

Abstract
Objectives
Animal and human myocytes demonstrate significant swelling and reduced contractility during exposure to stress (metabolic inhibition, hyposmotic stress, or hyperkalemic cardioplegia), and these detrimental consequences may be inhibited by the addition of diazoxide (adenosine triphosphate–sensitive potassium (KATP) channel opener) via an unknown mechanism. Both SUR1 and SUR2A subunits have been localized to the heart and mouse sarcolemmal KATP channels are composed of SUR2A/Kir6.2 subunits in the ventricle and SUR1/Kir6.2 subunits in the atria. This study was performed to localize the mechanism of diazoxide by directly probing sarcolemmal KATP channel current and by genetic deletion of channel subunits.
Methods
Sarcolemmal KATP channel current was recorded in isolated wild type ventricular mouse myocytes during exposure to Tyrode’s solution, Tyrode’s + 100µM diazoxide, hyperkalemic cardioplegia, cardioplegia + diazoxide, cardioplegia + 100µM pinacidil, or metabolic inhibition using whole cell voltage clamp. Ventricular myocyte volume was measured from SUR1(−/−) and wild type mice during exposure to control solution, hyperkalemic cardioplegia, or cardioplegia + 100µM diazoxide.
Results
Diazoxide did not increase sarcolemmal KATP current in wild type myocytes, although they demonstrated significant swelling during exposure to cardioplegia that was prevented by diazoxide. SUR1(−/−) myocytes also demonstrated significant swelling during exposure to cardioplegia but this was not altered by diazoxide.
Conclusions
Diazoxide does not open the ventricular sarcolemmal KATP channel, but provides volume homeostasis via an SUR1 – dependent pathway in mouse ventricular myocytes, supporting a mechanism of action distinct from sarcolemmal KATP channel activation.
Introduction
The adenosine triphosphate–sensitive potassium (KATP) channel opener diazoxide is cardioprotective and mimics ischemic preconditioning in animal models (1). Previously we have documented significant swelling of isolated myocytes and associated reduced contractility secondary to exposure to standard hypothermic hyperkalemic cardioplegia solution in human and other species (2,3). These detrimental consequences were ameliorated by the addition of diazoxide (2,3). As diazoxide is a KATP channel opener, we hypothesized that KATP channels may play a role in myocyte volume homeostasis and that myocyte swelling may be one mechanism of myocardial stunning (4). Myocyte swelling and reduced contractility are also observed following exposure to hyposmotic and ischemic stress (4,5). Interestingly, diazoxide ameliorates both the structural and functional derangements secondary to all three stresses: hyperkalemic cardioplegia, mild hyposmotic stress, and ischemic stress (2–5). The addition of 5-hydroxydecanoate, a claimed mitochondrial KATP (mKATP) channel blocker, or HMR 1098, a claimed sarcolemmal KATP (sKATP) channel blocker, to ischemic stress and hyperkalemic cardioplegia did not alter the beneficial observations noted with diazoxide alone (2,4). The cardioprotective mechanism of diazoxide remains unknown.
Diazoxide has been described as a mitochondrial KATP channel opener, with “selectivity” towards the mKATP channel and only weak sKATP channel activation at high doses (6). However, controversy regarding the specificity o f KATP channel openers and blockers suggests that pharmacologic manipulation of the KATP channel, using potassium channel openers or sulfonylurea receptor blockers, may be inadequate to definitively confirm ion flux across the sKATP channel (7–9), and it remains unclear whether diazoxide provides cardioprotection via the sKATP channel, the mKATP channel, or via a KATP channel-independent mechanism.
KATP channels are composed of a Kir inward rectifier channel forming subunit and a sulfonylurea sensitive regulatory subunit, SUR (Figure 1) (10). It is clear that sarcolemmal KATP channels require both Kir6.2 and SUR2A subunits in the ventricle, but Kir6.2 and SUR1 in the atria (11). Both SUR1 and SUR2A are expressed in mouse heart (11,12) and there is evidence that SUR2 and SUR1 subtypes are both present in cultured neonatal rat ventricular myocytes (13). Mice lacking the SUR1 subunit appear to tolerate ischemia/reperfusion injury better than wild type mice in a model of left anterior descending coronary artery ligation (12). While there is some evidence for SUR1 subunits in the ventricle of various species, KATP currents in mouse ventricle are unaltered in mice lacking the SUR1 subunit (11).
Figure 1.

Composition of the sarcolemmal KATP channel and location of various subunits in various tissues. The channel is a functional octamer of four Kir6.x subunits (generating the channel pore) and four sulfonylurea receptor (SUR) subunits that generate the regulatory subunit. TMD is transmembrane domain, NBF is nucleotide – binding fold, and M1 and M2 are transmembrane helices.
We initially hypothesized that the mechanism of action of diazoxide might involve opening the sarcolemmal KATP channel (SUR2A/Kir6.2). Previous work documented a beneficial effect of KATP channel opener diazoxide in ventricular myocytes that was unaltered by the pharmacologic inhibition of the KATP channel (2,4). The first part of this study was designed to definitively investigate the action of diazoxide by the direct measurement of sarcolemmal KATP channel current in wild type mice using whole cell voltage clamp.
After demonstrating that sKATP (SUR2A/Kir6.2) channel activity was not observed in wild type mice during exposure to diazoxide, and knowing that SUR1 subunits have been documented in ventricular tissue (11,12,13), we next hypothesized that diazoxide might act via SUR1 subunits of an alternative KATP channel or some other channel in ventricular cells. The second part of this study was therefore designed to determine if ventricular myocytes lacking the SUR1 subunit would be responsive to diazoxide during stress.
This study was designed to elucidate the location of action of diazoxide by the direct measurement sKATP channel activity (in wild type mice) and by response to stress in ventricular myocytes from (wild type mice and mice lacking the SUR1 subunit. The elucidation of diazoxide’s mechanism of action in this model will facilitate its future clinical use.
Materials and Methods
Myocyte Isolation
All animal procedures were approved by the Animal Studies Committee at Washington University School of Medicine and all animals received humane care in compliance with the “Guide to Care and Use of Laboratory Animals”.
Ventricular myocytes were utilized for all experiments and were isolated from adult mice (either wild type (WT) or SUR1 knockout (KO), either sex, 6 weeks to 5 months, 25 to 30 g body weight) as previously described (14). Rapid cardiectomy was performed in the anesthetized (2.5% Avertin) mouse and the aorta was cannulated using a 28-gauge needle. The heart was attached to a Langendorff apparatus, and solution A was perfused through the aorta for 5 minutes. The heart was then perfused at 37°C for 12 minutes with solution B. The left ventricle was removed and transferred into solution C, where it was gently dispersed by glass pipette at room temperature. The cells were allowed to centrifuge by gravity, and serial washings were performed every 10 minutes for a 30-minute period. Cells were used in experiments within 5 hours after isolation. A typical yield of viable myocytes was 65% to 75% per mouse.
Solution A consisted of (in mmol/L, except as noted) 116 NaCl; 5.36 KCl; 0.97 Na2HPO4; 1.47 KH2PO4; 21.10 HEPES (N-[2-hydroxyethyl]piperazine-N′-[4-butanesulfonic acid]); 11.65 glucose; 26.50 µmol/L phenol red (Sigma Chemical Co; St. Louis, MO); 3.72 MgCl2; 4.40 NaHCO3; essential vitamins (100×, 10 mL, GIBCO, Grand Island, NY); and amino acids (50×, 20 mL, GIBCO, Grand Island, NY). Solution B consisted of solution A plus 10 µmol/L CaCl2; 1.2 mg/mL collagenase (Type 2, Worthington Biochemical Corporation; Freehold, NJ). Solution C consisted of solution A plus 5 mg/mL bovine serum albumin (Sigma); 1.25 mg/mL taurine; and 150 µmol/L CaCl2.
The diazoxide (7-chloro-3-methyl-1,2,4-benzothiadiazine-1,1-dioxide [DZX]; Sigma, St. Louis, MO) dose of 100 µmol/L was utilized as it was effective in ameliorating cell swelling secondary to stress (hyperkalemic cardioplegia, hyposmotic stress, and metabolic inhibition) in pervious studies (2–5). A stock solution of DZX was made by dissolving the reagent in 0.1% dimethyl sulfoxide (DMSO), at which concentration DMSO has no effect on cell volume (15).
Cells were selected for viability using the following criteria: normal rod shape, smooth edges, sharp borders and clear striations, absence of vacuoles or blebbing, and lack of spontaneous beating (15). A maximum of two cells were utilized per each animal.
Electrophysiology in Wild Type Myocytes
Following isolation, wild type myocytes were placed in a recording chamber containing normal Tyrode’s solution. Macroscopic currents in isolated ventricular myocytes were recorded using standard whole cell voltage-clamp recording techniques (16). Patch-clamp electrodes (1–3MΩ when filled with electrode solution) were fabricated from soda lime glass microhematocrit tubes (Kimble 73813, Kimble Glass Co., Vineland, NJ). Electrode solution contained the following (in mmol/L): 140 KCl, 10 HEPES, and 10 EGTA; pH 7.3–7.4. Cell capacitance and series resistance were determined using a 5- to 10-mV hyperpolarizing square pulse from a holding potential of −70 mV after establishment of the whole cell recording configuration. PClamp 9.2 software and DigiData 1322 were used to generate command pulses and collect data. Data was filtered at 5 kHz. A 4 second ramp from −110 and 40 mV was used to isolate and detect sKATP channel current only (17).
Experimental protocol
Isolated wild type myocytes were exposed to normal Tyrode’s solution (NT) for baseline measurement for 1–2 minutes followed by exposure to test solution (5–10 min) followed by NT for 5–10 min. Test solutions included NT (n=8 cells), NT+100µM/L diazoxide (DZX) (n=7 cells), hyperkalemic cardioplegia in the form of St. Thomas’ solution (CPG, Plegisol, Abbott Laboratories, North Chicago, IL) (n=11 cells), CPG+100µM/L DZX (n=8 cells), CPG+100µM/L pinacidil (Sigma, St. Louis, MO) (n=12 cells), and metabolic inhibition (MI) (n=8 cells). NT consisted of the following (in mmol/L): 137 NaCl, 5.4 KCl, 25 NaH2PO4, 10 glucose, 0.5 MgCl2, 5 HEPES, and 3 NaHCO3; pH 7.3–7.4. St. Thomas’ solution consisted of: (in mmol/L): 110 NaCl, 10 NaHCO3, 16 KCl, 32 MgCl2, and 2.4 CaCl2 and was equilibrated with 95% O2 – 5% CO2 and titrated to correct to pH 7.3. MI solution consisted of (in mmol/L): 137 NaCl, 5.4 KCl, 3 NaHCO3, 0.16 NaH2PO4, 10 2-deoxyglucose, 0.5 MgCl2, 5 HEPES, 5% oligomycin, and 3 NaHCO3; pH 7.3–7.4. Isolated sarcolemmal KATP channel activity and cellular capacitance were recorded during exposure to NT and compared to channel activity during test solution exposure.
Metabolic inhibition was utilized as a positive control to document an increase in sarcolemmal KATP channel current. Myocytes which demonstrated an increase in potassium current were subsequently exposed to glibenclamide (10 µM/L, a known sKATP channel blocker, Sigma, St. Louis, MO) to confirm that the increase in potassium current during exposure to metabolic inhibition was through sKATP channels.
Myocyte Volume Imaging in Wild Type and SUR1 (−/−) Myocytes
An aliquot of isolated myocytes was placed in a glass-bottomed chamber on an inverted microscope stage (Leitz, Wetzlar, Germany) equipped with Hoffman modulation optics (Modulation Optics, Greenvale, NY). After a 5-minute stabilization period, the chamber was perfused at a rate of 3 mL/min with 37°C normal Tyrode’s solution (in mmol/L) 130 NaCl; 5 KCl; 2.5 CaCl2; 1.2 MgSO4; 24 NaHCO3; 1.75 Na2HPO4; and 10 glucose; buffered to a pH of 7.4 using 95% O2–5% CO2. Chamber temperature was controlled by a waterbath system (Thermo Haake, Karlsruhe, Germany). Cell images were displayed on a video monitor using a charge-coupled device camera (KPM1U; Hitachi Denshi, Tokyo, Japan). Digital images of cells were captured at a rate of 120 frames per second using a video-frame grabber (Scion Corporation, Frederick, MD) and were manually traced using Scion Image software (Scion Corporation). Length, width, and area were measured and recorded. To calculate cell volume, it was assumed that changes in cell width and thickness were proportional, and relative cell volume change was determined by the following formula (15):
where t and c refer to test and control, respectively. On the basis of repeated measurements of single images and measurements of multiple images of a cell, this methodology for estimating cell volume has been shown to be reproducible with an error of less than 1% (15).
SUR1(−/−) mice were created by removal of the 1-kbp gene segment containing both promoter and exon 1 sequences of SUR1 gene by cre-mediated recombination (18). Genotype was confirmed by PCR analysis (18). This deletion is not lethal, and the SUR1 (−/−) mice lack pancreatic β cell KATP channels (SUR1/Kir6.2) and lack glucose-stimulated insulin secretion and are mildly glucose intolerant (18).
Experimental Protocol
Myocytes were not subjected to ischemia or modified ischemia in this protocol to delineate changes caused by the stress of exposure to cardioplegia alone. Myocytes from WT and KO mice were perfused with 37°C normal Tyrode’s solution for 5 min to obtain baseline measurements. Any baseline changes in cell volume secondary to the isolation or imaging protocol would be evident during this period. Myocytes were then perfused for 5 min with test solution, followed by a 5-min reexposure period to 37°C normal Tyrode’s solution. Test solutions included NT (n=8 cells for KO; n=7 cells for WT), hyperkalemic cardioplegia (CPG) in the form of St. Thomas’ solution (n=10 cells for KO; n=7 cells for WT) or CPG+100µM/L DZX (n=8 cells for KO; n=7 cells for WT). Volume measurements were made after the end of each 5 minute period.
Statistical Analysis
Data were analyzed using SYSTAT 11 (SYSTAT software Inc., Point Richmond, CA). All data are presented as mean value ± standard error of the mean, with N equal to the number of cells in each group. For cell volume measurements a repeated-measures analysis of variance was used for sequential time-based measurements for each test solution against its own baseline value. Using Fisher’s least significant difference test, post hoc multiple comparisons between different test groups were made separately during the test solution and reexposure periods. For electrophysiology experiments, Student’s t test was used to compare data. Probability values less than 0.05 were considered significant.
Results
Sarcolemmal KATP Channel Current in Wild Type Myocytes
Wild type ventricular myocytes exposed to normal Tyrode’s solution exhibited no increase in sarcolemmal adenosine triphosphate–sensitive potassium current throughout the experiment (Figure 2A), and the addition of diazoxide to normal Tyrode’s solution was not associated with an increase in potassium current (Figure 2A). Sarcolemmal KATP current increased significantly in wild type cells exposed to metabolic inhibition (p=0.002 vs. Tyrode’s) (Figure 2B), and was inhibited by the addition of glibenclamide (Figure 2A).
Figure 2.
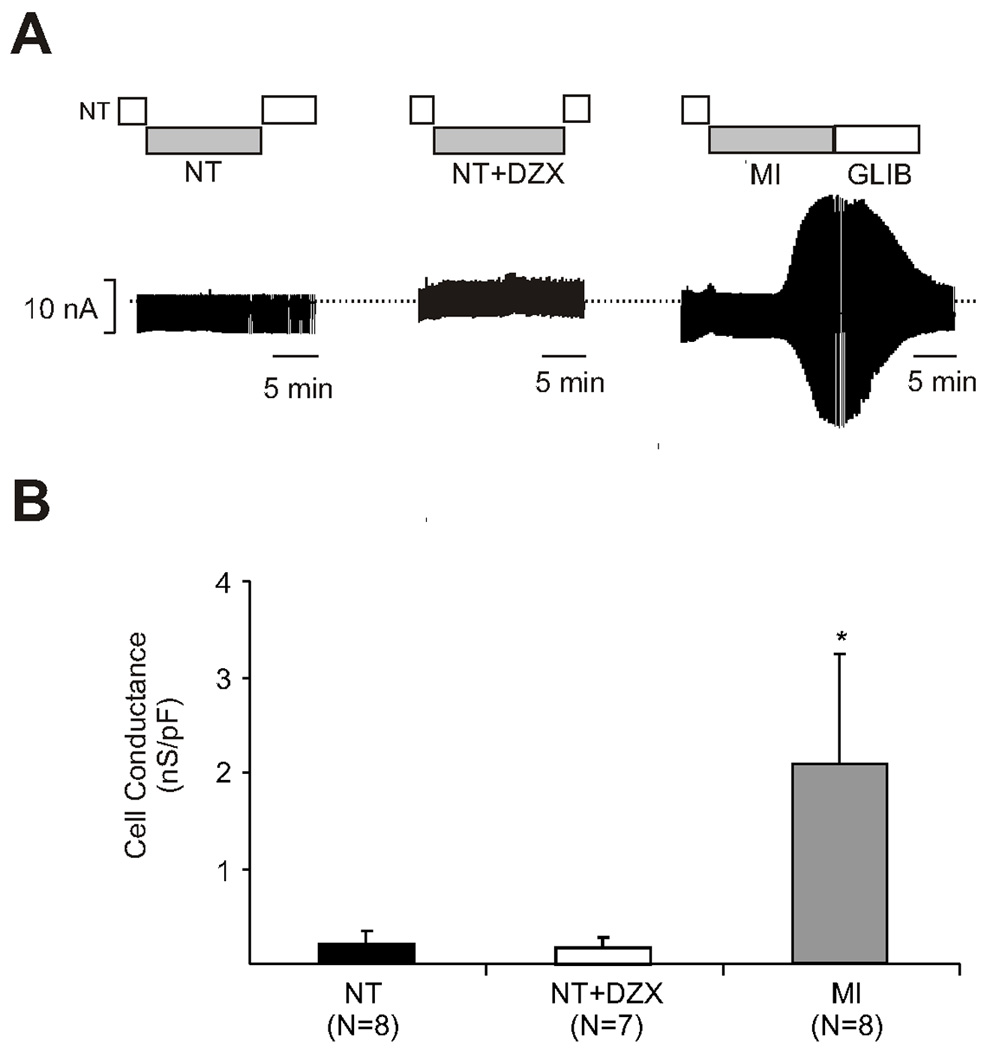
A. Diazoxide does not elicit sarcolemmal adenosine triphosphate–sensitive potassium current in wild type myocytes. The y-axis represents potassium current (nA) and the x-axis represents time. Wild type myocytes in were initially exposed to normal Tyrode’s solution for baseline potassium current measurement, followed by test solution, followed by reexposure to normal Tyrode’s solution. The first graph (far left) is a representative cell exposed to normal Tyrode’s solution throughout the experiment. The second graph (middle) is a representative cell exposed to normal Tyrode’s solution with diazoxide during the test period. The addition of diazoxide to normal Tyrode’s solution did not alter the potassium current. The third graph (far right) is a representative cell exposed to normal Tyrode’s solution followed by metabolic inhibition. Metabolic inhibition was associated with an increase in potassium current which is reversed by the addition of glibenclamide. DZX is diazoxide, GLIB is glibenclamide, MI is metabolic inhibition, and NT is normal Tyrode’s solution.
B. Diazoxide does not elicit sarcolemmal adenosine triphosphate-sensitive potassium current in wild type myocytes. Cell conductance is represented in nanoseimens/picofarads (nS/pF, y axis) and the test solution on the x axis. Test solutions included: normal Tyrode’s, normal Tyrode’s solution in addition to diazoxide, and metabolic inhibition. When correcting for cell conductance, there was a significant increase in potassium current in cells exposed to metabolic inhibition (*p= 0.002 vs. NT). DZX is diazoxide, MI is metabolic inhibition, NT is normal Tyrode’s solution, and N is number of myocytes.
Wild type myocytes exposed to cardioplegia alone after exposure to normal Tyrode’s solution exhibited no increase in sKATP current (Figure 3A), and the addition of diazoxide to cardioplegia had no effect on the potassium current when compared to cardioplegia alone (Figure 3A). The addition of pinacidil in cardioplegic solution was associated with an increase in sKATP current, which returned to baseline when cells were re-exposed to normal Tyrode’s solution (Figures 3A and 3 B). This increase in potassium current was comparable to the potassium current induced by metabolic inhibition.
Figure 3.
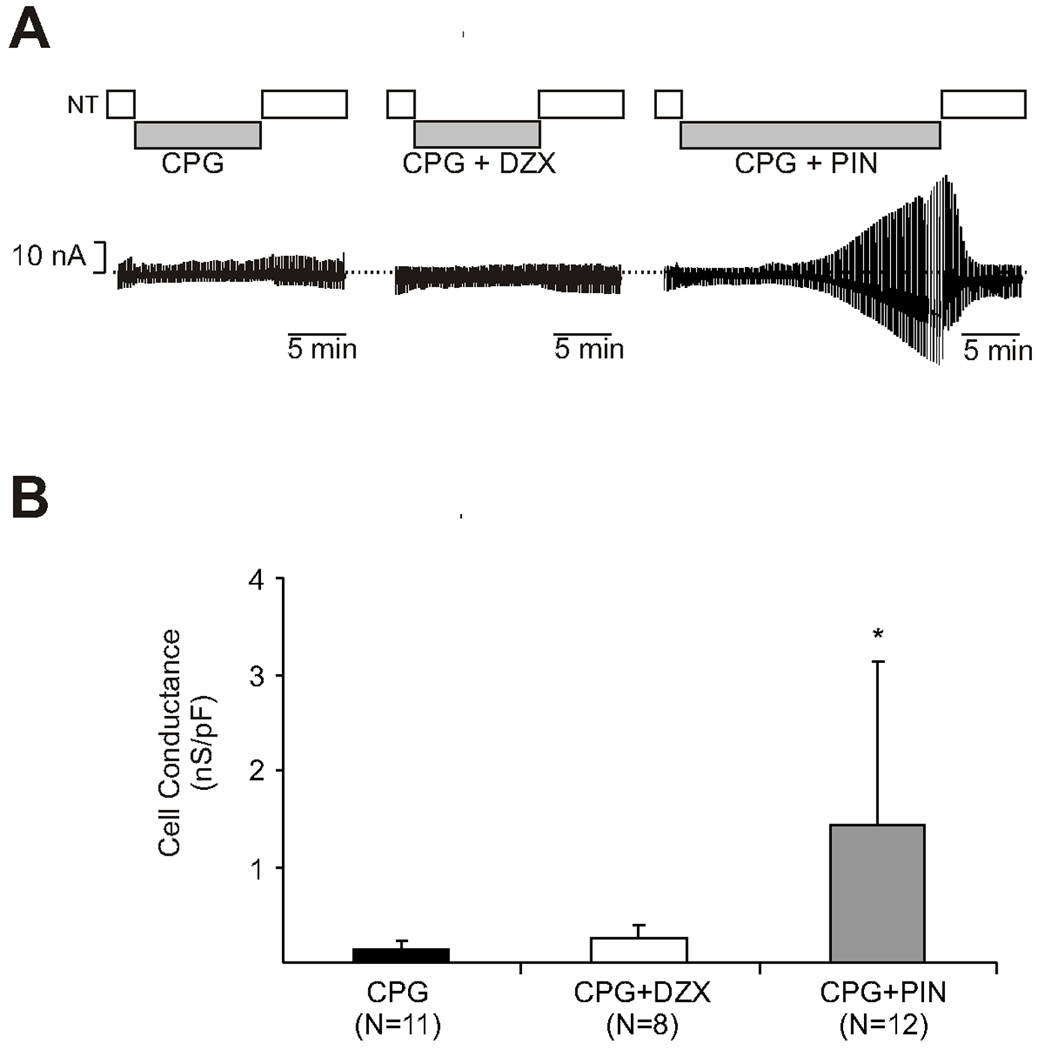
A. Pinacidil elicits an increase in adenosine triphosphate–sensitive potassium current in wild type cells exposed to cardioplegia while diazoxide does not. The y-axis represents potassium current (nA) and the x-axis represents time. Wild type myocytes were initially exposed to normal Tyrode’s solution for baseline potassium current measurement, followed by test solution, followed by reexposure to normal Tyrode’s solution. The first graph (far left) is a representative cell exposed to hyperkalemic cardioplegia (St. Thomas solution) alone during the test period. The second graph (middle) is a representative cell exposed to cardioplegia with diazoxide during the test period. The third graph (far right) is a representative cell exposed to cardioplegia with pinacidil during the test period. The addition of pinacidil to cardioplegia induced potassium current. CPG is hyperkalemic cardioplegia, DZX is diazoxide, NT is normal Tyrode’s solution, and PIN is pinacidil.
B. Pinacidil elicits an increase in adenosine triphosphate–sensitive potassium current in wild type cells exposed to cardioplegia while diazoxide does not. Cell conductance is represented in nanoseimens/picofarads (nS/pF, y axis) and test solution on the x axis. Test solutions included: hyperkalemic cardioplegia (St. Thomas solution), cardioplegia with diazoxide, and cardioplegia with pinacidil. When correcting for cell conductance, there was a significant increase in potassium current in cells exposed to pinacidil supplemented cardioplegia (*p= 0.025 vs. CPG). CPG is cardioplegia, DZX is diazoxide, PIN is pinacidil, and N is number of myocytes.
Myocyte Volume in Wild Type and SUR1(−/−) Mice
Wild type and SUR1(−/−) myocytes exposed to normal Tyrode’s solution had no significant change in volume throughout the experiment (Figure 4). As demonstrated previously, exposure to hyperkalemic cardioplegia resulted in significant (p<0.05 vs. control) wild type myocyte swelling (Figures 4 and 5), and diazoxide (100µM) significantly ameliorated the volume change secondary to cardioplegia (p<0.05). Although diazoxide is apparently without effect on mouse ventricular myocytes, and ventricular myocytes from SUR(−/−) animals contain normal sKATP density (11), there was a marked loss of diazoxide action on cardioplegic swelling in SUR(−/−) myocytes (Figures 4 and 5). As discussed below, this result indicates that diazoxide is indeed ameliorating cardioplegic swelling through the SUR1 subunit, but that this is independent of sarcolemmal KATP channels.
Figure 4.
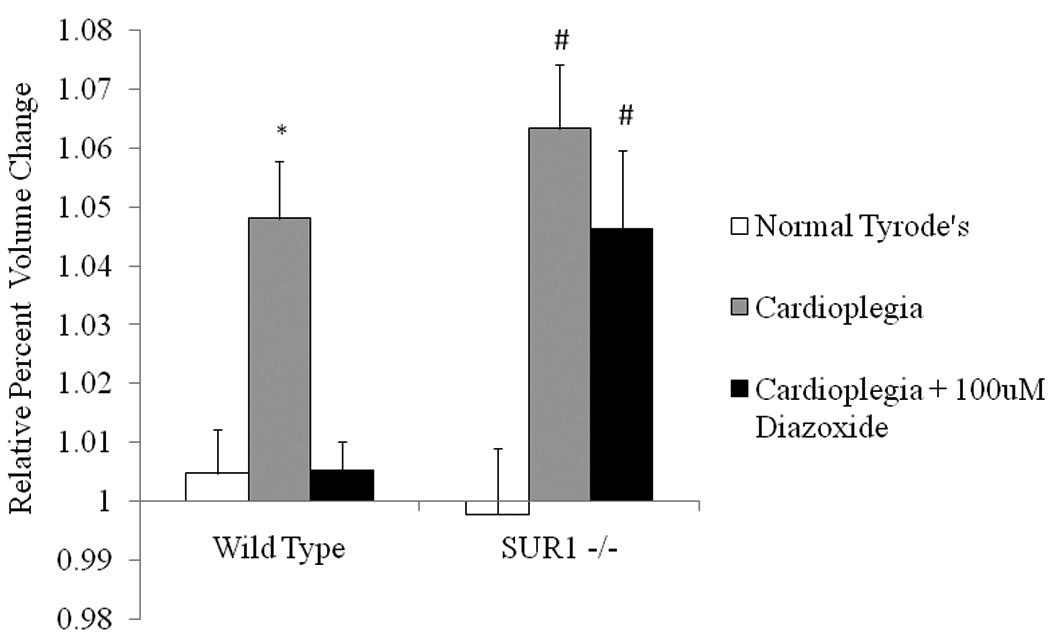
Diazoxide requires the SUR1 subunit of the KATP channel to maintain myocyte volume homeostasis in isolated ventricular myocytes. Myocyte volume change is represented as relative percent increase in volume for wild type and SUR1(−/−) cells on the y axis. Myocytes were initially exposed to normal Tyrode’s solution for 5 minutes for baseline volume measurement. Myocytes were then exposed to hyperkalemic cardioplegia or hyperkalemic cardioplegia + 100µM diazoxide. Wild type myocytes exposed to cardioplegia significantly swelled, and this was prevented by the addition of diazoxide (*p<0.05 vs. control and cardioplegia+DZX). SUR1(−/−) myocytes also significantly swelled when exposed to cardioplegia (#p<0.05 vs. control), but the addition of diazoxide did not prevent the swelling secondary to cardioplegia exposure (#p<0.05 vs. control). N= 8 cells for KO NT, N= 7 cells for KO WT, N=10 cells for KO CPG, N= 7 cells for WT CPG, N= 8 cells for KO CPG + DZX, and N= 7 cells for WT CPG + DZX.
Figure 5.

Representative isolated wild type or SUR1 (−/−) myocytes at baseline volume and following exposure to test solution via inverted microscopy. Myocytes were initially exposed to normal Tyrode’s solution for 5 minutes for baseline volume measurement. Myocytes were then exposed to test solution (hyperkalemic cardioplegia or hyperkalemic cardioplegia + 100µM diazoxide). The red line drawn on the representative test cell represents the outline of that cell’s baseline volume. Significant volume increase was noted in response to cardioplegia in the wild type (WT) and SUR (−/−) cells, and this was eliminated with the addition of diazoxide in the WT cells only. WT is wild type, CPG is cardioplegia, and DZX is diazoxide.
Discussion
Diazoxide is not Associated with an Increase in sKATP Channel Current in Wild Type Myocytes
Diazoxide did not induce a sarcolemmal KATP channel current in ventricular myocytes alone or in the presence of hyperkalemic cardioplegia. These experiments indicate that under the observed conditions, diazoxide (DZX, 100ųM/L) does not open the sKATP channel in isolated mouse ventricular myocytes, consistent with recent findings also in mouse (11), rabbit (1), and rat ventricular myocytes (19).
Previous experiments documented significant (3.2% from baseline) myocyte swelling secondary to hyperkalemic cardioplegia in myocytes isolated from mice lacking the Kir6.2 subunit of the sKATP channel (3). This swelling was not altered by the addition of diazoxide. Together with the findings of the present study, these data could be consistent with an action of diazoxide on a unique ventricular KATP channel composed of Kir6.2/SUR1. Such a composition is clearly the major composition in pancreatic, neuronal, and mouse atrial cells (20), but there is clear evidence that SUR2A, and not SUR1, is the critical SUR subunit in generation of mouse ventricular KATP (11,21).
Both Pinacidil and Metabolic Inhibition Produce an Increase in sKATP Channel Current in Wild Type Myocytes
Pinacidil, a non-specific KATP channel opener, was associated with a significant increase in sKATP channel current, consistent with the findings of previous studies (22). Metabolic inhibition is known to induce a large sKATP channel current, and this is consistent with the results of the present study (23).
Mechanism of Myocyte Swelling Secondary to Stress
In the present study, the myocyte volume derangement following exposure to hyperkalemic cardioplegia alone was evaluated as in previous work (2). Wild type and SUR1(−/−) myocytes both demonstrated a significant increase in size following exposure to hyperkalemic cardioplegia (4–7% increase in size vs. baseline volume). Similar volume derangements have been observed following the stress of mild hyposmotic stress (6% increase in volume) and metabolic inhibition (10% increase in volume) (4,5). Each stress exposed the myocyte to a unique osmotic challenge: hyperkalemic cardioplegia exposed the myocyte to a hyposmotic extracellular environment, hyposmotic stress exposed the myocyte to a different hyposmotic extracellular environment, and metabolic inhibition exposed the myocyte to a hyperosmotic intracellular environment. The KATP channel opener diazoxide prevented the volume derangement in response to all 3 stresses independently. The mechanism remains unknown; however, these data suggest a role in cellular volume regulation.
Maintenance of Myocyte Volume Homeostasis by Diazoxide Requires SUR1
Myocytes from mice lacking the SUR1 gene also swelled significantly when exposed to cardioplegia, however, diazoxide failed to reverse the effect. This finding indicates that diazoxide is in fact ameliorating cardioplegic swelling through the SUR1 subunit, but that this is independent of sarcolemmal KATP channels. This raises the possibility that SUR1 is actually present in mouse ventricle, but is not required for, or part of the sKATP channel.
Location of Action of Diazoxide
The results of the present study suggest that the observed protective mechanism of diazoxide in isolated myocytes (maintenance of volume homeostasis and contractility during stress) is dependent upon the SUR1 subunit, but is independent of the sarcolemmal KATP channel. The mechanism of action may be at the mitochondrial (mKATP) level or at a KATP channel-independent location in the ventricle. Many investigators attribute the myocardial protection provided by KATP channel openers to the opening of the purported mitochondrial KATP (mKATP) channel rather than the sarcolemmal KATP (sKATP) channel (6,25,26). However, much of the data claimed as support for the existence of a mitochondrial KATP channel have been indirect (27–29), including by measurement of mitochondrial flavoprotein oxidation. Critics have noted that diazoxide inhibits succinate dehydrogenase activity, which in turn will inhibit the tricarboxylic acid cycle leading to oxidation of mitochondrial flavoproteins (28). Other evidence supported by patch clamping of the inner mitochondrial membrane documenting the presence of mKATP channels has been criticized because of its lack of reproducibility and potential contamination by other cellular membranes (7,28,30). The determination of the structure of a mitochondrial KATP channel would thus facilitate localization of diazoxide’s mechanism of action.
Recent studies have documented RNA expression for SUR1 in left ventricular tissue from failing human hearts (24). The elucidation of the composition of the KATP channel specifically in humans will also increase the knowledge of the exact site of cardioprotection offered by diazoxide.
Study Limitations
This study investigated the action of diazoxide at the cellular level and in one species. This species was chosen because of the availability of a genetic knockout. Genetically tractable larger animal models are not available and pharmacologic methods have limitations as discussed. Potassium current at the cellular level was measured in order to definitively observe any action of diazoxide at the sKATP channel in isolation. Extrapolation to the whole organism level should therefore be taken with caution.
Clinical Relevance
The inverse relationship previously demonstrated between myocyte volume derangement and contractility in isolated myocytes suggested loss of myocyte volume homeostasis as a potential mechanism of myocardial stunning (4). The ability of diazoxide to prevent myocyte swelling and resultant contractile dysfunction secondary to 3 independent stresses in three species suggests that its use may be exploited for the reduction of myocardial stunning. Elucidation of the mechanism of action of diazoxide in the mouse at the cellular and subcellular levels will subsequently facilitate its acceptance and use at the clinical level.
Acknowledgments
Supported by the American Heart Association Beginning Grant in Aid 0565514Z (JSL) and the Thoracic Surgery Foundation for Research and Education Nina Starr Braunwald Career Development Award (JSL).
Footnotes
Presented at the 2010 American Association for Thoracic Surgery Meeting, Toronto, Canada
References
- 1.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2470. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 2.Mizutani S, Al-Dadah AS, Bloch JB, Prasad SM, Diodato MD, Schuessler RB, et al. Hyperkalemic cardioplegia – induced myocyte swelling and contractile dysfunction: prevention by diazoxide. Ann Thorac Surg. 2006;81:154–159. doi: 10.1016/j.athoracsur.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 3.Prasad SM, Al-Dadah AS, Byrd GD, Flagg TP, Gomes J, Damiano RJ, et al. Role of the sarcolemmal adenosine triphosphate-sensitive potassium channel in hyperkalemic cardioplegia-induced myocyte swelling and reduced contractility. Ann Thorac Surg. 2006;81(1):148–153. doi: 10.1016/j.athoracsur.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 4.Al-Dadah AS, Voeller RK, Schuessler RB, Damiano RJ, Lawton JS. Maintenance of myocyte volume homeostasis during stress by diazoxide is cardioprotective. Ann Thorac Surg. 2007;84(3):857–862. doi: 10.1016/j.athoracsur.2007.04.103. [DOI] [PubMed] [Google Scholar]
- 5.Mizutani S, Prasad SM, Sellitto AD, Schuessler RB, Damiano RJ, Jr, Lawton JS. Myocyte volume and function in response to osmotic stress: observations in the presence of an adenosine triphosphate-sensitive potassium channel opener. Circulation. 2005;112(9 Suppl):I219–I223. doi: 10.1161/CIRCULATIONAHA.104.523746. [DOI] [PubMed] [Google Scholar]
- 6.Hu H, Sato T, Seharaseyon J, Liu Y, Johns DC, O’Rourke B, et al. Pharmacological and histological distinctions between molecularly defined sarcolemmal KATP channels and native cardiac mitochondrial KATP channels. Mol Pharm. 1999;55:1000–1005. [PubMed] [Google Scholar]
- 7.Das M, Parker JE, Halestrap AP. Matrix volume measurements challenge the existence of diazoxide/glibenclamide-sensitive KATP channels in rat mitochondria. J Physiol. 2003;547(3):893–902. doi: 10.1113/jphysiol.2002.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’hahan N, Moreau C, Prost AL, Jacquet H, Alekseev AE, Terzic A. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc Natl Acad Sci. 1999;96:12162–12167. doi: 10.1073/pnas.96.21.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grover GJ, Garlid KD. ATP-sensitive potassium channels: A review of their cardioprotective pharmacology. J Mol Cell Cardiol. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- 10.Shyng S-L, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flagg TP, Kurata HT, Masia R, Caputa G, Magnuson MA, Lefer DJ, et al. Differential Structure of Atrial and Ventricular KATP: Atrial KATP Channels Require SUR1. Circ Res. 2008;103:1458–1465. doi: 10.1161/CIRCRESAHA.108.178186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elrod JW, Harrell M, Flagg TP, Gundewar S, Magnuson MA, Nichols CG, et al. Role of sulfonylurea receptor type 1 subunits of ATP-sensitive potassium channels in myocardial ischemia/reperfusion injury. Circulation. 2008;117:1405–1413. doi: 10.1161/CIRCULATIONAHA.107.745539. [DOI] [PubMed] [Google Scholar]
- 13.Yokoshiki H, Sunagawa M. Antisense Oligonucleotides of Sulfonylurea Receptors Inhibit ATP-Sensitive K+ Channels in Cultured Neonatal Rat Ventricular Cells. Pflugers Arch. 1999;437:400–408. doi: 10.1007/s004240050794. [DOI] [PubMed] [Google Scholar]
- 14.Flagg TP, Remedi MS, Masia R, Gomes J, McLerie M, Lopatin A, et al. Transgenic overexpression of SUR1 in the heart suppresses sarcolemmal K(ATP) J Mol Cell Cardiol. 2005;39(4):647–656. doi: 10.1016/j.yjmcc.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Drewnowska K, Clemo HF, Baumgarten CM. Prevention of Myocardial Intracellular Edema Induced by St. Thomas’ Hospital Cardioplegic Solution. J Mol Cell Cardiol. 1991;23:1215–1221. doi: 10.1016/0022-2828(91)90079-2. [DOI] [PubMed] [Google Scholar]
- 16.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 17.Flagg TP, Charpentier F, Manning-Fox J, Remedi MS, Enkvetchakul D, Lopatin A, et al. Remodeling of excitation-contraction coupling in transgenic mice expressing ATP-insensitive sarcolemmal KATP channels. Am J Physiol Heart Circ Physiol. 2004;286(4):H1361–H1369. doi: 10.1152/ajpheart.00676.2003. [DOI] [PubMed] [Google Scholar]
- 18.Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, et al. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002;277:37176–37183. doi: 10.1074/jbc.M206757200. [DOI] [PubMed] [Google Scholar]
- 19.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K channels: possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 20.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 21.Chutkow WA, Pu J, Wheeler MT, Wada T, Makielski JC, Burant CF, McNally EM. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 K(ATP) channels. J Clin Invest. 2002;110(2):203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arena JP, Kass RS. Enhancement in potassium-sensitive current in heart cells by pinacidil: Evidence for the modulation of the ATP-sensitive potassium channel. Circ Res. 1989;65(2):436–445. doi: 10.1161/01.res.65.2.436. [DOI] [PubMed] [Google Scholar]
- 23.Nichols CG, Ripol C, Lederer WJ. ATP-sensitive potassium channel modulation of the guinea pig ventricular action potential and contraction. Circ Res. 1991;68:280–287. doi: 10.1161/01.res.68.1.280. [DOI] [PubMed] [Google Scholar]
- 24.Soltysinska E, Olesen SP, Christ T, Wetter E, Varro A, Grunnet M, et al. Transmural Expression of Ion Channels and Transporters in Human Nondiseased and End-Stage Failing Hearts. Pflugers Arch. 2009;459:11–23. doi: 10.1007/s00424-009-0718-3. [DOI] [PubMed] [Google Scholar]
- 25.Toyoda Y, Levitsky S, McCully JD. Opening of mitochondrial ATP-sensitive potassium channels enhances cardioplegic protection. Ann Thorac Surg. 2001;71(4):1281–1288. doi: 10.1016/s0003-4975(00)02667-9. [DOI] [PubMed] [Google Scholar]
- 26.Sato T, Sasaki N, Seharaseyon J, O’Rourke B, Marban E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal KATP channels in ischemic cardioprotection. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- 27.Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. KATP channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002;542.3:735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim KH, Javadov SA, Das M, Clarke SJ, Suleiman M, Halestrap AP. The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J Physiol. 2002;545.3:961–974. doi: 10.1113/jphysiol.2002.031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rainbow RD, Lodwick D, Hudman D, Davies NW, Norman RI, Standen NB. SUR2A C-terminal fragments reduce KATP currents and ischaemic tolerance of rat cardiac myocytes. J Physiol. 2004;557.3:785–794. doi: 10.1113/jphysiol.2004.061655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorgato MC, Keller BU, Stuhmer W. Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature. 1987;330(3):498–500. doi: 10.1038/330498a0. [DOI] [PubMed] [Google Scholar]