

Abstract
To clarify the biological rationale for social regulation of gene expression, this study sought to identify the specific immune cell types that are transcriptionally sensitive to subjective social isolation (loneliness). Using reference distributions for the expression of each human gene in each major leukocyte subtype, we mapped the cellular origin of transcripts found to be differentially expressed in the circulating immune cells from chronically lonely individuals. Loneliness-associated genes derived primarily from plasmacytoid dendritic cells, monocytes, and, to a lesser extent, B lymphocytes. Those dynamics reflected per-cell changes in the expression of inducible genes and related more strongly to the subjective experience of loneliness than to objective social network size. Evolutionarily ancient myeloid antigen-presenting cells appear to have evolved a transcriptional sensitivity to socioenvironmental conditions that may allow them to shift basal gene expression profiles to counter the changing microbial threats associated with hostile vs. affine social conditions.
Keywords: social genomics, inflammation, bioinformatics, ecological immunology
Research in social genomics has linked adverse life circumstances to changes in the expression of hundreds of genes in circulating human immune cells (1–3). Those genes subject to socioenvironmental regulation do not represent a random cross-section of our ∼22,000 genes, however. Instead, in leukocytes sampled from people confronting a diverse array of adverse social conditions, including chronic loneliness (4), imminent bereavement (5), depression (6), and low socioeconomic status (7, 8), gene expression profiling shows a recurrent up-regulation of proinflammatory genes and down-regulation of genes involved in IFN-mediated antiviral responses and IgG antibody production (1–3). These dynamics appear to stem from coordinated changes in the activity of gene-regulating transcription factors, including reduced sensitivity of the glucocorticoid receptor (GR) and consequent activation of the proinflammatory NF-κB transcription factor that it would otherwise inhibit (4, 5, 7), as well as decreased activity of IFN response factors and modulation of GATA, EGR, and CREB/ATF transcription factors (3–5, 7, 9). The resulting transcriptional alterations appear to place socially stressed individuals at increased risk for chronic inflammation-related illnesses, such as heart disease, neurodegeneration, and some types of cancer (10, 11). Why would the immune system activate such a hazardous transcriptional program in response to social adversity?
To clarify how and why social environments regulate immune function (11–14), it would be helpful to know which specific immune cells mediate those effects. Circulating leukocytes are an aggregate population composed of several distinct cell subsets that express different genes and perform different functional roles in pathogen recognition, immune response, and tissue repair (15). In the present study, we sought to determine which of those cell types is most sensitive to socioenvironmental adversity (i.e., which specific type of leukocyte is predominately responsible for the change in aggregate gene expression profiles observed in the leukocyte pool as a whole). Is it the monocytes, which patrol the body surveilling for infectious agents and damaged tissue and coordinate the early inflammatory stage of an immune response? Perhaps it is the natural killer (NK) cells, which search out and destroy cells lacking the distinctive MHC molecular name tags that distinguish our own cells from foreign cells. Perhaps it is the T lymphocytes which are most sensitive, as they coordinate the development of immune responses (CD4+ helper T lymphocytes) or destroy our own cells that have been hijacked by viruses and other intracellular pathogens (CD8+ cytotoxic T cells). Maybe it is the B cells, which synthesize antibodies to help combat extracellular pathogens, such as parasitic organisms or viral particles trafficking from one cell to another. Another possibility is the dendritic cells, which, like monocytes, patrol for damage and initiate inflammatory responses but also play a unique role in activating T-cell responses. Determining the particular type of cell that is most sensitive to our macrolevel socioenvironmental conditions is, from an immunologist's perspective, the key to understanding the underlying logic of a socially regulated immune system (15).
This study seeks to identify the specific cell type responsible for the global leukocyte gene expression dynamics observed in one of the earliest major social genomics studies — an analysis identifying systematic differences in the expression of 209 gene transcripts in circulating leukocytes from people who experienced themselves as alone and distant from others consistently over the course of 3 y (i.e., chronically lonely) (4). Lack of close social ties is a well-established risk factor for diseases involving the immune system and inflammation (16, 17), and this study established a functional genomic framework for understanding those effects. This study also uncovered several major gene regulation themes that have subsequently reemerged in other studies of social adversity, including increased expression of inflammation-related genes and reduced expression of genes involved in Type I IFN responses and IgG antibody production (1–4). As such, this study provides a natural context for determining which leukocytes are most sensitive to our experienced social environment.
Results
To identify the specific cell type mediating any observed difference in gene expression within the circulating leukocyte pool, we first quantified the extent to which each human gene transcript was generally expressed predominately by monocytes, plasmacytoid dendritic cells, NK cells, CD4+ T lymphocytes, CD8+ T lymphocytes, or B lymphocytes (Eq. 1), based on independent reference data on the expression of all named human genes in isolated samples of each cell type (18). Transcript origin diagnosticity scores for each gene and cell type are presented in Dataset S1. Validation studies of five independent datasets capturing experimentally induced transcriptional alterations in isolated human monocytes, plasmacytoid dendritic cells, NK cells, T lymphocytes, and B lymphocytes confirmed that the transcript origin diagnostic score used here correctly identified the cellular origin of those genome-wide transcriptional alterations in each case (Table 1). Across different cell types, heterogeneous microarray platforms, and diverse experimental manipulations (including cytokine or neurotransmitter stimulation, transcription factor overexpression, and myeloid cell differentiation), transcript origin diagnosticity scores consistently reached the highest degree of statistical significance for the specific cell type known to have generated the observed data (all P < 0.01). Diagnosticity scores were also highly reliable as measured by split-half correlations computed within each study (average r = 0.91, P = 0.0004).
Table 1.
Transcript origin analysis of experimentally induced transcriptional alterations in isolated leukocyte subsets
| Isolated cell type (comparison) | Mean TOA diagnosticity score | Difference from genome mean TOA score* (±SE) | P |
| Monocyte (LPS + IFN-γ vs. IL-4) | |||
| Monocyte | 1.35 | 1.15 ± 0.09 | 0.0000 |
| Dendritic cell | 0.90 | 0.49 ± 0.14 | 0.0002 |
| NK cell | 0.67 | −0.25 ± 0.17 | 0.9325 |
| CD4+ T cell | 0.15 | −0.26 ± 0.05 | 0.9999 |
| CD8+ T cell | 0.05 | −0.16 ± 0.04 | 0.9999 |
| B cell | −0.76 | 0.11 ± 0.09 | 0.1082 |
| Dendritic cell (vs. monocyte) | |||
| Monocyte | −0.25 | −0.05 ± 0.04 | 0.8980 |
| Dendritic cell | 0.22 | 0.08 ± 0.03 | 0.0040 |
| NK cell | 0.60 | 0.05 ± 0.03 | 0.0588 |
| CD4+ T cell | 0.28 | −0.05 ± 0.02 | 0.9768 |
| CD8+ T cell | 0.15 | −0.03 ± 0.02 | 0.8852 |
| B cell | −1.30 | −0.02 ± 0.04 | 0.6579 |
| NK cell (untreated vs. IL-2 + IL-15) | |||
| Monocyte | 0.33 | 0.11 ± 0.09 | 0.3468 |
| Dendritic cell | 1.23 | 0.81 ± 0.08 | 0.0392 |
| NK cell | 2.38 | 1.45 ± 0.08 | 0.0080 |
| CD4+ T cell | −0.11 | −0.49 ± 0.06 | 0.9957 |
| CD8+ T cell | −0.18 | −0.37 ± 0.06 | 0.9924 |
| B cell | −0.90 | −0.01 ± 0.10 | 0.5254 |
| T lymphocyte (untreated vs. norepinephrine) | |||
| Monocyte | 0.36 | −0.01 ± 0.07 | 0.5547 |
| Dendritic cell | 0.46 | −0.02 ± 0.08 | 0.6143 |
| NK cell | 0.75 | −0.18 ± 0.17 | 0.8522 |
| CD4+ T cell | 0.45 | 0.10 ± 0.04 | 0.0029 |
| CD8+ T cell | 0.21 | 0.07 ± 0.03 | 0.0078 |
| B cell | −0.76 | 0.05 ± 0.09 | 0.3083 |
| B lymphocyte (untreated vs. EBNA-2†) | |||
| Monocyte | −0.17 | −0.38 ± 0.27 | 0.9240 |
| Dendritic cell | 0.86 | 0.46 ± 0.38 | 0.1130 |
| NK cell | 0.69 | −0.23 ± 0.48 | 0.6818 |
| CD4+ T cell | −0.05 | −0.45 ± 0.15 | 0.9983 |
| CD8+ T cell | −0.14 | −0.34 ± 0.12 | 0.9960 |
| B cell | 1.31 | 2.19 ± 0.27 | 0.0000 |
Positive diagnosticity indicates that differentially expressed genes originate predominately from the analyzed cell type. Negative values are uninformative, implying that transcripts originate from other cell types or from the analyzed cell type as well as other cell types. TOA, Transcript Origin Analysis.
*Positive values indicate that differentially expressed genes originate from the indicated cell type. Negative values are uninformative (transcripts are not distinctive to target cell type or are distinctive to other cells).
†Epstein–Barr virus nuclear antigen 2.
Primary discovery studies applied transcript origin analysis to identify the cellular source of 209 gene transcripts showing ≥30% difference in expression in circulating leukocytes from six chronically lonely individuals and eight demographically matched individuals reporting consistently high levels of social contact and support (4). Study participants were healthy older adults aged 55–72 y at leukocyte capture who had been sampled from the Chicago metropolitan area and broadly represented its demographic composition. Chronically lonely individuals were identified by the UCLA Loneliness Scale scores in the top 15% of the sample distribution consistently over the course of 3 y, whereas low-lonely individuals consistently scored in the bottom 15% of the distribution. Among the 209 differentially expressed mRNA species (corresponding to 144 named human genes), 78 (37%) were overexpressed in leukocytes from high-lonely individuals and 131 (63%) were underexpressed (i.e., relatively overexpressed in nonlonely individuals; specific transcripts are listed at http://genomebiology.com/content/supplementary/gb-2007-8-9-r189-s1.doc). Previous bioinformatic analyses identified general functional characteristics of differentially expressed genes, including up-regulation of transcripts involved in inflammation, cell proliferation, and transcription control and down-regulation of transcripts involved in innate antiviral responses, antibody production, and cell death (4).
Fig. 1A presents results showing that loneliness-associated transcripts derived predominately from plasmacytoid dendritic cells and monocytes. Transcripts expressed by B lymphocytes and NK cells appeared at approximately the same rate in the differentially expressed gene pool as they did across the genome as a whole, and transcripts expressed predominately by CD4+ and CD8+ T lymphocytes were markedly nondiagnostic (i.e., less frequently observed among loneliness-associated transcripts than expected in a random sample of all human genes).
Fig. 1.

Transcript origin analysis of genes differentially expressed in circulating leukocytes from chronically lonely vs. nonlonely individuals in a discovery sample of 14 individuals in study year 4 (A) and a confirmation sample of 93 individuals in study year 8 (B). Data represent mean ± SE cell type diagnosticity score, with P values testing overrepresentation relative to the genome-wide null distribution for each cell type. Positive diagnosticity indicates that differentially expressed genes originate predominately from the analyzed cell type. Negative values are uninformative, implying that transcripts originate from other cell types or from the analyzed cell type as well as other cell types. Chronically lonely individuals were identified by UCLA Loneliness Scale scores in the top 15% of the sample distribution for each of the study's first 3 y (43% of the analyzed subsample) (A) or in the top quartile for 3 y or more of the study's first 5 y (26% of the analyzed sample) (B). All participants are healthy older adults enrolled in the CHASRS (4).
To determine whether loneliness-associated transcriptional up-regulation vs. down-regulation might be occurring in different cell types, we carried out separate transcript origin analyses for each gene set. Results in Table 2 show that the genes up-regulated in circulating blood from lonely individuals were predominately expressed by dendritic cells, whereas down-regulated transcripts originated from dendritic cells, monocytes, and, to a marginally significant extent, B lymphocytes.
Table 2.
Cellular origin of transcripts up- and down-regulated in the circulating leukocyte pool of chronically lonely individuals
| Mean cell diagnosticity score | Difference from genome-wide mean diagnosticity* (±SE) | t† | P | |
| Genes up-regulated | ||||
| Monocyte | −0.33 | −0.14 ± 0.18 | −0.79 | 0.7485 |
| Dendritic cell | 0.52 | 0.39 ± 0.15 | 2.69 | 0.0042 |
| NK cell | 0.48 | −0.07 ± 0.15 | −0.44 | 0.6704 |
| CD4+ T cell | 0.31 | −0.01 ± 0.12 | −0.12 | 0.5472 |
| CD8+ T cell | 0.32 | 0.15 ± 0.11 | 1.33 | 0.0926 |
| B cell | −1.61 | −0.35 ± 0.20 | −1.77 | 0.9604 |
| Genes down-regulated | ||||
| Monocyte | 0.26 | 0.45 ± 0.09 | 4.75 | 0.0001 |
| Dendritic cell | 0.82 | 0.70 ± 0.08 | 9.20 | 0.0001 |
| NK cell | 0.44 | −0.11 ± 0.08 | −1.39 | 0.9175 |
| CD4+ T cell | −0.27 | −0.59 ± 0.06 | −9.52 | 0.9999 |
| CD8+ T cell | −0.38 | −0.54 ± 0.06 | −9.30 | 0.9999 |
| B cell | −1.09 | 0.17 ± 0.10 | 1.69 | 0.0459 |
Positive diagnosticity indicates that differentially expressed genes originate predominately from the analyzed cell type. Negative values are uninformative, implying that transcripts originate from other cell types or from the analyzed cell type as well as other cell types.
*Positive values imply that differentially expressed genes originate from the indicated cell type. Negative values are uninformative (transcripts originate from other cell types or from the indicated cell type as well as other cells).
†Up-regulated df = 94, down-regulated df = 350.
We next asked whether the “socially sensitive” cell types responded primarily to the subjective experience of social isolation or to the objective density of an individual's social network. Objective isolation, as measured by the social network index (SNI) (19), was only modestly correlated with subjective social isolation [r(12) = 0.26, P = 0.3629]. Simultaneous multivariate analyses showed that subjective social isolation was associated with a substantially greater number of differentially expressed genes than was objective social isolation [377 transcripts differed by ≥30% as a function of UCLA Loneliness Scale scores vs. 161 as a function of the SNI; difference: X2(1) = 86.97, P < 0.0001, odds ratio (OR) = 2.36]. In contrast to results for subjective social isolation, transcripts associated with objective social isolation did not originate disproportionately from either monocytes or dendritic cells (P = 0.5703 and P = 0.1937, respectively; both d < 0.10) but, instead, derived predominately from B lymphocytes [t(200) = 4.19, P < 0.0001, d = 0.29].
In a final set of discovery analyses, we asked whether the observed differences in loneliness-related gene expression stemmed from differing abundance of each cell type within the total leukocyte pool or whether they reflected per-cell changes in the intensity of gene expression. Initial analyses found no significant difference in the expression of any leukocyte subset-defining marker gene (CD14 for monocytes, BDCA-4/NRP1 for dendritic cells, CD56/NCAM1 for NK cells, CD4 for CD4+ T cells, CD8A for CD8+ T cells, and CD19 for B lymphocytes) (18) as a function of loneliness [all differences <8%, all t(12) < 1.22, P > 0.2462]. Transcript origin analyses also yielded similar results after gene expression data were adjusted for variations in the abundance of those cell type-defining marker transcripts using analysis of covariance [i.e., plasmacytoid dendritic cell and monocyte-derived transcripts remained overrepresented, both t(2,289) > 2.67, P < 0.0077, d > 0.05; genes predominately expressed by other cell types showed no differential contribution, all t(2,289) < 1.58, P > 0.1169, d < 0.05].
To verify discovery study results, we carried out parallel transcript origin analyses of circulating leukocyte gene expression profiles collected 4 y later from all 93 study participants for whom blood samples were available. Chronically lonely individuals were identified by scores in the top quartile of the UCLA Loneliness Scale distribution in 3 y or more of the study's first 5 y (25 individuals, or 26% of the sample), and all analyses controlled for age; gender; race/ethnicity; marital status; (log) household income; body mass index (BMI); and the relative percentage of granulocytes, monocytes, and lymphocytes in the assayed leukocyte sample. Microarray transcriptional profiling identified 98 genes showing a ≥15% difference in average expression in high-lonely individuals relative to the remainder of the sample [i.e., exceeding the 5% false discovery rate (FDR) reliability threshold; 25 up-regulated and 73 down-regulated, listed in Table S1]. Twenty-two (22.4%) of those transcripts were also identified as being differentially expressed in the discovery sample (significantly greater than the <0.01% concordance expected by chance; binomial P < 10−10; annotated in Table S1). Up-regulated transcripts included genes involved in leukocyte activation and inflammation (CCL4L1, EGR1, EGR2, FOSB, HLA-DR, and PTGS2/COX2) and cellular responses to oxidative stress (GSTM1 and GSTM2). Down-regulated genes were associated with Type I IFN innate antiviral responses (IFI27, IFI44, IFI44L, IFI6, IFIT1, IFIT2, IFIT3, ISG15, and MX1) and innate antimicrobial responses (ORM1 and RNASE3). Transcript origin analyses again found differentially expressed genes to derive predominately from plasmacytoid dendritic cells, monocytes, and B lymphocytes (Fig. 1B). Up-regulated transcripts derived predominately from dendritic cells [t(26) = 1.88, P = 0.0362, d = 0.36], whereas down-regulated transcripts were associated with B lymphocytes [t(99) = 2.40, P = 0.0091, d = 0.24], monocytes [t(99) = 2.72, P = 0.0038, d = 0.27], and dendritic cells [t(99) = 1.85, P = 0.0336, d = 0.19].
Multivariate analyses comparing the effects of objective vs. subjective social isolation also confirmed discovery study results, with those two variables again showing modest correlation [r(91) = 0.31, P = 0.0024] and subjective isolation associating with significantly more differentially expressed genes than objective isolation [81 vs. 38 transcripts; X2(1) = 15.59, P < 0.0001, OR = 2.14]. Transcripts distinctively associated with objective social isolation derived predominately from B lymphocytes [t(40) = 4.46, P < 0.0001, d = 0.69], with no other cell type contributing significantly [all t(40) < 1.57, P ≥ 0.0618, d = 0.25]. Transcripts associated with subjective social isolation, net of objective social isolation, derived from monocytes [t(112) = 3.91, P < 0.0001, d = 0.37], dendritic cells [t(112) = 2.83, P = 0.0028, d = 0.28], and, for down-regulated transcripts, B lymphocytes [t(112) = 1.88, P = 0.0312, d = 0.18].
A final validation analysis also confirmed that a standardized composite of the top 10 monocyte-diagnostic transcripts correlated significantly with the measured fraction of monocytes in the assayed leukocyte pool [r(85) = 0.61, P < 0.0001], and the sum of composite scores for CD4+ T cells, CD8+ T cells, NK cells, and B cells correlated significantly with the measured fraction of lymphocytes [r(85) = 0.47, P < 0.0001].
Discussion
These analyses identify plasmacytoid dendritic cells and monocytes as the key cellular mediators of the human immune system's transcriptional response to loneliness (4). Those two myeloid lineage antigen-presenting cells (APCs) contributed disproportionately to the set of transcripts differentially expressed in the circulating leukocytes of chronically lonely individuals, whereas genes expressed by other cell types showed little differential expression as a function of loneliness. Consistent with the hypothesis that CNS-mediated differences in neural or endocrine signaling are responsible for such effects (1–3, 11), differential expression of monocyte- and dendritic cell-derived transcripts was strongly associated with the subjective experience of social isolation but showed no significant relationship to objective social network size. Analyses also showed that the observed differences in APC gene expression profiles do not stem from differences in the prevalence of those cell types within the circulating leukocyte pool but, instead, reflect per-cell changes in the expression of inducible genes that are flexibly expressed depending on environmental conditions (1, 2, 4). Thus, among all the cell types within the circulating leukocyte pool, plasmacytoid dendritic cells and monocytes appear to show a unique degree of transcriptional sensitivity to the experienced social environment.
Transcript origin analyses also indicated that some of the transcriptional down-regulation associated with loneliness originates in B lymphocytes. That finding is consistent with previous data linking loneliness to decreased expression of genes involved in antibody production (which occurs in B-lymphocyte lineage cells) (15). Interestingly, reduced B-cell gene expression was the only cell-specific transcriptional dynamic linked to objective social network size, suggesting that this specific component of loneliness-related transcriptional inhibition might potentially stem from socially mediated differences in pathogen exposure (20). Thus, the overall leukocyte transcriptional response to loneliness may involve multiple cellular components that are activated through distinct biological pathways [e.g., APCs sensitive to threat-related neuroendocrine dynamics associated with subjective loneliness (21) and B-lymphocyte dynamics stemming from social-behavioral differences (20)].
Based on the identification of myeloid APCs as our most socially sensitive leukocytes, what can we infer about the potential consequences of experienced social isolation for human immune function? Both of these cell types mediate “first line of defense” innate immune responses (22, 23) and derive from the phylogenetically ancient myeloid cell lineage that is developmentally distinct from the lymphoid lineage cells that showed minimal transcriptional sensitivity to loneliness (i.e., NK cells, CD4+ T cells, CD8+ T cells, B lymphocytes) (15). APCs produce the immediate inflammatory response to tissue damage that initiates immune responses (15, 23). However, long-term or recurrent inflammation also promotes the chronic diseases that dominate “modern mortality,” including atherosclerosis in cardiovascular disease (24, 25), cancer initiation and metastasis (26, 27), and neurodegeneration (10). The APC transcriptional activation observed here is consistent with previous data showing increased inflammatory gene expression in lonely individuals (4) and in people confronting a diverse array of other social adversities (1–3), suggesting a molecular basis for epidemiological links between social adversity and inflammation-mediated heart disease (11, 28). Dendritic cells and monocytes also deploy genetically preprogrammed innate immune responses against evolutionarily conserved pathogens, such as viruses (15, 23). Social stress is known to suppress innate antiviral responses via neural inhibition of IFN gene transcription (4, 29–31). Such dynamics comprise a significant part of the APC transcriptional repression observed here (particularly in plasmacytoid dendritic cells, which are the primary IFN-producing leukocytes) (23), providing a cellular context for relationships between social adversity and viral infection (11, 28, 31, 32). As sentinels for damaged tissue and invading pathogens, APCs have also evolved a central role in the activation and guidance of T and B lymphocytes as they mount more complex adaptive immune responses (15, 23). Bidirectional dendritic cell regulation by social adversity is consistent with previous neural manipulation studies (33) showing a redirection of T lymphocytes away from “T-helper 1” (Th1) responses effective against viruses and other intracellular pathogens and toward Th2 responses targeting extracellular pathogens, such as bacteria (11, 15, 23). Monocytes and dendritic cells also orchestrate the long-term trophic development, maintenance, and repair of healthy tissue and incidentally contribute to cancer development and metastasis in the process (26, 27). Thus, the present results may also provide a cellular context for epidemiological relationships between the social circumstances of patients who have cancer and the risk for disease recurrence or progression (34). Based on the known functions of monocytes and dendritic cells, the social signal transduction pathways analyzed here (1) appear to target the leukocyte subsets that are the most evolutionarily ancient, most immediately responsive to tissue damage and invading pathogens, and most strategically positioned to direct the adaptive immune responses that emerge later in both our collective evolutionary history and our individual physiological responses to pathogens.
Given those biological implications, what might be the underlying teleological purpose for socially sensitive APCs? Given that (i) infectious disease has historically driven both immune system evolution and human natural selection (35) and (ii) social processes are central to the epidemiology of infections (36, 37), the immune system may have developed a molecular sensitivity to the neural and endocrine correlates of our social conditions (e.g., sympathetic nervous system and hypothalamic-pituitary-adrenal axis) (1–3, 11) to allostatically anticipate the types of pathogens we are most likely to encounter and optimally redeploy our molecular defenses in response (38, 39). Most viruses are highly species-specific and cannot survive long outside their host tissue environment (40). As such, viral infection is greatly facilitated by social contact with conspecifics in general and particularly by the long-term, recurrent, and physically intimate contact associated with affine social conditions (e.g., friendship, family, mate relationships). Under those circumstances, a forward-looking immune system would shift its basal transcriptional stance toward innate antiviral and Th1 immune responses to counter the increased threat of viral infection (i.e., the transcriptional profile observed in nonlonely individuals). In contrast, many bacteria and other extracellular pathogens can survive for long periods in the external environment and easily transmit across species boundaries. Infections with those agents are greatly facilitated by wounding and other types of tissue damage associated with both general threat (e.g., predation injury, to which socially isolated individuals are particularly vulnerable) (41) and hostile social interactions with conspecifics (e.g., social conflict, rejection). Under those adverse social circumstances, the likelihood of viral infection is reduced, the likelihood of bacterial infection increases, and a forward-looking immune system would shift its basal transcriptional stance away from antiviral responses and toward innate antibacterial and Th2 adaptive immune responses (as observed for lonely individuals). The association of social conditions with differential pathogen exposure is not perfect, of course, but even a moderate degree of correlation would provide selective pressure for the evolution of social signal transduction pathways that allow the immune system to forecast changes in the risk for socially mediated microbial threats (37, 39). As the key early decision makers in the immune response, dendritic cells and monocytes would be the optimal cellular targets for sociobiological redirection of the immune system's basal defensive positions. Transcriptional regulation of APCs may thus constitute an immunological form of vigilance against external social threats in the same sense as do psychologically triggered fight-or-flight stress responses in other organ systems (38, 39).
Limitations of this analysis include the correlational nature of relationships between loneliness and gene expression, which could reflect inflammatory influences on social experience (42, 43) in addition to casual effects of social adversity on gene expression (27, 31, 44, 45). Future studies will need to confirm the present bioinformatic attributions of cell-specific transcriptional dynamics using physically isolated monocytes and dendritic cells. Nevertheless, the pattern of differential gene expression observed here is consistent with that emerging from other analyses of social adversity (5, 7, 46, 47), including those using isolated monocytes (5). Finally, the teleological basis and health significance of these findings remain to be validated in future studies. Despite those limitations, this study's bioinformatic dissection of leukocyte gene expression profiles into their constituent cellular components deepens our insight into the origins and functional significance of the human immune system's transcriptional response to social deprivation. Identification of APCs as the primary targets of those dynamics provides both an evolutionary framework and a defined cellular context for future research on the interplay between social conditions and the molecular architecture of human health.
Methods
Transcript Origin Analysis.
To identify the cellular source of differentially expressed genes in genome-wide transcriptional profiles, we defined a cell type diagnosticity score, Scg, quantifying the extent to which each individual gene transcript (indexed g = 1 to G, g ∈ 22,283 human gene transcripts assayed by the Affymetrix U133A microarray) is predominately expressed by each major leukocyte cell type (indexed c = 1 to C, c ∈ {monocyte, plasmacytoid dendritic cell, CD4+ T cell, CD8+ T cell, B cell, NK cell}). Reference data on basal expression of all named human genes in distinct leukocyte subsets come from the publicly available Human Gene Atlas [Gene Expression Omnibus (GEO) series GSE1133; http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE1133] (18). Scg quantifies the average level of gene g’s expression in cell type c (denoted 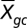 ) as a Z-score computed relative to the mean and SD of the same gene's average level of expression across all other cell types [excluding cell type c (i.e., i = 1 to C, i ≠ c)]:
) as a Z-score computed relative to the mean and SD of the same gene's average level of expression across all other cell types [excluding cell type c (i.e., i = 1 to C, i ≠ c)]:
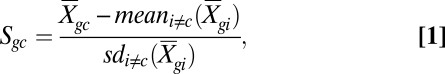 |
where mean and sd represent the mean and SD computed over the indexed cell types (48). The target cell type c is excluded from the computation of the reference mean and SD because in cases in which gene g is predominately expressed in a single cell type (i.e., is highly diagnostic), inclusion of that cell type in the reference distribution would introduce an extreme positive outlier that spuriously inflates both the reference mean and SD (48). To detect cell type-diagnostic transcripts most efficiently, this score focuses on differences in the mean expression level across cell types and intentionally excludes information about variation in expression within cell types (49).
Given any arbitrary set of differentially expressed genes, the mean diagnosticity score for those genes can be computed for each potential originating cell type and tested for statistically significant elevation above the population average score for that cell type across all human genes (e.g., using a single-sample t test) (48). This accommodates the fact that population average diagnosticity scores differ across cell types (49) and the fact that the total set of assayed genes approximates the entire human genome (i.e., the population mean and variance of diagnosticity scores are essentially known and need not be estimated from the much smaller and possibly unrepresentative subset of differentially expressed genes). Sample average diagnosticity scores provide a unipolar measure of the extent to which the sample gene set is uniquely characteristic of a given cell type, with negative values indicating nondiagnosticity (i.e., not predominately expressed by that cell type alone). Negative scores are nonprobative, and statistical tests thus focus on the one-tailed statistical significance of high positive scores (i.e., the extent to which the observed transcripts are distinctively expressed by a given cell type). Negative diagnosticity scores do not provide information about the cellular origin of down-regulated genes. The cellular origin of down-regulated transcripts is identified by significant positive diagnosticity scores computed over the set of down-regulated genes.
Validation Studies.
Transcript origin analysis was tested for empirical accuracy in five independent transcriptional profiling datasets involving isolated human leukocyte subsets. CD14+ monocytes were assessed for differential gene expression following stimulation with LPS + IFN-γ or IL-4 using Affymetrix U133A high-density oligonucleotide arrays (GEO accession no. GSE5099) (50). BDCA4+ plasmacytoid dendritic cells were surveyed for differential gene expression relative to monocytes using Affymetrix U133A high-density oligonucleotide arrays (GEO accession no. GSE11943) (51). CD16+/CD56+ NK cells were cultured in medium alone or stimulated with IL-2 + IL-15 before transcriptional profiling by Amersham CodeLink Human 20K I spotted cDNA arrays (GEO accession no. GSE1511) (52). CD3+ T lymphocytes activated with antibodies to CD3 + CD28 were exposed to norepinephrine or vehicle before transcriptional profiling by Affymetrix HuGene FL high-density oligonucleotide arrays (53). B-lymphocyte cell lines were subject to gene expression profiling by Affymetrix U133A 2.0 high-density oligonucleotide arrays following culture in the absence or presence of the viral transcription factor Epstein–Barr nuclear antigen 2 (GEO accession no. GSE4525) (54). Across all studies, differential gene expression thresholds were optimized to maintain FDRs ≤10% (55) and Affymetrix expression values were floored at 100 to suppress spurious fold-change estimates (53). In each dataset, diagnosticity scores were computed for each cell type and the predicted cellular origin was taken as the cell type showing the highest degree of statistical significance (lowest P value). Reliability of transcript diagnosticity scores was assessed by split-half correlations computed on random partitions of samples in each dataset.
Discovery Studies.
Characteristics of the study sample and measurement methodology have been reported previously (4). Briefly, genome-wide transcriptional profiling was carried out in peripheral blood mononuclear cells (PBMCs) isolated by standard Ficoll density gradient centrifugation of 10 mL of whole blood from 14 participants in the Chicago Health, Aging, and Social Relations Study (CHASRS), 6 of whom had consistently scored in the top 15% of the UCLA Loneliness Scale (56) score distribution over the previous 4 y (chronically lonely) and 8 age-, gender-, and ethnicity-matched individuals who consistently scored in the bottom 15% (nonlonely). Objective social contact was measured by the SNI (19). Gene expression profiling was carried out on total RNA from 107 PBMCs using Affymetrix U133A high-density oligonucleotide arrays in the UCLA DNA Microarray Core. Low-level transcript abundance was quantified by Robust Multiarray Averaging (57), and differentially expressed transcripts were identified by a ≥30% difference in mean expression level in samples from low- vs. high-lonely individuals (corresponding to a 10% FDR), as estimated in a general linear model analysis of log2-transformed expression data (55). Among the total 22,283 mRNA transcripts analyzed, 78 were up-regulated in chronically lonely individuals and 131 were down-regulated (4).
Confirmation Studies.
Genome-wide transcriptional profiles were obtained on PBMC samples from all 93 CHASRS participants who provided leukocyte specimens in study year 8. Chronic loneliness was identified by a UCLA Loneliness Scale score ≥41 (top 25%) in 3 y or more of the study's first 5 y, and objective social isolation was measured by average SNI score over the same period. Gene expression profiling was carried out on total RNA from 107 Ficoll-separated PBMCs using Illumina Human Ref 8 v3.0 BeadArrays in the UCLA Southern California Genotyping Consortium core laboratory following the manufacturer's standard protocol. Transcript abundance values for 18,630 assayed genes were quantile-normalized (57), and differentially expressed genes were identified by a ≥15% difference in average expression level in samples from high-lonely individuals compared with the remainder of the sample (corresponding to a 5% FDR), as estimated by a general linear model analysis of log2-transformed expression values controlling for age; gender; ethnicity; marital status; (log) household income; BMI; the fractional composition of granulocytes, monocytes, and lymphocytes within the assayed leukocyte pool (complete blood cell count performed at the University of Chicago Medical Center Clinical Laboratories); and, where indicated, (standardized) SNI scores. Ancillary analyses showed no significant difference in prevalence of smoking, alcohol consumption, or drug use in chronically lonely individuals compared with the remainder of the sample. Data are deposited as GEO accession no. GSE25837.
Acknowledgments
We thank Emma Adam, Brigitte Kudielka, and the UCLA Southern California Genotyping Consortium and DNA Microarray Cores for technical assistance. This study was supported by the National Institutes of Health (Grants P01 AG19811, P20 RR020645, P30 AG028748, R01 AI52737, R01 CA116778, and R01 AG033590); the Mind, Body, Brain, and Health Initiative of the John D. and Catherine T. MacArthur Foundation; the Norman Cousins Center at UCLA; the John Templeton Foundation; and the James B. Pendleton Charitable Trust.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The microarray gene expression data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE25837).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1014218108/-/DCSupplemental.
References
- 1.Cole SW. Social regulation of human gene expression. Curr Dir Psychol Sci. 2009;18:132–137. doi: 10.1111/j.1467-8721.2009.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller G, Chen E, Cole SW. Health psychology: Developing biologically plausible models linking the social world and physical health. Annu Rev Psychol. 2009;60:501–524. doi: 10.1146/annurev.psych.60.110707.163551. [DOI] [PubMed] [Google Scholar]
- 3.Cole SW. Elevating the perspective on human stress genomics. Psychoneuroendocrinology. 2010;35:955–962. doi: 10.1016/j.psyneuen.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole SW, et al. Social regulation of gene expression in human leukocytes. Genome Biol. 2007;8:R189. doi: 10.1186/gb-2007-8-9-r189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller GE, et al. A functional genomic fingerprint of chronic stress in humans: Blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 2008;64:266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole SW, et al. Computational identification of gene-social environment interaction at the human IL6 locus. Proc Natl Acad Sci USA. 2010;107:5681–5686. doi: 10.1073/pnas.0911515107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller GE, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci USA. 2009;106:14716–14721. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen E, Miller GE, Kobor MS, Cole SW. Maternal warmth buffers the effects of low early-life socioeconomic status on pro-inflammatory signaling in adulthood. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.53. 10.1038/mp.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen E, et al. Genome-wide transcriptional profiling linked to social class in asthma. Thorax. 2009;64:38. doi: 10.1136/thx.2007.095091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finch CE. The Biology of Human Longevity: Inflammation, Nutrition, and Aging in the Evolution of Lifespans. Burlington, MA: Academic; 2007. [Google Scholar]
- 11.Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: Implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- 12.McDade TW. Life history theory and the immune system: Steps toward a human ecological immunology. Am J Phys Anthropol. 2003;122(Suppl 37):100–125. doi: 10.1002/ajpa.10398. [DOI] [PubMed] [Google Scholar]
- 13.Idaghdour Y, Storey JD, Jadallah SJ, Gibson G. A genome-wide gene expression signature of environmental geography in leukocytes of Moroccan Amazighs. PLoS Genet. 2008;4:e1000052. doi: 10.1371/journal.pgen.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibson G. The environmental contribution to gene expression profiles. Nat Rev Genet. 2008;9:575–581. doi: 10.1038/nrg2383. [DOI] [PubMed] [Google Scholar]
- 15.Janeway CA, Travers P, Walport M, Shlomchik MJ. Immunobiology. 5th Ed. New York: Garland Science; 2001. [Google Scholar]
- 16.Seeman TE. Social ties and health: The benefits of social integration. Ann Epidemiol. 1996;6:442–451. doi: 10.1016/s1047-2797(96)00095-6. [DOI] [PubMed] [Google Scholar]
- 17.Uchino BN, Cacioppo JT, Kiecolt-Glaser JK. The relationship between social support and physiological processes: A review with emphasis on underlying mechanisms and implications for health. Psychol Bull. 1996;119:488–531. doi: 10.1037/0033-2909.119.3.488. [DOI] [PubMed] [Google Scholar]
- 18.Su AI, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berkman LF. Berkeley, CA: University of California; 1977. Social networks, host resistance and mortality: A follow-up study of Alameda County residents. PhD dissertation. [Google Scholar]
- 20.Hamrick N, Cohen S, Rodriguez MS. Being popular can be healthy or unhealthy: Stress, social network diversity, and incidence of upper respiratory infection. Health Psychol. 2002;21:294–298. [PubMed] [Google Scholar]
- 21.Cacioppo JT, Hawkley LC. Perceived social isolation and cognition. Trends Cogn Sci. 2009:13, 447–454. doi: 10.1016/j.tics.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Millar DA, Ratcliffe NA. The evolution of blood cells: Facts and enigmas. Endeavour. 1989;13:72–77. doi: 10.1016/0160-9327(89)90005-7. [DOI] [PubMed] [Google Scholar]
- 23.Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soehnlein O, Drechsler M, Hristov M, Weber C. Functional alterations of myeloid cell subsets in hyperlipidaemia: Relevance for atherosclerosis. J Cell Mol Med. 2009;13:4293–4303. doi: 10.1111/j.1582-4934.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimada K. Immune system and atherosclerotic disease: Heterogeneity of leukocyte subsets participating in the pathogenesis of atherosclerosis. Circ J. 2009;73:994–1001. doi: 10.1253/circj.cj-09-0277. [DOI] [PubMed] [Google Scholar]
- 26.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sloan EK, et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010;70:7042–7052. doi: 10.1158/0008-5472.CAN-10-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. JAMA. 2007;298:1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- 29.Cole SW, Korin YD, Fahey JL, Zack JA. Norepinephrine accelerates HIV replication via protein kinase A-dependent effects on cytokine production. J Immunol. 1998;161:610–616. [PubMed] [Google Scholar]
- 30.Collado-Hidalgo A, Sung C, Cole S. Adrenergic inhibition of innate anti-viral response: PKA blockade of Type I interferon gene transcription mediates catecholamine support for HIV-1 replication. Brain Behav Immun. 2006;20:552–563. doi: 10.1016/j.bbi.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Sloan EK, et al. Social stress enhances sympathetic innervation of primate lymph nodes: Mechanisms and implications for viral pathogenesis. J Neurosci. 2007;27:8857–8865. doi: 10.1523/JNEUROSCI.1247-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capitanio JP, Mendoza SP, Lerche NW, Mason WA. Social stress results in altered glucocorticoid regulation and shorter survival in simian acquired immune deficiency syndrome. Proc Natl Acad Sci USA. 1998;95:4714–4719. doi: 10.1073/pnas.95.8.4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grebe KM, et al. Sympathetic nervous system control of anti-influenza CD8+ T-cell responses. Proc Natl Acad Sci USA. 2009;106:5300–5305. doi: 10.1073/pnas.0808851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kroenke CH, Kubzansky LD, Schernhammer ES, Holmes MD, Kawachi I. Social networks, social support, and survival after breast cancer diagnosis. J Clin Oncol. 2006;24:1105–1111. doi: 10.1200/JCO.2005.04.2846. [DOI] [PubMed] [Google Scholar]
- 35.Finch CE. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: Roles of infection, inflammation, and nutrition. Proc Natl Acad Sci USA. 2010;107(Suppl 1):1718–1724. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson RM. The Population Dynamics of Infectious Diseases. New York: Chapman and Hall; 1982. [Google Scholar]
- 37.Cole SW. The complexity of dynamic host networks. In: Deisboeck TS, Kresh JY, editors. Complex Systems Science in BioMedicine. New York: Kluwer Academic–Plenum; 2005. pp. 605–629. [Google Scholar]
- 38.Sterling P. In: Allostasis, Homeostasis, and the Costs of Physiological Adaptation. Schulkin J, editor. Cambridge, UK: Cambridge Univ Press; 2004. [Google Scholar]
- 39.Cole SW. Social regulation of gene expression in the immune system. In: Segerstrom S, editor. Oxford Handbook of Psychoneuroimmunology. New York: Oxford Univ Press; 2011. [Google Scholar]
- 40.Knipe DM, et al. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 41.Hamilton WD. The genetical evolution of social behaviour. II. J Theor Biol. 1964;7:17–52. doi: 10.1016/0022-5193(64)90039-6. [DOI] [PubMed] [Google Scholar]
- 42.Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eisenberger NI, Inagaki TK, Mashal NM, Irwin MR. Inflammation and social experience: An inflammatory challenge induces feelings of social disconnection in addition to depressed mood. Brain Behav Immun. 2010;24:558–563. doi: 10.1016/j.bbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avitsur R, Powell N, Padgett DA, Sheridan JF. Social interactions, stress, and immunity. Immunol Allergy Clin North Am. 2009;29:285–293. doi: 10.1016/j.iac.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 45.Thaker PH, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- 46.Cole SW, Mendoza SP, Capitanio JP. Social stress desensitizes lymphocytes to regulation by endogenous glucocorticoids: Insights from in vivo cell trafficking dynamics in rhesus macaques. Psychosom Med. 2009;71:591–597. doi: 10.1097/PSY.0b013e3181aa95a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cole SW. Social regulation of leukocyte homeostasis: The role of glucocorticoid sensitivity. Brain Behav Immun. 2008;22:1049–1055. doi: 10.1016/j.bbi.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller RG. Beyond ANOVA: Basics of Applied Statistics. New York: Wiley; 1986. [Google Scholar]
- 49.Abbas AR, et al. Immune response in silico (IRIS): Immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 2005;6:319–331. doi: 10.1038/sj.gene.6364173. [DOI] [PubMed] [Google Scholar]
- 50.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 51.Wrzesinski S, Uhlenhake J, Fisher J, Ernstoff M. 2008. Gene Expression Omnibus GSE11943: Human dendritic cell subtype gene arrays. Available at http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE11943. Accessed December 1, 2010.
- 52.Hanna J, et al. Novel insights on human NK cells’ immunological modalities revealed by gene expression profiling. J Immunol. 2004;173:6547–6563. doi: 10.4049/jimmunol.173.11.6547. [DOI] [PubMed] [Google Scholar]
- 53.Cole SW, Galic Z, Zack JA. Controlling false-negative errors in microarray differential expression analysis: A PRIM approach. Bioinformatics. 2003;19:1808–1816. doi: 10.1093/bioinformatics/btg242. [DOI] [PubMed] [Google Scholar]
- 54.Maier S, et al. Cellular target genes of Epstein-Barr virus nuclear antigen 2. J Virol. 2006;80:9761–9771. doi: 10.1128/JVI.00665-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 56.Russell D, Peplau LA, Cutrona CE. The revised UCLA Loneliness Scale: Concurrent and discriminant validity evidence. J Pers Soc Psychol. 1980;39:472–480. doi: 10.1037//0022-3514.39.3.472. [DOI] [PubMed] [Google Scholar]
- 57.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]