

Abstract
Parthenogenesis is the biological phenomenon by which embryonic development is initiated without male contribution. Whereas parthenogenesis is a common mode of reproduction in lower organisms, the mammalian parthenote fails to produce a successful pregnancy. We herein describe in vitro parthenogenetic development of monkey (Macaca fascicularis) eggs to the blastocyst stage, and their use to create a pluripotent line of stem cells. These monkey stem cells (Cyno-1 cells) are positive for telomerase activity and are immunoreactive for alkaline phosphatase, octamer-binding transcription factor 4 (Oct-4), stage-specific embryonic antigen 4 (SSEA-4), tumor rejection antigen 1-60 (TRA 1-60), and tumor rejection antigen 1-81 (TRA 1-81) (traditional markers of human embryonic stem cells). They have a normal chromosome karyotype (40 + 2) and can be maintained in vitro in an undifferentiated state for extended periods of time. Cyno-1 cells can be differentiated in vitro into dopaminergic and serotonergic neurons, contractile cardiomyocyte-like cells, smooth muscle, ciliated epithelia, and adipocytes. When Cyno-1 cells were injected into severe combined immunodeficient mice, teratomas with derivatives from all three embryonic germ layers were obtained. When grown on fibronectin/laminin-coated plates and in neural progenitor medium, Cyno-1 cells assume a neural precursor phenotype (immunoreactive for nestin). However, these cells remain proliferative and express no functional ion channels. When transferred to differentiation conditions, the nestin-positive precursors assume neuronal and epithelial morphologies. Over time, these cells acquire electrophysiological characteristics of functional neurons (appearance of tetrodotoxin-sensitive, voltage-dependent sodium channels). These results suggest that stem cells derived from the parthenogenetically activated nonhuman primate egg provide a potential source for autologous cell therapy in the female and bypass the need for creating a competent embryo.
The use of human embryos to derive embryonic stem cells (ES cells) is viewed by some sectors of our society as ethically problematic. In nonhuman primates, there are currently three methods for deriving pluripotent stem cells: from embryos produced by in vitro fertilization (1-4), parthenogenesis (5), and from adult tissues such as cells derived from the bone marrow (6). We have previously reported the creation of a line of nonhuman primate stem cells from parthenogenetically activated eggs (5). By using this technique, ES cells were derived without the need to create or destroy a viable embryo.
Parthenogenesis, the process by which a single egg can develop without the presence of the male counterpart, is a common form of reproduction in nature. Flies, ants, lizards, snakes, fish, birds, reptiles, amphibians, honeybees, and crayfish routinely reproduce in this manner. Eutherians (placental mammals) are not capable of this form of reproduction. However, chimeras of parthenogenetic cells coupled with biparentally derived embryonic tissues have generated apparently normal offspring, and the parthenogenetic origin of several tissues has been confirmed in such chimeric animals (7). In a reported case of a human parthenogenetic chimera, contribution to several tissues has been demonstrated, including blood where 100% of the leukocytes were found to be of parthenogenetic origin (8).
Eutherian oocytes, on the other hand, can undergo parthenogenesis in vitro with variable success. When mammalian oocytes are activated (emulating the fertilization process) and transferred to a surrogate mother, they are capable of surviving to day 10 of development in the mouse, day 21 for sheep, day 29 in pigs, and day 11.5 in rabbit (9-12).
The reason for this halted development is believed to be due to genetic imprinting. It has been shown that maternal and paternal genomes are epigenetically different, and that both sets are required for successful development (13-15). In parthenotes (activated eggs), all of the genetic material is of maternal origin, and hence lacking paternal imprinting. It is believed that parthenotes are not capable of developing to term because they fail to develop a trophectoderm and primitive endoderm-extraembryonic tissues (9). They resemble ovarian teratomas and consist of only embryonic tissue. Androgenotes (created by the fusion of two sperm nuclei or diplodization of one sperm in the absence of female counterpart) are of purely paternal origin and develop into a structure consisting of a trophoblast and yolk sac (16). These resemble hydatidiform moles (solely trophoblastic tissue), which are formed when a sperm fertilizes an enucleated egg (17).
In the present report, we describe the parthenogenetic activation of cynomolgus macaque eggs in vitro and the derivation of a pluripotent cell line (Cyno-1). When cultured under selective conditions, these cells have divided for >2 yr, and, on induced differentiation, cell derivatives from all three germ layers were obtained.
Materials and Methods
Superovulation, Oocyte Retrieval, and Oocyte Maturation/Activation. Monkeys were injected (i.m.) with 1,000 units of pregnant mare serum gonadotrophin 5 days before surgery and then injected with 500 units of human chorionic gonadotropin 24 h before surgery. For ovary isolation, monkeys were tranquilized with ketamine (10 mg/kg of body weight), intubated endotracheally, and anesthetized with isoflurane (monitored to effect: no pal-pebral reflex, no deep pain response). Ovaries were removed by midline laparotomy incision.
Oocytes were manually harvested under a dissecting microscope. Oocyte maturation was performed in CMRL-1066 media (Sigma) with 20% FCS (HyClone), 10 units/ml pregnant mare serum (Sigma), 10 units/ml human chorionic gonadotropin (Sigma), 0.05 mg/ml penicillin, and 0.075 mg/ml streptomycin (Sigma). Eggs were incubated for 36 h at 37°C, in 5% CO2 and 20% O2. Mature metaphase II eggs were subsequently activated by incubation with 10 μM ionomycin for 8 min, followed by culture with 2 mM 6-dimethylaminopurine for 4 h. The inner cell masses (ICM) were isolated by immunosurgery as described (20) and cultured on a feeder layer of mitotically inactive mouse embryonic fibroblasts in Dulbecco's minimal essential medium (GIBCO) with 15% FCS (HyClone).
Cell Culture Conditions. Neural progenitor-ES cells were plated in flasks coated with fibronectin/laminin or fibronectin/BSA/collagen with NPMM (Clonetics, East Rutherford, NJ) and maintained at 37°C in 5% CO2. Media were changed every 3 days. Differentiation was induced by removing basic fibroblast growth factor (bFGF) and epidermal growth factor, with the addition of 200 μM ascorbic acid.
Immunohistochemical Staining. A variety of markers of stem cells and stem cell differentiation were assessed by immunocytochemistry. Antibodies and staining conditions were as follows. For surface markers, cells were incubated with primary antibodies [stage-specific embryonic antigen-4 (1:20; Developmental Hybridoma Bank), tumor rejection antigen 1-81 (1:80), and tumor rejection antigen 1-60 (a gift from P. Andrews, Sheffield, U.K.)]. For immunocytochemisrty of embryonic markers, primary antibodies were diluted in PBS supplemented with 0.5% BSA. After washing with PBS-BSA, cells were fixed with 2% formaldehyde for 30 min and washed three times in PBS-BSA, followed by incubation with 10% normal goat serum in PBS at room temperature. Subsequently, primary antibody was added for 30 min at room temperature; cells were then washed with PBS-BSA three times, followed by incubation with secondary antibody for 30 min, washed, stained with 4′,6-diamidino-2-phenylindole (DAPI), and mounted. For immunocytochemistry of differentiated cells, cells were fixed in 4% paraformaldehyde at room temperature for 20 min, followed by permeabilization for 2 min in 100% ethanol. After fixation, cells were washed with PBS, blocked with 10% normal goat serum in PBS at room temperature for 2 h, followed by incubation at room temperature for 2 h with nestin antibody (1:200, Chemicon), TH polyclonal 1:200 (Pel-Freez Biologicals) or TH monoclonal 1:1000 (Sigma) β-tubulin type III (TuJ1) monoclonal (1:500, Babco, Richmond, CA), in PBS. After washing, cells were incubated with a rabbit secondary antibody in PBS-BSA at room temperature for 30 min. Cells were then washed in PBS and mounted.
Alkaline Phosphatase. Alkaline phosphatase was determined as described (5). Briefly, culture medium was removed from the plates, and cells were fixed with 4% paraformaldehyde for 20 min. Cells were washed three times in Tris-maleate buffer [(3.6 g of Trizma base (Sigma), in 1 liter of water, pH raised to 9.0 with 1 M maleic acid)] for 10 min each wash. The last wash was removed, and the staining solution [(Tris-maleate buffer: 200 μl of a 5 mM MgCl naphthol AS-MX phosphate (Sigma), 0.4 mg/ml; Fast red (Sigma), 1 mg/ml)] was added to the cells for 15 to 20 min. Once red colonies were detected, the reaction was stopped by adding PBS and bringing the pH to 7.4.
Antigen Profiling. Peripheral blood lymphocytes (PBLs) were isolated from whole blood by flotation on Ficoll-Hypaque. Cells were harvested from the medium:Ficoll-Hypaque interface, and the remaining red cells were hypotonically lysed. After additional washes, cells were labeled with FITC-conjugated anti-HLA-A,-B,-C [clone G46 52.6, Pharmingen) and phycoerythrin (PE)-conjugated anti-HLA-DR (clone G46.6, Pharmingen). Isotype-matched control antibodies were included as negative controls (shaded curves in Fig. 7). Cyno-1-derived neural cells were cultured, harvested, and stained with FITC-labeled anti-HLAA,-B,-C and PE-labeled anti-HLA-DR as above and compared with cells stained with isotype-matched control antibodies (shaded curves in Fig. 7). Treatment of Cyno-1-derived neural cells with IFN-γ involved overnight incubation with human IFN-γ (40 ng/ml). Stained cells were analyzed with a FACScan flow cytometer and CELLQUEST software (Becton Dickinson).
Fig. 7.
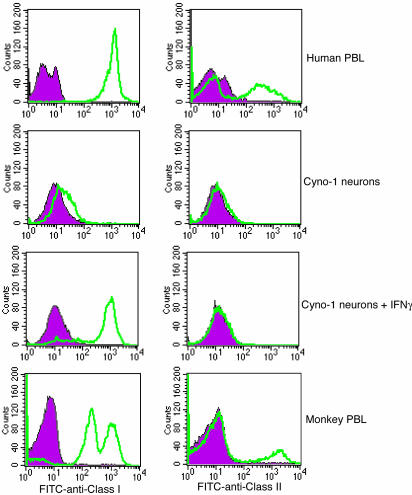
Immunological profile of Cyno-1 cells. PBLs and Cyno-1-derived neural cells were analyzed by flow cytometry to quantitate expression of M. fasicularis class I (anti-HLA-A,-B,-C) and class II (anti-HLA-DR) antigens and compared with cells stained with isotype-matched control antibodies (shaded curves). PBLs express both class I and class II (DR) antigens whereas differentiated Cyno-1-derived neurons do not express either class of antigen unless treated with IFN-γ.
Telomerase Activity Measurement. Telomerase activity was measured by using the TRAPeze kit (Intergen, Purchase, NY) as recommended by the manufacturer. Control template, buffer, and control extract were supplied by the TRAPeze kit. Extracts from the mouse feeder cells, the Cyno-1 cells (maintained on mouse feeder layer), and the differentiated Cyno-1 cells (grown for 14 days without mouse feeder layer) were normalized to the protein concentration. Heat inactivated extracts were boiled for 3 min before the assay.
Electrophysiology. The whole cell patch clamp technique was performed on differentiated stem cells that were continuously perfused with a Hepes-buffered saline (HBS) solution (containing, in mM: 150 NaCl, 10 Hepes, 2.5 KCl, 2.5 CaCl2, 1.0 MgCl2, 10 D-glucose, pH 7.4, with NaOH, osmolality 320 mmol/kg adjusted with sucrose). Tetrodotoxin (Calbiochem) was diluted from concentrated stocks into HBS and applied within 100 μm of the cell by using a linear array of fused-silica tubes (150 mm I.D., Hewlett-Packard) mounted on a manipulator. A Cs+-based internal solution (in mM: 130 CsCl, 10 Hepes, 10 EGTA, 1 CaCl2, 4 Mg-ATP, pH 7.2 with CsOH, osmolality 305 mmol/kg adjusted with sucrose) was used in the patch electrode.
Recordings were performed at room temperature according to published procedures by using an Axopatch ID amplifier (Axon Instruments, Foster City, CA) in voltage-clamp mode as described (18, 19). Whole-cell capacitance and series resistance were determined by fits of the capacitive transients during square-wave voltage steps by using standard software procedures contained within PCLAMP 7.0 software (Axon Instruments) and monitored throughout the recordings. Resting membrane potentials were -70 mV. Voltage-gated currents were elicited by square-wave membrane depolarizations to 0 mV.
Results
Creation and Characterization of Monkey Parthenogenetic Stem Cells. Stem cells were created via parthenogenetic activation of eggs as described (5). Briefly, 77 eggs were isolated from the ovaries of three different cynomolgus monkeys (Macaca fascicularis, ≈18 yr of age) after hormone-induced superovulation. The oocytes were then maintained in maturation medium for 36 h. Twentyeight eggs reached metaphase II stage and were subsequently activated by incubation with 10 μM ionomycin for 8 min, followed by culture with 2 mM 6-dimethylaminopurine for 4 h. Four embryos developed to the blastocyst stage after 8 days in culture (14%) (Fig. 1A). Immunosurgically isolated ICMs (20) were cultured on a feeder layer of mitotically inactive mouse embryonic fibroblasts in Dulbecco's minimal essential medium with 15% FCS (HyClone). Three ICM showed outgrowth within 1 week of plating, and one stable cell line (Cyno-1) was successfully derived (Fig. 1B). Cyno-1 cells displayed many features that are typical for ES cells: cytoplasmic lipid bodies, small cytoplasmic/nuclear ratio, and clearly distinguishable nucleoli. These cells were immunoreactively positive for alkaline phosphatase, stage-specific embryonic antigen 4, tumor rejection antigen 1-60, and tumor rejection antigen 1-81 and were positive for octamer-binding transcription factor 4 mRNA (Fig. 1 C-G) and negative for stage-specific embryonic antigen-1 and -3 (data not shown). These cells have been propagated for >2 yr maintaining their undifferentiated state. Karyotype analysis revealed 40 + 2 chromosomes, in accordance with the species of origin, M. fascicularis (data not shown).
Fig. 1.
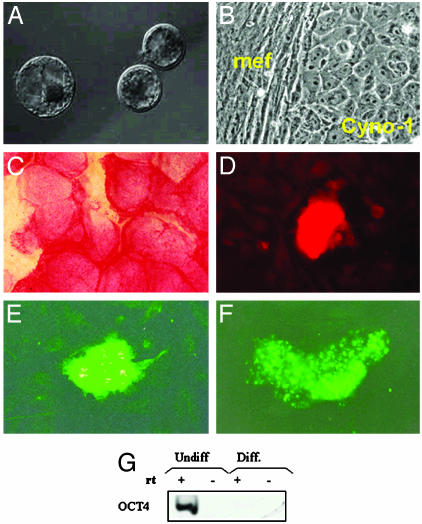
Characterization of parthenogenetic embryos and derived cell lines. (A) Parthenogenetically activated eggs at day 8 of development before ICM isolation. (B) Phase contrast of Cyno-1 stem cells growing on top of mitotically inactivated mouse feeder layer (mef). (C) Alkaline phosphatase staining. (D) Stage-specific embryonic antigen 4. (E) Tumor rejection antigen 1-60. (F) Tumor rejection antigen 1-81 staining. (G) RT-PCR octamer-binding transcription factor 4 expression in undifferentiated Cyno-1 cells. (Scale bars = 50 μm in A, 10 μm in B and D-F, and 4 mm in C.)
Parthenogenetic Stem Cell Differentiation. In vitro differentiation was induced by isolating the cells from the mouse feeder layer and culturing them in the presence of Dulbecco's minimal essential medium with 15% FCS; in some instances, 1,000 units of leukemia inhibitory factor was added to the media. A large variety of specialized cell types could be generated in vitro, such as spontaneously beating cardiomyocyte-like cells and ciliated epithelium, smooth muscle cells and cytokeratin-positive cells, as well as neuronal cells (data not shown).
To assess the differentiation capacity of Cyno-1 cells, we injected them into the peritoneal cavity of immunocompromised severe combined immunodeficient mice. Eight to 15 weeks after injection, teratomas were isolated and histologically analyzed. Microscopic observations revealed the presence of mature tissues and low frequency of mitotic figures, indicating their benign nature. Furthermore, derivatives of all three germ layers were observed, including cartilage, neurons, skin, and hair follicles (ectoderm), intestinal epithelia (endoderm), and muscle and bone (mesoderm) (Fig. 2).
Fig. 2.
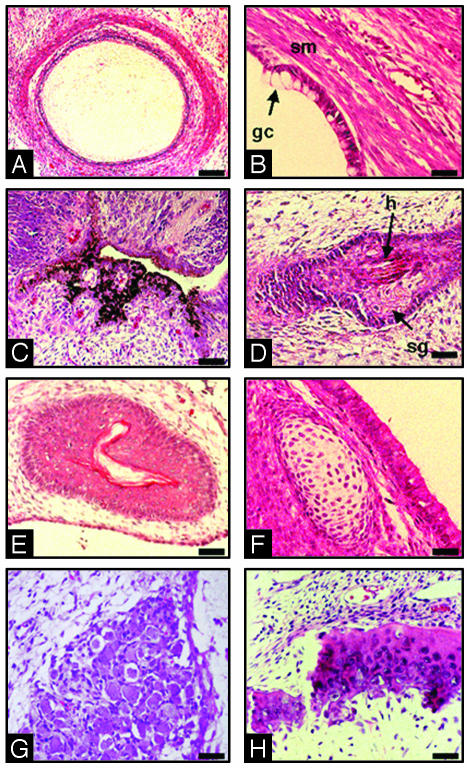
In vivo differentiation of Cyno-1 cells. Cells were injected i.p. in severe combined immunodeficient mice. Eight and 15 weeks after injection, teratomas 12 and 30 mm in diameter, respectively, were isolated, fixed with 10% paraformaldehyde, and paraffin-embedded. Sections were stained with hematoxylin/eosin. The following complex structures were observed: gut (A), intestinal epithelium with typical goblet cells (gc) and smooth muscle (sm) (B), neuronal tissue with melanocytes (C), hair follicle complex with evident hair (h) and sebaceous gland (sg) (D), skin (E), cartilage (F), ganglion cells (G), and bone (H). (Scale bars = 40 μmin A,10 μmin B and D-H, and 20 μmin C.)
Telomerase activity is often correlated with replicative immortality and is typically expressed in germ cells, cancer cells, and a variety of stem cells, including ES cells, but absent in most somatic cell types (21-23). Undifferentiated Cyno-1 cells displayed high levels of telomerase activity as detected by the TRAP assay (TRAPeze kit). However, no telomerase activity could be detected in differentiated progeny of Cyno-1 cells (Fig. 3A). These data indicate a physiologically normal control of telomerase activity in Cyno-1 cells.
Fig. 3.
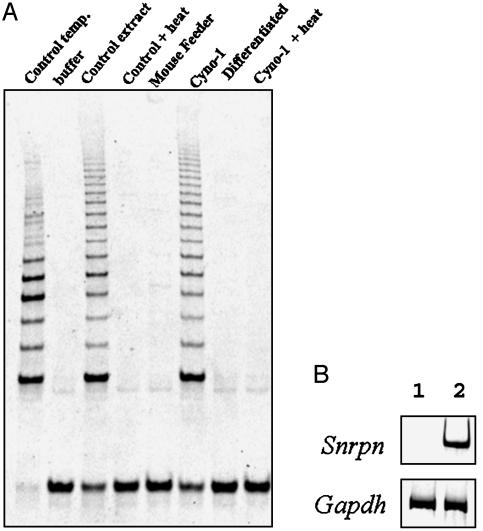
Telomerase activity. (A) Cyno-1 cells, maintained in the undifferentiated state on mouse feeder layers, express telomerase activity that diminishes to undetectable levels in differentiated Cyno-1 cells. (B) RT-PCR to detect expression of the paternally expressed imprinted gene Snrpn in Cyno-1 cells (lane 1) and in adult fibroblasts (lane 2) from the same species. The housekeeping gene Gapdh is used as a control to demonstrate that equal amounts of mRNA were used.
Genomic imprinting is initiated at gametogenesis and further modified during development. The small nuclear ribonucleoprotein polypeptide N (Snrpn) gene is an example of an imprinted gene that is expressed solely from the paternal allele (24, 25). It is monoallelically expressed from the onset of its expression at the four-cell stage (25). RT-PCR analysis confirmed the absence of Snrpn expression in Cyno-1 cells, whereas it is readily detected in heterozygous fibroblast cell cultures from the same species (Fig. 3B). Whereas biparental ES cells from M. fascicularis were unavailable to us for analysis, the Snrpn gene was readily detectable in biparental mouse ES cells under these same conditions (data not shown). These results indicate that the imprinting profile of at least one gene is consistent with the parthenogenetic origin of the Cyno-1 cells.
Nestin-Positive Neural Precursors. Cyno-1 ES cells were plated in flasks coated with fibronectin/laminin and NPMM (supplemented with bFGF, epidermal gowth factor, and neural survival factor-1). These cells begin to differentiate into a neuronal-like morphology. Within 10 days, they do not differentiate further and proliferate at a rate of 5- to 8-fold increase over a 10-day period (Figs. 4 and 5 A and B). Their stage of development seems to be similar to those described by Ying et al. (26). Specifically, these cells express a high amount of nestin, which is an intermediate filament found in the developing CNS, mesenchymal tissue of the developing pancreas, and immature skeletal muscle (National Center for Biotechnology Information Locus Link).
Fig. 4.
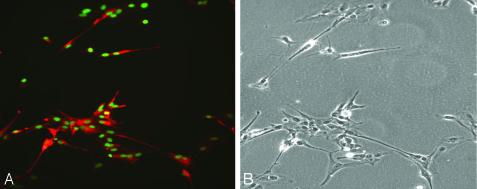
Nestin-positive neural precursors derived from Cyno-1 cells. (A) Neural precursors stained for nestin (green). The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). (B) Phase contrast of nestin precursors.
Fig. 5.
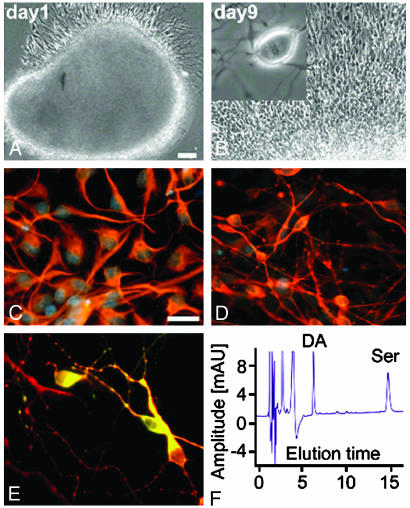
Neural differentiation of Cyno ES cells in vitro. (A) Phase contrast microscopy of proliferating Cyno1-derived neural precursors at day 1 in vitro (DIV1). (B) Same cluster of precursors shown at DIV9. The total cell number has increased by 5- to 8-fold over a 9-day period. (Inset) One of many mitotic figures. Immunohistochemical analyses after 5 days of neural differentiation in the absence of bFGF and epidermal growth factor and the presence of ascorbic acid revealed positive staining for glial fibrillary acidic protein (GFAP), an astrocytic marker seen in C, and TUJ1, a neuronal marker seen in D. (E) Sequential exposure to sonic hedgehog, FGF8b, and ascorbic acid yielded an average of 25% TUJ1+ neurons coexpressing tyrosine-hydroxylase (TH), a marker for dopamine neurons. (F) HPLC revealing the release of dopamine (DA) and serotonin (Ser).
With the removal of bFGF and epidermal growth factor and the addition of ascorbic acid, we are able to generate a high percentage of dopaminergic-like neurons (25% of TUJ+), glial, and epithelial cells. Immunocytochemistry stained positive for TUJ1, dopamine transporter (DAT), and microtubule associated protein-2, and negative for acetylcholine transferase, dopabeta-hydroxylase, and NeuN (Fig. 5 C-E and data not shown). Cyno-1-derived neurons exhibited both basal and KCl-evoked synaptic release of dopamine and serotonin (Fig. 5F). Single-cell electrophysiology was also performed on these differentiated neurons. At around day 20, they begin to express voltage-dependent sodium channels. The identities of these channels were confirmed by blocking with Tetrodotoxin. By day 30, 50% of neuron-like cells express these channels although none have yet to be identified in the neural precursors (Fig. 6). The differentiating cells assume morphological characteristics of neurons before functional ion channels become evident. This result suggests that there is a cascade of differentiation signals and/or precise coordination of differentiation events.
Fig. 6.
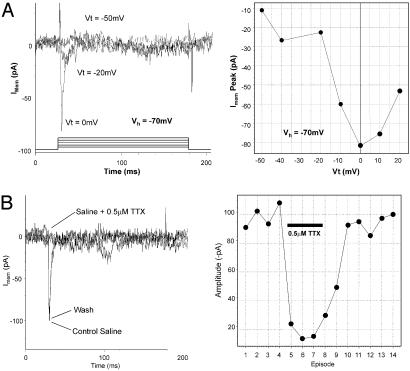
Single-cell electrophysiology. (A) Neurons derived from Cyno-1 express voltage-dependent inward currents that are blocked by tetrodotoxin. Currents were elicited by membrane depolarizations to 0 mV every 15 s from a holding potential of -70 mV. Application of 0.5 μM tetrodotoxin inhibited >90% of these currents. (B) Inhibition was complete within 30 s of tetrodotoxin application and washed completely in <1 min.
DNA Profiling of Cyno-1 Cells. To confirm their autologous origin, DNA profiling was performed. Total genomic DNA from the cynomolgus monkey oocyte donor (no. 5571) and from a preparation of cultured stem cells (Cyno-1; derived from no. 5571) were genotyped and compared by using seven simple sequence repeat (SSR) human markers (Research Genetics, Huntsville, AL) that had been shown previously to amplify monkey DNA and to discriminate between two individuals. The markers represent seven different chromosomes (nos. 3, 6, 7, 10, 11, 16, and 17) and in all cases except one (marker D16S403), alleles for no. 5571 were identical in number and size to the alleles for the Cyno-1 cells. An additional test was performed on DNA from no. 5571 and from the Cyno-1 cells (as well as two control animals). Micro SSPTM Generic HLA Class II DNA typing was performed in a 96-well tray format through the Wake Forest University-Baptist Medical Center Histocompatibility Laboratory. The data demonstrated that Cyno-1 stem cells and somatic cells from no. 5571 were indistinguishable from each other and therefore should be considered autologous (data not shown).
Histocompatibility Antigen Profile of Cyno-1. The histocompatibility antigen profile of Cyno-1-derived neurons was investigated and compared with lymphocytes from the oocyte donor by investigating polymorphic genes within the MHC that encode class I and class II cell surface proteins. These proteins present immunogenic peptides to CD8+ and CD4+ T cells, respectively. We have analyzed the Cyno-1-derived neural cells by flow cytometry for the expression of Mafa (MHC of M. fascicularis) class I and class II antigens. PBLs from the original cell donor expressed class I and class II antigens detected by antibodies specific for monomorphic HLA-A,-B,-C and HLA-DR antigens, respectively (Fig. 7). Seventy-five percent of PBLs were positive for class I, and 14% of PBLs were positive for class II. However, Cyno-1-derived neural cells were negative for both Mafa class I and class II antigens (Fig. 7), consistent with observations that these CNS cell types are class I- and class II-negative in normal murine CNS (27, 28). Viral infection or treatment with IFN-γ stimulates up-regulation of class I and class II expression in murine CNS cells. The ability of IFN-γ to up-regulate class I and class II expression by Cyno-1-derived neural precursor cells was investigated by preculturing these cells with IFN-γ (40 ng/ml) overnight before staining and flow cytometry. As shown in Fig. 7, pretreatment of Cyno-1 derived cells resulted in class I-specific staining with an intensity that was comparable to staining of normal human PBLs. However, IFN-γ treatment did not increase class II expression. These results support the prediction that an in vivo inflammatory response, expectedly involving IFN-γ expression, would up-regulate class I expression on transplanted Cyno-1-derived neural cells. Accordingly, in the event of transplantation into a nonisogenic animal, these cells should not escape surveillance by CD8+ cytotoxic T lymphocytes.
Discussion
We have generated a primate parthenogenetic cell line (Cyno-1) with ES cell-like properties that can be propagated in vitro in an undifferentiated state for at least 2 yr. These cells express telomerase activity consistent with their extended lifespan property. The in vitro derivation of large numbers of specific cell lineages from Cyno-1 cells, including the generation of unlimited numbers of dopaminergic neurons, is of particular interest. In the present context, we have demonstrated that these cells (i) express TH, (ii) release neurotransmitter (dopamine and serotonin), and (iii) are electrophysiologically active. For these reasons, we believe that neurons, differentiated from parthenogenetic stem cells, may provide an important source of therapeutic treatments. Clinical transplantation of specific fetal neurons has shown promise in the treatment of Parkinson's disease (29) and Huntington's disease (30), but obtaining such cells from animals or human fetal brain remains problematic. Neurons derived in vitro from a renewable source, such as CNS precursors (31), ES cells (32, 33), or stem cells of parthenogenetic origin, could alleviate some of the ethical and technical concerns of human cell therapy.
Although the Cyno-1 parthenogenetic stem cell seems, in all respects, to be similar to traditional ES cells, it is reasonable to question their viability and utility. They are, after all, exclusively derived from maternal DNA. When trying to understand how these parthenogenetic stem cells are capable of developing into functional tissue, it is important to remember the following characteristics of genetic imprinting. First, ES cells are isolated from the blastocyst stage, this stage exhibits low DNA methylation levels, and the effects of imprinting could be minimized (34). Second, imprinting, in some cases, has been shown to not be completely silent: there are reports of mRNA expressed from imprinted genes that should not have been transcribed (24). Third, Surani and Barton (9) suggest that parthenogenetic embryos do not develop to term because of a high frequency of errors in X chromosome inactivation that occurs in extraembryonic tissues when both X chromosomes are derived only from the female. One might conclude that the effects of imprinting have a significant effect on extraembryonic tissue, and not the ICM from which our stem cells are derived.
One could speculate that these parthenogenetically derived stem cells are capable of differentiating into a high percentage of electrophysiologically active dopaminergic neurons due to the effects of genetic imprinting. It is believed, for example, that 0.1-1% of all mammalian genes are imprinted (35). However, only ≈50 imprinted genes have been identified in mice, some of which are conserved in humans (36). One of the most interesting characteristics of imprinting is that it occurs in clusters. The imprinting cluster on mouse chromosome 7 and the corresponding human chromosome 11p15.5 contains 14 imprinted genes (37). Tyrosine hydroxylase (TH), the rate-limiting enzyme of dopamine synthesis (38), is located on chromosome 11p15.5 and resides in the middle of a very well-characterized imprinted region that spans 1 Mb. There are in this cluster some of the best characterized imprinted genes, such as H19, insulin growth factor 2, and insulin growth factor antisense (37). Whereas tyrosine hydroxylase is biallelically expressed in mice, its imprinting status in humans has yet to be determined (37). Could this high expression level of tyrosine hydroxylase (in maternally derived parthenote cells) suggest it as an imprinted gene in nonhuman primates? Moreover, are these epigenetic modifications associated with imprinting permitting the robust expression of TH in Cyno-1-derived neural cells?
Further data analysis of functional genomic studies at the neural precursor and differentiated stage show normal gene expression of housekeeping genes such as the mRNAs of the 60S ribosomal subunit, the glycolytic pathway, and the tricarboxylic acid (TCA) cycle. Most interesting, when the “Stemness” genes recently described by Ramalho-Santos et al. (39) were converted into their human orthologs, 96 of 216 were expressed in the parthenogenetically derived monkey neural precursor cells (J.D.H., J. C. Mychaleckyj, and K.E.V., unpublished results).
We report here the isolation and further characterization of nonhuman primate parthenogenetic stem cells. These cells may provide a novel tool for assessing the effects of genomic imprinting on cell differentiation and function during development in primates. Their striking differentiation capabilities (electrophysiologically active, dopamine-secreting neurons) indicate their therapeutic potential and suggest a valid alternative to biparentally derived ES cells.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Regenerative Medicine,” held October 18-22, 2002, at the Arnold and Mabel Beckman Center of the National Academies of Science and Engineering in Irvine, CA.
Abbreviations: ES cell, embryonic stem cell; Cyno-1 cell, parthenogenetically derived stem cell line from the cynomolgus macaque; bFGF, basic fibroblast growth factor; Snrpn, small nuclear ribonucleoprotein polypeptide N; TUJ1, β-tubulin III; PBL, peripheral blood lymphocytes; ICM, inner cell mass.
References
- 1.Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S. & Jones, J. M. (1998) Science 282, 1145-1147. [DOI] [PubMed] [Google Scholar]
- 2.Thomson, J. A., Kalishman, J., Golos, T. G., Durning, M., Harris, C. P., Becker, R. A. & Hearn, J. P. (1995) Proc. Natl. Acad. Sci. USA 92, 7844-7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomson, J. A., Kalishman, J., Golos, T. G., Durning, M., Harris, C. P. & Hearn, J. P. (1996) Biol. Reprod. 55, 254-259. [DOI] [PubMed] [Google Scholar]
- 4.Suemori, H., Tada, T., Torii, R., Hosoi, Y., Kobayashi, K., Imahie, H., Kondo, Y., Iritani, A. & Nakatsuji, N. (2001) Dev. Dyn. 222, 273-279. [DOI] [PubMed] [Google Scholar]
- 5.Cibelli, J. B., Grant, K. A., Chapman, K. B., Cunniff, K., Worst, T., Green, H. L., Walker, S. J., Gutin, P. H., Vilner, L., Tabar, V., et al. (2002) Science 295, 819. [DOI] [PubMed] [Google Scholar]
- 6.Jiang, Y., Jahagirdar, B. N., Reinhardt, R. L., Schwartz, R. E., Keene, C. D., Ortiz-Gonzalez, X. R., Reyes, M., Lenvik, T., Lund, T., Blackstad, M., et al. (2002) Nature 418, 41-49. [DOI] [PubMed] [Google Scholar]
- 7.Boediono, A., Suzuki, T., Li, L. Y. & Godke, R. A. (1999) Mol. Reprod. Dev. 53, 159-170. [DOI] [PubMed] [Google Scholar]
- 8.Strain, L., Warner, J. P., Johnston, T. & Bonthron, D. T. (1995) Nat. Genet. 11, 164-169. [DOI] [PubMed] [Google Scholar]
- 9.Surani, M. A. & Barton, S. C. (1983) Science 222, 1034-1036. [DOI] [PubMed] [Google Scholar]
- 10.Hagemann, L. J., Peterson, A. J., Weilert, L. L., Lee, R. S. & Tervit, H. R. (1998) Mol. Reprod. Dev. 50, 154-162. [DOI] [PubMed] [Google Scholar]
- 11.Ozil, J. P. & Huneau, D. (2001) Development 128, 917-928. [DOI] [PubMed] [Google Scholar]
- 12.Kure-bayashi, S., Miyake, M., Okada, K. & Kato, S. (2000) Theriogenology 53, 1105-1119. [DOI] [PubMed] [Google Scholar]
- 13.Surani, M. A. (1998) Cell 93, 309-312. [DOI] [PubMed] [Google Scholar]
- 14.Monk, M. (1988) Genes Dev. 2, 921-925. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki, H., Jones, P. A., Chaillet, J. R., Ferguson-Smith, A. C., Barton, S. C., Reik, W. & Surani, M. A. (1992) Genes Dev. 6, 1843-1856. [DOI] [PubMed] [Google Scholar]
- 16.McGrath, J. & Solter, D. (1984) Cell 37, 179-183. [DOI] [PubMed] [Google Scholar]
- 17.Berkowitz, R. S. & Goldstein, D. P. (1996) N. Engl. J. Med. 335, 1740-1748. [DOI] [PubMed] [Google Scholar]
- 18.Hamill, O. P., Marty, A., Neher, E., Sakmann, B. & Sigworth, F. J. (1981) Pflugers Arch. 391, 85-100. [DOI] [PubMed] [Google Scholar]
- 19.McCool, B. A. & Farroni, J. S. (2001) Eur. J. Neurosci. 14, 1082-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brigit Hogan, Beddington, R., Constantini, F. & Lacy, E. (1994) Manipulating the Mouse Embryo (Cold Spring Harbor Lab. Press, Planview, NY).
- 21.Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, M. D., Ho, P. L., Coviello, G. M., Wright, W. E., Weinrich, S. L. & Shay, J. W. (1994) Science 266, 2011-2015. [DOI] [PubMed] [Google Scholar]
- 22.Armstrong, L., Lako, M., Lincoln, J., Cairns, P. M. & Hole, N. (2000) Mech. Dev. 97, 109-116. [DOI] [PubMed] [Google Scholar]
- 23.Amit, M., Carpenter, M. K., Inokuma, M. S., Chiu, C. P., Harris, C. P., Waknitz, M. A., Itskovitz-Eldor, J. & Thomson, J. A. (2000) Dev. Biol. 227, 271-278. [DOI] [PubMed] [Google Scholar]
- 24.Szabo, P. & Mann, J. R. (1994) Development 120, 1651-1660. [DOI] [PubMed] [Google Scholar]
- 25.Mann, M., Latham, K. E. & Varmuza, S. (1995) Dev. Genet. 17, 223-232. [DOI] [PubMed] [Google Scholar]
- 26.Ying, Q. L., Stavridis, M., Griffiths, D., Li, M. & Smith, A. (2003) Nat. Biotechnol. 21, 183-186. [DOI] [PubMed] [Google Scholar]
- 27.Altintas, A., Cai, Z., Pease, L. R. & Rodriguez, M. (1993) J. Immunol. 151, 2803-2812. [PubMed] [Google Scholar]
- 28.Rodriguez, M., Pierce, M. L. & Howie, E. A. (1987) J. Immunol. 138, 3438-3442. [PubMed] [Google Scholar]
- 29.Olanow, C. W., Kordower, J. H. & Freeman, T. B. (1996) Trends Neurosci. 19, 102-109. [DOI] [PubMed] [Google Scholar]
- 30.Bachoud-Levi, A., Bourdet, C., Brugieres, P., Nguyen, J. P., Grandmougin, T., Haddad, B., Jeny, R., Bartolomeo, P., Boisse, M. F., Barba, G. D., et al. (2000) Exp. Neurol. 161, 194-202. [DOI] [PubMed] [Google Scholar]
- 31.Studer, L., Tabar, V. & McKay, R. D. G. (1998) Nat. Neurosci. 1, 290-294. [DOI] [PubMed] [Google Scholar]
- 32.Lee, S. H., Lumelsky, N., Studer, L., Auerbach, J. M. & McKay, R. D. (2000) Nat. Biotechnol. 18, 675-679. [DOI] [PubMed] [Google Scholar]
- 33.Kawasaki, H., Mizuseki, K., Nishikawa, S., Kaneko, S., Kuwana, Y., Nakanishi, S., Nishikawa, S. I. & Sasai, Y. (2000) Neuron 28, 31-40. [DOI] [PubMed] [Google Scholar]
- 34.Allen, N. D., Barton, S. C., Hilton, K., Norris, M. L. & Surani, M. A. (1994) Development 120, 1473-1482. [DOI] [PubMed] [Google Scholar]
- 35.Reik, W., Santos, F. & Dean, W. (2003) Theriogenology 59, 21-32. [DOI] [PubMed] [Google Scholar]
- 36.Lopes, S., Lewis, A., Hajkova, P., Dean, W., Oswald, J., Forne, T., Murrell, A., Constancia, M., Bartolomei, M., Walter, J. & Reik, W. (2003) Hum. Mol. Genet. 12, 295-305. [DOI] [PubMed] [Google Scholar]
- 37.Reik, W. & Walter, J. (2001) Nat. Rev. Genet. 2, 21-32. [DOI] [PubMed] [Google Scholar]
- 38.Kumer, S. C. & Vrana, K. E. (1996) J. Neurochem. 67, 443-462. [DOI] [PubMed] [Google Scholar]
- 39.Ramalho-Santos, M., Yoon, S., Matsuzaki, Y., Mulligan, R. C. & Melton, D. A. (2002) Science 298, 597-600. [DOI] [PubMed] [Google Scholar]