupredone acetate, fludrocortisone acetate, thioguanosine, oxyphenbutazone and gibberellic acid, that more than doubled the neurite mass primed by 2 ng/ml NGF. This simple procedure represents an important routine approach in high throughput screening of large chemical libraries using the neurite outgrowth phenotype as a measure of the effects of chemical molecules on neuronal cells.
Keywords: Nerve growth factor, neurite outgrowth, high content screening, PC12, NS-1, SU6656.
INTRODUCTION
The development of rapid screening methods to detect and quantify the effect of biochemical substances on a variety of cell types, including neurons, is an extremely active area of research [1, 2]. In both toxicology and drug development, in vitro assays have used transformed and primary neurons to evaluate the effect of compounds on neurite outgrowth and cell survival [3-5].
Neurite measurement assays rely on photomicrography combined with manual or software-based analytical methods to measure neurite formation, elongation, and regression to determine the effect of compounds on differentiated and undifferentiated neurons. For the most part, these methods are cumbersome and unsuitable for even low throughput screening (LTS) of compounds. The morphometric methods for assessment of neurite outgrowth can be classified into two broad categories: manual analysis [6] and newer techniques which employ automated imaging technologies [7].
Traditionally, manual analysis of photomicrographic images has been standard procedure for quantifying phenotypic changes in cultured neurons. Although the process in some cases employs limited automation, it is labor intensive, time consuming, and error prone, making it unsuitable for screening of chemical libraries. Nonetheless, manual methods are still utilized extensively in many research laboratories due to the relatively low cost of reagents, analytical instruments and ease of implementation [8, 9]
In an effort to automate neurite imaging and analysis, algorithms specifically designed to measure various aspects of neurite outgrowth have been developed [4, 10-13]. These programs have enabled semi-automated processing of photomicrographs taken by inverted microscope, significantly reducing the time spent on analysis of imaging data. Even so, there are limitations inherent in the use of these types of algorithms. Because of their highly interactive nature, the need for frequent operator intervention, and a limited capacity to process large numbers of images, throughput and precision both suffer.
As the current research trend in neurodegenerative diseases continues to shift towards translational medicine, the screening of large chemical libraries for disease modulators becomes essential. To meet the need for phenotype-dependent cell based high throughput screening (HTS) various commercial instrument platforms for high resolution imaging of cellular processes have become available to satisfy the demand for robust HT systems. Despite their availability, these instruments require the commitment of significant financial resources for acquisition, maintenance and operation. In addition, existing assay methods for recognition of fine cellular process are generally non-homogenous and require expensive immunofluorescence detection reagents and multiple handling steps. Despite these disadvantages, there has been a proliferation of high resolution imaging platforms in academic core laboratories and government research institutions.
To address the need for inexpensive, homogeneous and robust HTS methods for the assessment of neurite outgrowth across a range of imaging platforms, we have developed two protocols using HCS CellMask™ Red and GFP expression as alternatives to the widely used non-homogeneous and more expensive antibody-based labeling method. Our method uses NeuroScreen-1 (NS-1) cells, a neuronal model derived from the well-characterized PC12 pheochromocytoma line [14] which has been used successfully in high content neurite outgrowth screens of neurotoxins [5, 15]. In addition, we have developed a simplified measurement algorithm that takes fewer data points per sample, thus reducing data processing times and storage space, ultimately increasing throughput by increasing the rate of data input and decreasing the data volume acquired per sample. These protocols accurately measured known agonist (NGF) and antagonist (SU6656) dose-responses and successfully identified neurite-growth promoting compounds from a 1,120 member Prestwick library. Although we used NS-1 as test case, this method can be readily applied to other neuronal cell models.
MATERIALS AND METHODOLOGY
Cell Culture
NS-1 cells purchased from Cellomics (Cellomics Inc., Pittsburgh, PA) were maintained in culture medium consisting of RPMI 1640 containing 10% fetal bovine serum (FBS), 2mM glutamine and 100 µg/mL penicillin/streptomycin (pen/strep) at 37°C in a 5% CO2 humidified incubator.
Establishment of GFP-Expressing NS-1 Cells
To establish a GFP-positive cell line, actively growing cells were split to ~20% confluence in 175cm2 tissue culture flasks and cultured for 3 days prior to transfection. On the day of transfection, the cells were detached with versene, counted and transfected with the green fluorescent protein (GFP) expression vector, pMAXGFP using the Amaxa Nucleofector® (Amaxa GmbH, Koln, Germany). The transfection was performed using the protocol for PC12 cells as detailed in the Amaxa cell transfection database. Stable colonies expressing GFP were selected with G418 and sorted by expression analysis based on the intensity of GFP with a FACSAria™ cell sorter (Becton Dickinson, San Jose, CA) using WinList™ software (Verity, Topsham, ME) for data analysis. The cell populations were expanded and stored under cryogenic conditions to ensure reproducible testing results. Cell exhibiting moderate levels of GFP expression in the cell cytoplasm and neuritic processes was designated NS1-GFP medium and used for experiments described herein.
Assay Plate Preparation
Becton Dickinson 96-well imaging plates (BD Falcon™ 353219) were coated with 50µL/well collagen type I solution (10µg/mL) at 23°C for 1 h. The collagen solution was removed by vacuum aspiration and the plates were washed twice with 100µL calcium and magnesium free (CMF) Dulbecco’s phosphate buffered saline (DPBS) (Invitrogen, Carlsbad, CA), air dried for 20 min in a biosafety cabinet under ultraviolet (UV) light and used immediately. Untransfected and GFP expressing NS-1 cells were seeded in the 96-well collagen coated imaging plate in growth medium at 4x104 cells/well for 18 to 24 h prior to compound treatment.
Compound Treatment
NS-1 or NS1-GFP cells were exposed for 48 h to 2.5S murine NGF (Millipore, Billerica, MA) serially diluted to concentrations ranging from 0.05 ng/mL to 25 ng/mL in RPMI containing 2%FBS (treatment medium). Half maximal effective concentration (EC50) values for each lot of NGF were determined and used as positive controls and as the stimulus concentration for NGF-induced neurite outgrowth screens. For inhibition assays, cultured cells were treated with SU6656 (EMD Biosciences, San Diego, CA) at concentrations ranging from 0.04 μM to 20 µM in 25 µL volumes. After 30 min incubation at 37°C, NGF was added in 25 µL to yield a final concentration of 20ng/mL, and SU6656 concentration from 20 nM to 10 μM. The cells were then incubated for 48 h and assayed by the HCS CellMask Red™ (HCMR) method described below. The half maximal inhibitory concentration (IC50) value was determined by fitting the normalized inhibitor dose response curve in GraphPad Prism. The response data were normalized to 0ng/mL NGF for minimal neurite outgrowth and 20ng/mL NGF as maximum neurite outgrowth.
For screening of the Prestwick library, cells were seeded overnight in culture medium. The next day 2mM library compounds in 100% DMSO delivered to 96-well compound plates (#3359, Corning, Lowell, MA,) were diluted with serum-free medium (SFM) to 5µM and 25 µL of the diluted compounds was exchanged for the culture medium. The cell plates were returned to the 37°C incubator for 45 min. Thereafter, 25µL of 4ng/mL NGF in RPMI containing 4% FBS was added to the culture wells to produce a final concentration of 2.5 μM compound, 2ng/ml NGF and 2% FBS in the assay. The plates were then incubated at 37°C for 48h before fixing, staining and analysis.
Fixation and Staining
GFP-labeled NS-1cells were permeabilized and stained for nuclear identification in the dark at 23°C for 30 min using 4% buffered formaldehyde (50µl/well) containing 0.2µg/ml Hoechst 33342. This process coupled cell permeabilization to nuclear staining in a single step. For the labeling of untransfected cells, a solution containing 0.2µg/ml Hoechst dye and 1.5µg/ml HCS CellMask Red™ (Invitrogen, Eugene OR) was prepared in 4% formaldehyde. Treatment media was aspirated from the cells and replaced with 50 μL of staining solution. Thereafter, plates were sealed and incubated in the dark at 23°C for 30 min. This process yielded fixed samples with Hoechst-stained nuclei and HCMR-stained cell bodies and extensions.
For comparison, standard immunofluorescence-based labeling of NS-1 cells was performed using the Thermo Scientific Cellomics® Neurite Outgrowth Kit according the manufacturer’s instructions. The kit includes a mouse anti- βIII-tubulin primary antibody and a DyLight™ 488-conjugated goat anti-mouse secondary antibody. Briefly, medium in the test plates was aspirated and the cells were incubated in prewarmed fixation/Hoechst solution for 1.5 h. The wells were washed three times with 1X neurite outgrowth buffer and incubated for 1 h with primary anti-βIII-tubulin antibody. The wash step was repeated once more and the cells incubated for 1 h with fluorescent secondary antibody. Final washes were performed using twice each with 1X neurite outgrowth buffer and wash buffer-M prior to image analysis.
Image Data Collection
Images were collected on a BD Pathway 855™ Bioimaging System using a high resolution cooled CCD camera (12 bit, high QE, effective pixels 1344 x 1040). A 3x3 montage data was collected for each well using a 20x objective (Olympus, Semi-Plan Apochromat; NA 0.4; 6.9 mm WD). Data collection was automated to include autofocus during montage collection and separate images for nuclei, neurites and neuronal bodies were collected. Hoechst fluorescence was collected at 380/10nm excitation and 435 nm long pass (LP) emission, whereas GFP and DyLight 488 fluorescence were collected at 488/10nm excitation and 530/25nm emission. CellMask Red™ fluorescence was collected using 555/28nm excitation and 645/75nm emission filters. The CCD camera chip binning mode was set to 2x2. This processing step reduced data volume by 75% and analysis time by 60% (Table 1) over standard methods.
Table 1.
Data Volume Reduction by Camera Binning
| Image Montage | CCD Bin Size | File Size (MB) | Total Data Volume (GB)/Plate | Analysis Time (min) |
|---|---|---|---|---|
| 3x3 | 1x1 | 24 | 4.5 | 170 |
| 3x3 | 2x2 | 6 | 1.125 | 66 |
Image Data Analysis
Image data from 96-well data sets were analyzed using BD AttoVision™ 1.6 software which included the “Neurite Outgrowth” module. Hoechst-stained nuclei were segmented with a watershed algorithm following the application of shading and sharpening filters. Nuclear object size parameters were limited to minimum and maximum values to minimize artifacts in analysis. Likewise, segmentation was applied to the identification of “cytoplasm” objects stained with HCMR. Again, object size limitations were employed to minimize artifacts in automated analysis. The proprietary Neurite Outgrowth module in AttoVision™ allowed the assignment of limiting gates (in pixels) to neurite length and we chose values which correlated to 1.5X the average cell body width for images generated with our detection system. In addition, this analysis module provided for specific dilation of the cytoplasmic objects to minimize artifacts resulting from weak staining of cellular edges. The module produced data sets which included maximum neurite length, root count, total neurite length and average neurite length for each cell identified in the image. The resulting treatment data files were averaged and normalized using “0 NGF” and “EC50 NGF” concentrations as the minimum and maximum normalizing controls respectively for determination of a percent response. We used the average total neurite length as our primary analysis parameter in the normalization since it provided a broader range of values for data normalization compared to values such as “neurites per cell”. A comparative analysis of the acquired data and measurement parameters derived through this method and other published protocols are presented in the Table 2.
Table 2.
Comparison of Data Acquisition and Analysis Method Described Herein with Other Published Methods
| Ramm (Ref. #24) | Price (Ref. #25) | Mitchell (Ref. #4) | Radio (Ref. #5) | Yeyeodu (This paper) | |
|---|---|---|---|---|---|
| Instrument | IN Cell Analyser 1000 (Amersham) | Automated microscope stage (Prior Scientific) | Ludl stage | ArrayScan V (Cellomics) | BD Pathway 855 |
| Acquisition software | (included) | (Media Cybernetics) | VolumeScan (Vaytek) | (included) | BD AttoVision™ 1.6 |
| Autofocus | yes | yes | yes | yes | yes |
| Image intensity resolution | 12 bit | NR | 24 bit | NR | 12 bit |
| Analysis software | (included) | custom algorithm (Advansoft) in ImagePre Plus 4.1 (Media Cybernetics) | custom algorithm (detailed in text) in ImagePro Plus | (included) | BD AttoVision™ 1.6 with Neurite Outgrowth Module |
| Neurite defined | ~2x cell diam. (30µm) | NR | NR | ~2x total neurite length in cells w/o NGF (> 20 µm) | >1.5x cell width |
| Parameters measured | |||||
| Total neurite length | yes | yes | yes | ||
| Total cell body area | yes | ||||
| Num. neurites | yes | yes | |||
| Avg cell diameter | yes | ||||
| Num. nuclei or cells | yes | yes | yes | yes | |
| Individual neurite length | yes | ||||
| Num. branches | yes | yes | yes | ||
| Maximum neurite length | yes | yes | |||
| Neurites/cell | yes | ||||
| Cell body area | yes | ||||
| Parameters derived | |||||
| % neurite bearing cells | yes | yes | |||
| Num. cells | yes | yes | |||
| Mean neurite length | yes | yes | yes | yes | |
| Total neurite length/cell | yes | yes | yes | ||
| Mean neurite length/cell | yes | yes |
NR=not reported.
RESULTS
We sought to generate a simple robust method with which we could visualize and measure activities of large chemical compound libraries on neurite processes rapidly and inexpensively in a high throughput environment. Since NS-1 cells are a subclone of PC12 cells that have been used successfully in screening the effects of neurotoxins on neurite outgrowth with immunofluorescence tags [5, 15] we used this line to explore the utility of green fluorescent protein (GFP) expression and HCS CellMask™ Red dye as alternatives to the antibody staining method.
To generate a GFP line we stably transfected NS-1 cells with pMAX-GFP expression vector. The expression of GFP was heterogeneous, represented as three major spectral populations of cells. The heterogeneity prevented accurate enumeration of the cells due to failure of the camera to auto focus on cells as a result of the distortion in fluorescence caused by the intensity gradient. In addition, the cells gradually lost GFP expression with passage number, resulting in increased heterogeneity, and the appearance of revertants from low GFP expressing cells.
In order to minimize the GFP signal gradient and ensure reproducibility of the data, we sorted the cell populations by FACS analysis and collected samples based on fluorescence intensity-dependent gating (Fig. 1). About 80% of the sorted cell population exhibited fluorescence intensity of at least 10-fold compared to untransfected cells.
Fig. (1).

Fluorescence intensity profile of GFP expressing NS-1 cells. A flow cytometry histogram showing the profile of a mixed population of GFP expressing NS-1 cells (green shading) relative to the population of untransfected NS-1 cells (gray shading). In this example, 80% of the NS1-GFP population exhibited an intensity of at least 10 times that of the control population.
The cell fractions, which were relatively homogeneous after sorting, were tested in the neurite outgrowth assay. At this point the camera was able to autofocus with precision on the cells due to uniformity of the GFP fluorescence in the cell populations. The cell fractions were rapidly expanded and frozen for future use and we chose to continue our work with the cell population which showed moderate GFP fluorescence, because of slow growth characteristics observed with the high expressors. In cells with moderate expression, GFP fluorescence was clearly visible throughout the cytoplasm and neurite processes when viewed by fluorescence microscopy (Fig. 2A).
Fig. (2).

A comparison of fluorescent images generated by the three staining methods. GFP-NS-1cells (A), βIII-tubulin immunofluorescence (B) and HCS CellMask™ Red (C) fluorescence images of cell bodies and neurites were acquired on the BD Pathway 855 Bioimager. Unaltered, exemplar TIFF files were retrieved with “ImageJ” software and the “Yellow Hot” look-up table was used to determine relative intensities; no other data manipulations were applied. A gray scale ramp is shown in panel C.
In addition to the recombinant GFP assay, we developed an alternative dye method to visualize elongated neurites by modifying the one-step fixation/stain method of Mitchell et. al. [4]. To the fixative and nuclear stain Hoechst 33342, we added HCS CellMask Red™, a stain to identify overall cell morphology, including neurite extensions. Cells double stained with Hoechst and CellMask Red™ (HCMR), were effectively visualized as shown in Fig. (2C).
Compared with the standard indirect immunofluorescent detection of neurites with βIII-tubulin (Fig. 2B), both GFP expression and HCMR staining of NS-1 cells offered comparable images (Fig. 2A&C) that were generated in a much shorter timeframe than other published neurite outgrowth detection and analysis methods (Table 3).
Table 3.
Comparison of Sample Preparation and Assay Conditions
| Ramm (Ref. #24) | Price (Ref. #25) | Mitchell (Ref. #4) | Radio (Ref. #5) | Yeyeodu (This paper) | |
|---|---|---|---|---|---|
| Detection method(s) | Abs: primary + FITC-secondary | Coomassie blue G-250 | Hoechst + Ab: primary + AlexaFluor + 488-secondary | Hoechst + Ab: primary + AlexaFluor + 488-secondary | a) Hoechst + GFP expressing cells or b) Hoechst + CellMask Red dye |
| Plate/pretreat | 8 h | 5 d | 4-6 d | 1-4 d | 16-24 h |
| Treat | 3 d | 4 d | 16-24 h | 4 d | 2 d |
| Total time in culture | >3 d | 9 d | >4.5 d | 5 d | <3 d |
| Cell preparation for imaging | |||||
| Fix(/stain) | 2 h | 1 h | 30 min | 20 min | 30 min |
| Permeabilize/wash | 30-60 min | - | 30 min | 30 min | - |
| Primary Antibody | 1 h | - | 18 h | 1 h | - |
| Secondary Antibody | 1 h | - | 1 h | 1 h | - |
| Final stain | - | ON | - | - | - |
| Total preparation time | 4.5-5 h | ON | ON | 3 h | 30 min |
ON=overnight; Abs=Antibodies; d=day(s).
In order to establish that GFP expression and CellMask™ Red staining accurately reflected the pharmacological response of NS-1 cells to NGF, we treated cells with varying doses of NGF and measured percent neurite response as a function of NGF concentration. Following NGF treatment, the cells were imaged by both direct methods described in this paper, and the dose-response curves were compared to the curve generated with the standard antibody labeling method (Fig. 3). The resulting curves produced comparable EC50 values of 3.3 ng/ml for GFP expressing cells, 2.1 ng/ml for anti βIII-tubulin labeled cells, and 1.4 ng/ml for HCMR stained cells. These results are in agreement with each other and data reported for antibody detection of NGF induced neurite outgrowth in NS-1 in the vendor technical bulletin (Cellomics, Pittsburg, PA). The data also showed that NS-1 cells are approximately 5-fold to 8-fold more sensitive to recombinant mouse 2.5S NGF compare to literature values reported for the parent PC12 cells [16].
Fig. (3).

Comparison of NGF dose on neurite outgrowth response measured by three visualization methods. NS-1 cell neurites were visualized by transfection with GFP (GFP), staining with anti-βIII-tubulin followed by DyLight 488-conjugated secondary antibody (anti-βIII tubulin) or staining with CellMask Red (CellMask Red). Data were normalized using responses to 0ng/ml and 50ng/ml NGF as minimum and maximum total neurite length/cell, respectively. Calculated EC50 values were 3.3, 2.1 and 1.4 ng/ml using GFP, anti-βIII-Tubulin and CellMask Red, respectively. Values plotted are means ± S.E.M. (n=4).
Because the HCMR dye method presented a much faster alternative to GFP expression for enabling neurite outgrowth assays and is potentially applicable to other neuronal cells, we used the HCMR assay for the remainder of our studies. To further validate HCMR as a viable method for accurate assessment of neurite outgrowth, we tested whether the method could accurately model the dose-response profile of SU6656, a Src-family kinase inhibitor [17] for the inhibition of neurite outgrowth [18, 19]. As shown in the sigmoidal curve (Fig. 4), SU6656 caused a dose-dependent inhibition of neurite outgrowth induced by 20 ng/mL NGF. This concentration of NGF, which correlates to the EC90 of NGF for neurite outgrowth in NS-1 cells, was determined from a dose response assay with escalating concentrations of NGF. The calculated SU6656 IC50 of 64 nM is well within the range of SU6656 IC50s for various Src-kinase family members.
Fig. (4).

Inhibition of NGF-induced neurite outgrowth by Src-family kinase inhibitor SU6656. NS-1 cells were treated with varying doses of SU6656, and then NGF was added to a final concentration of 20ng/ml. Neurite outgrowth was analyzed at 48h by HCMR. The curve was fitted in GraphPad PRISM using a 4-parameter logistic model with recursive least squares weighting. Each point represents an average of three experiments in triplicate. The bars represent the standard deviation of the data from the means.
Finally, to further validate the suitability of the HCMR assay for high throughput screening of neurite outgrowth modulators, we used the assay to screen a 1,120-member Prestwick library of FDA-approved small molecules for enhancers of NGF-dependent neurite-outgrowth. NS-1 cells were pretreated with 3 μM library compounds for 45 min at 37o C prior to NGF addition. The final concentration of NGF and compound is 2 ng/ml and 1.5 μM, respectively. Compound effect on neurite outgrowth was measured after 48 h of incubation.
The assay identified several compounds with ≥ 150% response compared to 2 ng/ml NGF alone in two separate runs. Among these were four compounds which have previously been shown to promote neurite outgrowth, namely zaprinast [20], etoposide [21], corticosterone [22] and serotonin [23]. Of these, six compounds yielded a response greater than 200%, our threshold for the assay, compared to background outgrowth in the presence of 2 ng/ml NGF (Fig. 5). The activity values of these compounds in the two independent tests were very similar. Using a threshold of two-fold enhancement of total neurites as cut off, the actives rate in this assay is 0.5%. Compiled values of the average window (defined as the plate mean of NGF-stimulated control values divided by the plate mean of non-stimulated control values) and the average of Z primes from a representative plate set (n =14) were 4.8 and 0.57 respectively.
Fig. (5).

Active compounds exhibiting greater than 200% activity in two separate experiments. NS1 cells were treated with 2 ng/mL NGF in the presence of 1.5 µM compounds for 48 h and stained with a cocktail of Hoechst and HCS CellMask Red™ for 30 min. Data was acquired on the BD Pathway855 imaging station and analyzed with BD Attovision™ 1.6 software. The histograms represent the compound activity measured as total neurites per cell over background of 2 ng/mL NGF in two independent experiments; the error bars represent standard deviations.
Besides etoposide which is already known to promote neurite outgrowth at sublethal concentrations, the assay identified the steroidal (isoflupredone acetate) and non-steroidal (oxyphenbutazone) anti-inflammatory agents, an adrenocorticoid (fludrocorticone acetate) that promotes Na+ retention and K+ excretion in renal tubules, a guanosine analog (thioguanosine), and a plant hormone (gibberellic acid) known to stimulate rapid stem and root growth. A list of the most active compounds, their structures and functional classification is presented in Table 4.
Table 4.
Active Potentiators of NGF-Induced Neurite Outgrowth Identified from Prestwick Compound Set
| Name | Structure | Formula | Molecular weight | Classification/ function |
|---|---|---|---|---|
| Isoflupredone acetate |  |
C23H29FO6 | 420.5 | Anti-inflammatory sterol |
| Fludrocortisone acetate |  |
C23H31FO6 | 422.5 | Aldosterone analog |
| Thioguanosine |  |
C10H13N5O4S | 299.3 | Purine antagonist cell stressor used in the treatment of acute nonlymphocytic leukemias |
| Etoposide |  |
C29H32O13 | 588.6 | Topoisomerase II inhibitor shown to promote neurite outgrowtha |
| Gibberellic acid | 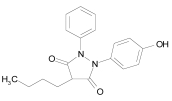 |
C19H22O6 | 346.4 | Growth-inducing Plant hormoneb |
| Oxyphen-butazone | 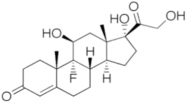 |
C19H20N2O3 | 324.4 | NSAID with multiple molecular targets |
NSAID: non-steroid anti-inflammatory.
(Aoki et al., 2003).TJ
(Halliday and Fankhauser, 2003).
DISCUSSION
We report a rapid high throughput screen to identify compounds which affect intrinsic functions of neurons that lead to phenotypic changes in neurite extension or neuronal morphology. This assay significantly improves on currently available neurite outgrowth assays. Like the method described by Ramm et al. [24], the HCMR assay is fully automated. However, it does not require costly reagents to visualize neurites and allows for direct measurement of cell numbers. In comparison with the method described by Price et al. [25], HCMR features three advantages; it 1) increased throughput, 2) utilizes stains which can be multiplexed with additional assays, and 3) avoids the use of DNA polymerase inhibitors which might obscure the effects of neuroactive drugs which are cell-cycle dependent or sensitive to autocrine feedback. HCMR is also an improvement over the methods described in the past [4, 5] by virtue of its single-step fix/double stain which represents a truly compliant homogeneous high throughput method for neurite analysis. Also improved is the significant reduction of data volume achieved by applying 2x2 camera binning to reduce data acquisition and analysis times as well as the size of space required for data storage (Table 1).
The use of GFP-expressing NS-1 cells or CellMask Red™ as a universal fluorescent cell stain allowed us to visualize (Fig. 2) and quantitate (Fig. 3) neurite outgrowth on par with the standard immunofluorescence method which detects βIII-tubulin, a cytoskeletal protein enriched in neurites [26]. However, we found HCMR more convenient because of its simplicity and potential for universal application in various neuronal cell lines. The most significant advantage of HCMR rendered the cloning of GFP-expressing cell lines unnecessary, thus eliminating several weeks of cell line development.
We validated the HCMR method in two ways. First, we compared HCMR with the standard βIII-tubulin immunofluorescence method in dose-response assays (Figs. 3&4) using agonist (NGF) and antagonist (SU6656) of neurite outgrowth, and obtained EC50 and IC50 values that were comparable to those reported in the literature. Second, we used HCMR to screen a 1,120-member Prestwick library in order to identify compounds that enhance the actions of NGF for neurite outgrowth. We identified 30 FDA-approved drugs capable of inducing more than 50% neurite mass above background activity elicited by 2 ng/ml NGF. Four of these compounds have already been reported as enhancers of neurite outgrowth, but the majority has not been reported to be involved in neurite outgrowth.
The known functions of these active compounds are not inconsistent with the biochemical mechanisms involved in neurite extension as most actives on the list induce stress and/or apoptotic responses in cultured cells, likely promoting stress dependent neurite outgrowth in neuronal cells at sublethal concentrations. Cell stressors have been widely reported to induce neuronal differentiation and neurite outgrowth via stress-activated protein kinases and p38 MAP kinase [27-31]. Even in the case of the non-steroidal anti-inflammatory compound class with multiple actions and reported neurotoxicity, there is precedent for neuroactive members, which induce either neurite outgrowth or neurotoxicity, depending on the experimental condition. One example is staurosporine which promotes neurite extension in a dose-dependent manner [32, 33] but also induces apoptosis [34, 35]. Corticosterone is another compound that either promotes or inhibits neurite outgrowth depending on the timing and dose administered [22]. Along these lines, it is of interest to note that we identified other sterols or sterol-like compounds with neurite outgrowth promoting properties, including two with activities greater than twice that of the NGF control. It is also not surprising to find a guanosine analog since NGF-induced neurite outgrowth involves the activation of the G proteins Ras and Rap1 [36]. Finally, the mechanism of stem and root growth induced by the neuroactive plant hormone may share a similar molecular mechanism to those which form extensions in various animal cells, including mammalian neurons.
Although the active compounds were identified by implementing our HCMR screen using the potentiator pharmacology approach, some of the actives could be functional agonists of neurite outgrowth and elongation acting independently of NGF. Future studies will establish the pharmacological profiles of the actives as agonists or potentiators of neurite outgrowth upon implementation of HCMR in the absence of NGF. As a whole, the number and identity of the positive compounds reported herein clearly support HCMR as a rapid and viable screening method for modulators of neurite outgrowth.
CONCLUSION
HCMR is a strategic low-cost, rapid and reliable HTS assay for compounds which affect neurite extension. When combined with mechanistic assays such as that described by Laketa et al. [37], and further in vitro and in vivo studies, HCMR presents a valuable and cost effective alternative to present methods to test large amounts of compound libraries in a reasonable timeframe to identify compounds that support, protect, enhance, or are toxic to neuronal cell development and differentiation.
ACKNOWLEDGEMENTS
The project was supported by a grant from the National Institute of General Medical Sciences (SC3GM081092) (S.Y.T.) and funds from the State of North Carolina (S.M.W. and N.G). The authors would also like to acknowledge the support of Golden LEAF Foundation. The authors wish to thank Susan Peacock, J.D., Ph.D., for assistance in reviewing this manuscript.
REFERENCES
- 1.Dragunow M. High-content analysis in neuroscience. Nat Rev Neurosci. 2008;9(10):779–88. doi: 10.1038/nrn2492. [DOI] [PubMed] [Google Scholar]
- 2.Thomas N. High-content screening: a decade of evolution. J Biomol Screen. 2010;15(1):1–9. doi: 10.1177/1087057109353790. [DOI] [PubMed] [Google Scholar]
- 3.Das KP, Freudenrich TM, Mundy WR. Assessment of PC12 cell differentiation and neurite growth: a comparison of morphological and neurochemical measures. Neurotoxicol Teratol. 2004;26(3):397–406. doi: 10.1016/j.ntt.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell PJ, Hanson JC, Quets-Nguyen AT, Bergeron M, Smith RC. A quantitative method for analysis of in vitro neurite outgrowth. J Neurosci Methods. 2007;164(2):350–62. doi: 10.1016/j.jneumeth.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 5.Radio NM, Breier JM, Shafer TJ, Mundy WR. Assessment of chemical effects on neurite outgrowth in PC12 cells using high content screening. Toxicol Sci. 2008;105(1):106–18. doi: 10.1093/toxsci/kfn114. [DOI] [PubMed] [Google Scholar]
- 6.Ventimiglia R, Jones BE, Moller A. A quantitative method for morphometric analysis in neuronal cell culture: unbiased estimation of neuron area and number of branch points. J Neurosci Methods. 1995;57(1):63–6. doi: 10.1016/0165-0270(94)00126-2. [DOI] [PubMed] [Google Scholar]
- 7.Federico FSS, Alberto D, editors. Cell Biology. 3rd. USA: Elsevier; 2006. The state of the art in biological image analysis; pp. 201–6. [Google Scholar]
- 8.Sano M, Kanefusa K, Tsuyoshi T, Katoh-Semba R. A convenient bioassay for NGF using a new subline of PC12 pheochromocytoma cells (PC12D) Brain Res. 1988;459(2):404–6. doi: 10.1016/0006-8993(88)90660-9. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto T, Oshimaa K, Miyamotoa A, Sakuraia M, Gotoa M, Hayashia S. Image analysis of CNS neurotrophic factor effects on neuronal survival and neurite outgrowth. J Neurosci Methods. 1990;31(2):153–62. doi: 10.1016/0165-0270(90)90160-h. [DOI] [PubMed] [Google Scholar]
- 10.Masseroli M, Bollea A, Forloni G. Quantitative morphology and shape classification of neurons by computerized image analysis. Comput Methods Prog Biomed. 1993;41(2):89–99. doi: 10.1016/0169-2607(93)90068-v. [DOI] [PubMed] [Google Scholar]
- 11.Siklos L, Kuhnt U. Algorithm for semi-automatic sorting of objects to specified tissue domains. An aid for co-ordinating morphometric data with identified tissue components. J Neurosci Methods. 1993;46(3):217–24. doi: 10.1016/0165-0270(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 12.Bilsland J, Rigby M, Young L, Harper S. A rapid method for semi-quantitative analysis of neurite outgrowth from chick DRG explants using image analysis. J Neurosci Methods. 1999;92(1-2):75–85. doi: 10.1016/s0165-0270(99)00099-0. [DOI] [PubMed] [Google Scholar]
- 13.Christina M, Weavera JD, Pinezichb W, Brent L, Marcelo EV. An algorithm for neurite outgrowth reconstruction. J Neurosci Methods. 2003;124(2):197–205. doi: 10.1016/s0165-0270(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 14.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73(7):2424–8. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radio NM, Freudenrich TM, Robinette BL, Crofton KM, Mundy WR. Comparison of PC12 and cerebellar granule cell cultures for evaluating neurite outgrowth using high content analysis. Neurotoxicol Teratol. 2010;32(1):25–35. doi: 10.1016/j.ntt.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Katzir I, Shani J, Regev K, Shabashov D, Lazarovici P. A quantitative bioassay for nerve growth factor, using PC12 clones expressing different levels of trkA receptors. J Mol Neurosci. 2002;18(3):251–64. doi: 10.1385/JMN:18:3:251. [DOI] [PubMed] [Google Scholar]
- 17.Robert AB, Martin AB, Xiangdong L, et al. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20(23):9018–27. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu G, Beggs H, Jürgensen C, et al. Netrin requires focal adhesion kinase and Src family kinases for axon outgrowth and attraction. Nat Neurosci. 2004;7(11):1222–32. doi: 10.1038/nn1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meriane M, Tcherkezian J, Webber CA, et al. Phosphorylation of DCC by Fyn mediates Netrin-1 signaling in growth cone guidance. J Cell Biol. 2004;167(4):687–98. doi: 10.1083/jcb.200405053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Messina E, Lupi F, Barile L, Giacomello A. Cyclic nucleotides and neuroblastoma differentiation. Nucleosides Nucleotides Nucleic Acids. 2004;23(8-9):1551–4. doi: 10.1081/NCN-200027775. [DOI] [PubMed] [Google Scholar]
- 21.Aoki S, Wei H, Matsui K, Rachmat R, Kobayashi M. Pyridoacridine alkaloids inducing neuronal differentiation in a neuroblastoma cell line, from marine sponge Biemna fortis. Bioorg Med Chem. 2003;11(9):1969–73. doi: 10.1016/s0968-0896(03)00086-5. [DOI] [PubMed] [Google Scholar]
- 22.Tsai SY, Chiu PY, Yang CP, Lee YH. Synergistic effects of corticosterone and kainic acid on neurite outgrowth in axotomized dorsal root ganglion. Neuroscience. 2002;114(1):55–67. doi: 10.1016/s0306-4522(02)00261-0. [DOI] [PubMed] [Google Scholar]
- 23.Homma K, Kitamura Y, Ogawa H, Oka K. Serotonin induces the increase in intracellular Ca2+ that enhances neurite outgrowth in PC12 cells via activation of 5-HT3 receptors and voltage-gated calcium channels. J Neurosci Res. 2006;84(2):316–25. doi: 10.1002/jnr.20894. [DOI] [PubMed] [Google Scholar]
- 24.Ramm P, Alexandrov Y, Cholewinski A, Cybuch Y, Nadon R, Bohdan JS. Automated screening of neurite outgrowth. J Biomol Screen. 2003;8(1):7–18. doi: 10.1177/1087057102239779. [DOI] [PubMed] [Google Scholar]
- 25.Price RD, Oe T, Yamaji T, Matsuoka N. A simple, flexible, nonfluorescent system for the automated screening of neurite outgrowth. J Biomol Screen. 2006;11(2):155–64. doi: 10.1177/1087057105283344. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan KF. Structure and utilization of tubulin isotypes. Annu Rev Cell Biol. 1988;4(4):687–716. doi: 10.1146/annurev.cb.04.110188.003351. [DOI] [PubMed] [Google Scholar]
- 27.Eom DS, Choi WS, Ji S, Cho JW, Oh YJ. Activation of c-Jun N-terminal kinase is required for neurite outgrowth of dopaminergic neuronal cells. Neuroreport. 2005;16(8):823–8. doi: 10.1097/00001756-200505310-00009. [DOI] [PubMed] [Google Scholar]
- 28.Eom DS, Choi WS, Oh YJ. Bcl-2 enhances neurite extension via activation of c-Jun N-terminal kinase. Biochem Biophys Res Commun. 2004;314(2):377–81. doi: 10.1016/j.bbrc.2003.12.094. [DOI] [PubMed] [Google Scholar]
- 29.Kano Y, Nakagiri S, Nohno T, et al. Heat shock induces neurite outgrowth in PC12m3 cells via the p38 mitogen-activated protein kinase pathway. Brain Res. 2004;1026(2):302–6. doi: 10.1016/j.brainres.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 30.Kano Y, Nohno T, Shimada K, et al. Osmotic shock-induced neurite extension via activation of p38 mitogen-activated protein kinase and CREB. Brain Res. 2007;1154:1–7. doi: 10.1016/j.brainres.2007.03.087. [DOI] [PubMed] [Google Scholar]
- 31.O'Driscoll CM, Gorman AM. Hypoxia induces neurite outgrowth in PC12 cells that is mediated through adenosine A2A receptors. Neuroscience. 2005;131(2):321–9. doi: 10.1016/j.neuroscience.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 32.Hashimoto S, Hagino A. Blockage of nerve growth factor action in PC12h cells by staurosporine, a potent protein kinase inhibitor. J Neurochem. 1989;53(6):1675–85. doi: 10.1111/j.1471-4159.1989.tb09230.x. [DOI] [PubMed] [Google Scholar]
- 33.Yamada S, Hakamada K, Munekata T, et al. A system for analyzing the event timing profile of single cells by using a model of neurite maturation in PC12D cells. Biosens Bioelectron. 2009;24(5):1493–7. doi: 10.1016/j.bios.2008.07.042. [DOI] [PubMed] [Google Scholar]
- 34.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 35.Williams B, Dwyer DS. Structure-based discovery of low molecular weight compounds that stimulate neurite outgrowth and substitute for nerve growth factor. J Neurochem. 2009;110(6):1876–84. doi: 10.1111/j.1471-4159.2009.06291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaudry D, Stork PJS, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296(5573):1648–9. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- 37.Laketa V, Simpson JC, Bechtel S, Wiemann S, Pepperkok R. High-content microscopy identifies new neurite outgrowth regulators. Mol Biol Cell. 2007;18(1):242–52. doi: 10.1091/mbc.E06-08-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]