

Abstract

From a scientific perspective, efforts to understand biology including what constitutes health and disease has become a chemical problem. However, chemists and biologists “see” the problems of understanding biology from different perspectives, and this has retarded progress in solving the problems especially as they relate to health and disease. This suggests that close collaboration between chemists and biologists is not only necessary but essential for progress in both the biology and chemistry that will provide solutions to the global questions of biology. This perspective has directed my scientific efforts for the past 45 years, and in this overview I provide my perspective of how the applications of synthetic chemistry, structural design, and numerous other chemical principles have intersected in my collaborations with biologists to provide new tools, new science, and new insights that were only made possible and fruitful by these collaborations.
In the spring of 1965 I made a fateful decision. I decided to change courses in my scientific career from synthetic chemistry and theoretic organic chemistry and accept an offer to join Vincent du Vigneaud’s group at Cornell University Medical College as an Instructor in Biochemistry (I had not taken a course in Biochemistry). In doing so I accepted a challenge he had been working on for many years, the structure of acetone oxytocin (an inactive form of oxytocin which contained 1 mol of acetone and 1 mol of oxytocin) using a combination of synthetic/mechanistic organic chemistry and a relatively new structure tool for solving organic structures (in this case a structure of over 1000 Da), nuclear magnetic resonance spectroscopy. I loved spectroscopy and mechanistic organic chemistry and thought it would be fun to try. It turned out to be a difficult problem, but fortunately we were successful.1 Thus began my career in what is now called Chemical Biology. Though both of my Professors, A. T. Blomquist, and V. du Vigneaud, were exceptionally supportive and gave me unparalleled freedom in both my Ph.D. and postdoctoral work, respectively, little did I realize the resistance this field would elicit from chemists. However, du Vigneaud, certainly the most future-looking scientist I have known, and the father of modern Chemical Biology in peptide and protein chemistry, provided all the incentives I have needed. “Victor, we just keep at it, eventually they will catch on.” I also learned that collaboration with biologists and medical doctors, which du Vigneaud had been doing for many years, was essential. This is one of the main reasons I chose The University of Arizona to begin my independent science career: the Medical School is less than a mile from the Chemistry Department. That, and the presence of Carl “Speed” Marvel, one of the giants of organic chemistry and the father of polymer chemistry, who wanted me to come to the University of Arizona and was my strongest advocate.
Indeed, I have always had outstanding collaboration in the Medical School and in the Biology Departments of the University. I got off to a great start at the University of Arizona by taking advantage of what organic chemists can do best, making things that no one else can make, and applying them to my state of the art abilities in spectroscopy, especially NMR spectroscopy, and by doing the same with my collaborators.2,3 Fortunately, I quickly got invitations to give talks at several international meetings, first in Europe and then in America. Though not from mainstream chemistry, they were in the emerging fields at the interface of chemistry and biology. To maximize my synthetic organic chemistry, I became the first group, not previously associated with Merrifield, to build instrumentation to do solid-phase synthetic chemistry both automated4 and by hand. This was met with criticism from synthetic chemists, but I persisted in no small part because Bruce Merrifield gave me much help and encouragement and because both du Vigneaud and Marvel supported my efforts. I thought they were wise and right, and they were! I have always felt that criticism can be a great stimulus for success, and I was able to exploit this synthetic advantage to do chemical biology research which quickly took on a life of its own. In the latter regard, it is interesting to note that I have been asked to write many reviews over the years, at first more by biologists than chemists, and this has turned out to be a blessing too, because as a result several ligands we designed have been used extensively by biologists in hundreds of laboratories worldwide, which provided great incentive to continue in a direction that had impact in both chemistry and biology.
The major focus of this overview will be to illustrate how direct collaboration with biologists and medical doctors can lead to new chemical considerations on the one hand and to new insights into biology on the other hand that otherwise might not have occurred. I believe that to maximize one’s creativity in Chemical Biology it is essential to collaborate with biologists. They have a different perspective than chemists and know things chemists need to know that inevitably lead to new directions in research that otherwise would never be taken. Of course, in order to do this we did an enormous amount of synthetic peptide and peptide mimetic chemistry, heterocyclic chemistry, and asymmetric synthesis of novel amino acids, β-turn mimetics, and other scaffolds. A discussion of these will have to wait until a later date.
A Few Principles in Chemical Biology
In synthetic and mechanistic organic chemistry we generally seek to develop methods that can make or break covalent bonds in three-dimensional structural space. This basic idea has dominated synthetic chemistry for two centuries. Though there are some parallels in Chemical Biology, for example, in enzymology, things generally are very different when addressing most chemical biology problems. Ultimately, to understand the chemistry of the biological system as it relates to health or disease, the chemistry must be considered in the context of the cell, or an organ, or even a whole organism or animal. This is particularly the case with the organic compounds I have studied in biology, peptide and peptide mimetic hormones, and neurotransmitters. These compounds affect and modulate behaviors related to life of multicellular organisms (animals), including humans. Thus, they are involved in virtually all behaviors including those associated with all our major diseases including cardiovascular diseases, reproductive diseases, cancer, diabetes, obesity, pain including prolonged neuropathic pain, etc. They usually do this without making or breaking covalent bonds. To understand the chemical basis for the biology of health or disease, it is critical to design molecules with the properties needed to understand the chemical biology in a biological context.
Furthermore, it is critical to do state of the art analysis on structure, conformation, chemical/physical properties, etc. In addition binding studies and related mechanistic studies (second messenger analysis, enzymatic properties, etc.) using purified receptor/acceptors, membrane fragments of tissues for assays of G-protein coupled receptors, ion channels, cytokines, electron transfer systems, etc., are needed. X-ray crystal structures can be very useful. However, results from such “in vitro” studies must be viewed with considerable caution when translating the findings to a cellular system, much less a whole animal. In examining the chemistry using cells or whole animals you are now dealing with a complex system, often without understanding all of the chemical components that may be involved in modulating the activity of your ligand by its interaction with a particular receptor/enzyme/acceptor, etc. Furthermore, you are not at equilibrium, and depending on the animal or cell used, your observations may only be relevant to a particular phenotype. Collaboration with an expert biologist is essential to interpret in vivo results and, in many cases, even to know what observations are relevant. In this regard, it is important to point to the need (indeed the requirement) for proper controls. Interestingly, here the chemist can play a critical, in fact essential role, which I have had the privilege to play in virtually all of my collaborations with biologists. In order to have appropriate positive and negative controls, molecules must be designed and synthesized that can enforce for example, agonist and antagonist functional activity, and (in many cases) suitable selectivity for a receptor/acceptor/enzyme type or subtype. There are a few chemists who can choose, much less develop, the best animal models to evaluate ligands that will be useful for the treatment of diseases. On the other hand, there are few biologists who have sufficient knowledge and expertise in synthetic chemistry to be able to design, synthesize, and evaluate, from a three-dimensional structure and chiral point of view, a potent and highly selective agonist or antagonist ligand for a particular disease state. Thus, collaboration without arrogance of knowledge or expertise is necessary to solve these problems.
The Consideration and Use of Conformational Constraint To Understand and Develop Structure–Biological Activity Relationships for Peptide Hormones and Neurotransmitters
An evaluation of the evolution of multicellular animal life from a chemical perspective demonstrates how multicellular living systems that acquired cooperation and collaboration between their different cell types obtained a competitive advantage in evolution. For this purpose, Nature chose peptides and protons as its major modulators of cooperative intercellular communication in multicellular life and dropped much of the chemistries related to small molecules (terpenes, alkaloids, etc.) which are used in unicellular life, perhaps because of the inherent toxicities of most of these scaffolds to multicellular life. (The inherent toxicities of these small molecules undoubtedly helped protect unicellular life from other unicellular life.) So what are the important structures to be considered? Much attention and discussion has been given in recent years to “chemical space” as a major consideration in Chemical Biology and Drug Design. This clearly is a critical issue and an unresolved problem. However, much of the discussion appears from my perspective to be misplaced, since Nature has chosen peptides and proteins to modulate and control most aspects of multicellular life. As we have pointed out previously,5 the chemical space for peptides is available in such a variety in which all the matter and time in the universe we know is not sufficient to have even synthesized, much less evaluated, all possible 100 amino acid peptides. Given the relative recent origin of our earth, and the very small biosphere from which life developed and was maintained, it seems clear that the real issue is to determine what is the structural bias that Nature has chosen to develop? I think the answer is clear; it has chosen peptides and proteins as the central players in life processes, nucleic acids as its principle storehouse of information necessary for life, and lipids/membranes as the principal organizers of life. Hence, the central hypothesis we have chosen is to design bioactive peptides in two- and three-dimensional space that reflects the bias of these organic compounds.6 From my perspective, the key insight into this bias is that of Ramachandran and co-workers. They pointed out, based on simple chemical principals, and before there were many high-resolution X-ray structures of peptides and proteins, that the low energy conformations of peptides and proteins were the α-helix, β-sheets, β-turns, and extended structures, all of which were readily interchangeable.7 Now thousands of X-ray and NMR structures later, this is exactly what has been found. Our design of bioactive conformationally constrained peptides has been based from the beginning on this central insight into the biological relevant structures of peptides and proteins. Of equal consideration is the fact that dynamic conformational changes are of critical importance to most biologically active peptide hormones and neurotransmitters in the course of their biological lifetime, i.e., a typical peptide hormone and neurotransmitter will need to exhibit different conformations during its biologically relevant existence, and this consideration should be an important part of design. This working hypothesis has been of central importance to nearly all successes we have had in chemical biology.
Another key hypothesis that has directed and informed our research from its earliest was that structure and biological activity have a reciprocal relationship that could inform molecular design for a biological goal. In terms of the peptide hormones and neurotransmitters, this basic principle is that in biology, changes in structure are necessary to change a biological response, and vice versa, a change in biological activity requires a change in structure. Thus, in the case of peptide hormones and neurotransmitters, design of an antagonist from an agonist would mean that some specific structural change would be needed so that the ligand would interact in a fundamentally different way with its receptor to display antagonist activity. To obtain an antagonist ligand for the same receptor requires that design of the ligand generally will require three-dimensional structure changes and will lead to different ligand–receptor interactions that produce a different conformation in antagonist ligand–receptor complex from that of the agonist ligand–receptor complex. These ideas and their development from our earliest studies8 have been critical in the development of biological collaborations and helped inform the discussions we had with biologists regarding the specific in vitro and in vivo assays that would be needed to develop biologically relevant ligands that would be useful for biological studies and for drug design and development.
Structural, Conformational, and Dynamic Considerations in the Design of Peptide Hormones, Neurotransmitters, and Related Compounds
Often, the structure of the starting ligand for such studies is a peptide whose structure is known, and the approaches for obtaining such ligands will not be discussed here. Equally critical for such studies is the availability of appropriate in vitro, and in many cases in vivo, assays (Table 1). A comprehensive discussion of Table 1 is not possible, but important points that are of critical importance to the chemist should be mentioned. The first is that one should get as a collaborator the very best biologist who is a leader in the biology of the system to be studied and who has interest in both normal and disease states associated with the target of interest. Second, though often it is possible to make critically important progress with in vitro assays, inevitably if one is to have an important impact in the biology, in vivo studies are necessary with appropriate animal models of both normal and disease states. Third, with a few exceptions, most peptide hormones and neurotransmitters in mammalian systems target more than one subtype of receptor, and this needs to be addressed from the beginning. As a typical example, there are three opioid receptor subtypes that are known to date, the mu (μ), delta (δ), and kappa (κ) receptors, and because of their location in the body they can have quite different bioactivity profiles. Interestingly, most of the mammalian hormone and neurotransmitter ligands have only modest selectivity for one or the other of those receptors. Thus, there is a need to develop both selective agonists and antagonists as positive and negative controls. Fourth, it has become increasingly clear that in both normal and disease states both native ligands and many analogues and derivatives that have been developed can induce more than one signaling pathway. It often is important to be able to determine this by appropriate assays.
Table 1.
Necessary Assays for Development of SAR for Peptide Hormones and Neurotransmitters
|
Conformational Constraint or Bias, Enhanced Potency and Selectivity, and Stabilization of Peptides from Proteolysis and for Enhanced Bioactivity
Most peptide hormones and neurotransmitters act as biological switches and/or modulators of biological action. Thus, peptide hormones or neurotransmitters once released into the bloodstream or CNS from their storage granule or neurosecretory granule are designed to have a limited half-life at elevated concentrations where they are substrates for proteases. Chemists and biologists have a need to measure what they study. In the case of peptide hormones and neurotransmitters, though tremendous progress has been made in enhancing detection limits, generally we can only detect endogenous peptides in the nanomolar to picomolar range. It is important to realize however, that many peptide hormones and neurotransmitters often have in vivo activities at concentrations where their presence cannot be measured. This biological phenomenon is often referred to as high efficacy (for example, see refs 10 and 11). Since these bioactivities often occur at very low receptor occupancies, standard binding and second messenger assays will not detect them, and in vivo assays are needed. For example, MT-II (an α-MSH analogue) has binding affinities of 0.1–1 nM, but Mac Hadley, my biological colleague, could detect it in vitro and in vivo at 10−14M.
These caveats aside, for many peptide hormones and neurotransmitters, it is critical to develop both agonist and antagonist analogues that are potent, are receptor selective, and have stability against proteolytic breakdown. Though peptides can and are protected from breakdown in most in vitro binding and second messenger assays by use of protease inhibitor cocktails as part of the assay procedure, for most in vivo studies this does not work, and instead, peptide ligands that are stable to proteolytic breakdown are needed. We have developed ways to do this from the beginning.
The development of highly receptor subtype selective ligands also is often critically necessary to establish that a particular response to a hormone or neurotransmitter actually is due to a particular ligand–receptor interaction. Again, this is particularly true when agonists are being examined in in vivo assay, because often even a few percent occupancy of one receptor relative to another can result in a full agonist biological response.
Designing peptides hormone and neurotransmitter analogues with specific backbone conformational preferences has been a central theme of our research from the beginning (see ref 12 for early reviews). A major early catalyst for this research was the observation from NMR studies of the constrained cyclic oxytocin antagonist c-[1-penicillamine]oxytocin, versus the agonist oxytocin, that they had different conformational and dynamic properties that could account for their different biological activities,8a and later the X-ray crystal analysis of deamino-oxtocin supported this conclusion.13,14 Continued ligand development led to a prolonged acting oxytocin antagonist, and with biological collaborator, Professor Walter Chan, it was shown that this prolonged action in vivo could be used for the treatment of preterm labor.15
The use of conformation constraint in linear peptides can be approached in many ways including use of d-amino acids,16,18 α-substituted amino acids,17 and cyclization of linear peptides to cyclic peptides (vide infra).
As an early example, we replaced the Phe7 position in the peptide hormone and neurotransmitter α-MSH (Figure 1, 1) with a d-Phe7 residue to stabilize a putative β-turn.16,18,19
Figure 1.
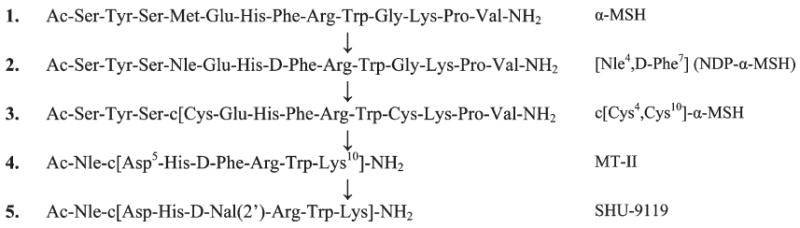
Evolution of a peptide hormone and neurotransmitter from a flexible linear peptide with short half-life to cyclic conformationally constrained receptor-selective analogues that cross the blood–brain barrier.
In addition, an isosteric replacement of the Met4 residue in α-MSH with Nle4 was achieved (Figure 1, 2). We showed with Mac Hadley that these simple substitutions had three dramatic effects: (1) a much more potent analogue was obtained that also had highly prolonged biological activities both in vitro and in vivo. (2) The compound had a dramatic increase in serum stability from minutes to hours. This peptide still serves as the standard peptide for hundreds of scientists who study melanotropin biology. (3) It could be used in experiments not possible with α-MSH to demonstrate the requirement for Ca2+ for α-MSH activity at melanophores.18b
To further examine our hypothesis that a β-turn structure was important biologically, we turned to cyclization, i.e., going from a linear to a cyclic peptide while maintaining the pharmacophore. The approach that needs to be taken depends on a model of the three-dimensional structure of the pharmacophore. One of the great advantages of peptides is their inherent three-dimensional properties, even in small peptides due to their chirality and the chiral bias of Nature. Except for glycine, all of the α-amino acids in most peptides and proteins have an l-configuration and therefore have distinct preferences in 3-D (Ramachandran) space. (It is important to recognize that glycine serves often as a d-amino acid in peptide and protein folded structures). Once this pharmacophore is determined (or proposed), then one can use other residues as the sites of cyclization (we have referred to these as ancillary sites) for side chain to side chain, backbone to backbone, side chain to backbone, etc. cyclization. In this case, we hypothesized that the His-Phe-Arg-Trp sequence was the key pharmacophore (later proved to be the case20,21 with caveats). Using model building and early computational methods, we designed a cyclic disulfide structure to stabilize the β-turn (3, Figure 1). The compound c-[Cys4, Cys10]-α-MSH was found to be a super agonist and more stable in vivo, though it contains all l-amino acids.22 At this time, more powerful computational methods were being developed, and I was fortunately able to obtain a Guggenheim Fellowship and took a sabbatical year at Harvard with Martin Karplus, David Evans, and others to incorporate these new methods into our research for design in Ramachandran space. At that time, I also became interested in design in (chi) χ space (side chain conformations of amino acid residues in peptides, discussed later). Eventually, using a combination of SAR studies and computational chemistry, we designed a cyclic lactam truncated analogue of α-MSH (4–10) Ac-Nle-c-[Asp5, d-Phe7, Lys10]-α-MSH-(4–10)-NH2 (4, Figure 1, MT-II).23,24 This compound was found to be superpotent with prolonged biological activity in vitro and in vivo, and it also was found to have very high stability agonist enzymatic breakdown, to cross the blood–brain barrier, and to have a number of other biological properties that have been exploited by ourselves and many laboratories both in academia and in industry. Its design was based on computational studies, and though efforts to obtain crystals have thus far been unsuccessful, NMR studies with caveats are consistent with a β-turn conformation. 122 Most importantly, this structure has become the basic template for the design of many peptide and nonpeptide ligands for the more recently discovered melanocortin 4 receptor (MC4R), and of other melanocortin receptor subtypes in our laboratory and in many other academic, pharmaceutical, and biotechnology companies.
These conformational considerations led to another critical discovery when we further considered topographical constraints in χ space in our design. All of the melanotropins we have discussed so far have been agonists. However, as previously discussed, one of the truisms of Chemical Biology is that to properly evaluate any biological function you need both positive and negative controls, that is, both agonists and antagonists. The development of peptide hormone and neurotransmitter antagonists has been a major aspect of our research, and we have written extensively on the approaches that can be taken to develop antagonists (e.g., refs 8d and 25). Efforts to obtain melanocortin receptor antagonists had been only modestly successful (e.g., ref 26). However, we found that by replacing the d-Phe7 residue in MT-II with a more bulky residue such as d-2′-naphthylalanine (Nal(2′)) the highly potent hMC3R, hMC4R, and frog MC1R antagonist Ac-Nle4-c-[Asp5,d-Nal(2′)7,Lys10]-α-MSH(4–10)-NH2 (SHU-9119) was obtained27 (Figure 1, 5). Interestingly the d-Nal(1′)7-analogue was an agonist. This antagonist (SHU-9119) has been a powerful tool for understanding the biology of melanocortin receptors.
In the meantime, in collaboration with Mac Hadley and medical colleagues in the University of Arizona Medical Center and later with other colleagues, we began to examine the application of these peptides to biological and medical questions, first in animals and then in human beings. I will briefly summarize some of the results of over 20 years of effort to emphasize again the power of chemistry to promote and examine important questions in biology and medicine.
The availability of NDP-α-MSH and later MT-II and SHU-9119 led us to examine the comparative endocrinology of melanotropin peptides in a wide variety of animals ranging from frogs and fish, to amphibians, to animals including mammals. These studies will not be discussed here except to say that they firmly established the comparative biology of pigmentation and established unambiguously its universality in a wide variety of animals. More specifically, however, the enhanced stability and bioavailability of NDP-α-MSH (MT-I) and later the ability of MT-II and SHU-9119 to also cross the blood–brain barrier (BBB) were of critical importance. Just a few highlights will be mentioned here, and a more comprehensive discussion can be found elsewhere.29
A very early discovery regarding the mechanism of melanocyte dispersion (skin darkening) was made possible by the stable prolonged biological activity of NDP-α-MSH when it was found that Ca2+ was essential for melanocyte dispersion. 18b Subsequently, it has been found that Ca2+ is essential or very important for the biological activity of many hormones and neurotransmitters. Similarly, the early development of an adenylate cyclase assay30 gave mechanistic insights. Furthermore, it was shown unambiguously using this ligand that hair pigmentation,31 tyrosinase stimulation32 but not cancer cell growth,33 and follicular melanogenesis in a mouse model34 were melanotropin dependent. Eventually, these and other biological studies including a variety of preclinical toxicity studies led to its application to human beings for pigmentation without sun and to protection from V radiation in prevention of melanoma cancer.35 In interesting parallel studies with the newly discovered melanin concentration hormone, we suggested that the two hormones might be evolutionarily and structurally related.36
In a similar manner, the development of MT-II and its ability to cross the BBB led in a parallel series of preclinical studies with our biological and medical colleagues to the discovery of central melanocortin receptors mediated erectile function, the first clinical trials in humans for erectile dysfunction,37,38 and the demonstration that melanotropins were also involved in sexual motivation,37c,39 in female sexual function, and other related biological sexual functions.40
Another major use of NDP-α-MSH was the preparation in the early 1990s of a number of multivalent constructs on a soluble polymer backbone that also had a fluorescent probe that could be utilized in conjunction with fluorescence microscopy to directly detect cancer cells including amelanotic cancer cells which had very low numbers of melanocortin receptors,41,42 whereas previous efforts using the radiolabeled ligand had given variable results. In addition, large beads (much larger than cancer cells) and small beads (much smaller than cells) used in solid-phase peptide chemistry could be prepared with multiple copies of NDP-α-MSH. For the larger beads, numerous cancer cells clustered around the bead by binding to the beads.43 In the case of the small beads, many bound to the cell surface, and some were sequestered into the cell by internalization.44 Furthermore, it was possible to follow the fluorescently labeled soluble multivalent NDP-α-MSH construct as the ligand bound to the cell surface, formed clusters on the cell surface, internalized into the cell, was partioned to various cellular organelles, and returned to the cells after several hours. More recently two photon fluorescence laser microscopy45a and confocal laser microscopy45b could be used to examine agonist and antagonist trafficking in cells. Furthermore, the use of multivalency has become a major tool for the development of a variety of molecular probes to examine many aspects of biology and for detection of disease.e.g.46
New Melanocortin Receptor, New Biological Function, and New Insights Utilizing Melanotropin Agonist and Antagonists
During this time, another major chemical revolution, the molecular biology revolution, had occurred. A major development in this area was the ability to clone specific receptors into cells in quantities sufficient for extensive biochemical, molecular pharmacological, and biophysical studies. In addition, this also allowed one to determine the structure of proteins directly from their gene sequences without the need to isolate, purify, and determine the protein sequence directly. This technology has been particularly useful in our collaborative research with biologists. A striking example has been in the area of melanocortin receptors and melanotropin peptides. In the early to mid-1990s, new mammalian, including human, melanocortin receptors were cloned and expressed (e.g., ref 47). In addition to the pigmentary (α-MSH) receptor (referred to now as the melanocortin 1 receptor MC1R) and ACTH receptor (MC2R) which were the first cloned, these were followed by the cloning of three other melanocortin previously unknown receptors referred to as the MC3R, MC4R, and MC5R receptors. It was quickly established that the MC3R and MC4R were primarily central receptors (though they also are found in specific peripheral sites) and the MC5R receptor was found throughout the body of mammals (initially all the cloned receptors have been mammalian receptors including the human receptors). This provided the opportunity for the discovery of significant new insights in chemical biology, and we were particularly well suited to participate with our biological colleagues in the discovery of the physiological, pharmacological, and medical significance of these new melanocortin receptors and the endogenous ligands for them, α-MSH, γ-MSH, and agouti-related protein (AGRP). We quickly established with our biological colleagues (e.g., ref 21) using radiolabeled NDP-α-MSH that the endogenous agonist ligand for all three of these new receptors was the melanotropin peptides in particular α-MSH and perhaps for the MC3R, γ-MSH. Furthermore, we quickly determined that NDP-α-MSH (MT-I) and MT-II had potent agonist activity at all three of these new receptors. Furthermore, with Roger Cone, we demonstrated that SHU-9119 the melanotropin antagonist (Figure 1) was an antagonist at the mammalian MC3R and MC4R27 but an agonist at the MC1R and MC5R (all of these melanotropin peptides have no binding at the MC2R as expected).
In collaboration with Roger Cone and using MT-II (which crosses the BBB) and SHU-9119 (a MC4R and MC3R) antagonists, we demonstrated that MT-II reduced food intake over several hours,48 and later it was shown that prolonged administration over several days led to significant weight loss. Many pharmaceutical and biotech companies have been developing MC4R agonists as a treatment for obesity (reviews),49 and the study of melanotropins and the MC4R and MC3R in various aspects of feeding behavior including obesity, cachexia, and energy balance is a huge area of current biological research. MT-II and SHU-9119 or SHU-9119 to block endogenous melanotropins were used with several biological collaborators to establish the role for melanotropin peptide and melanocortin receptor in antipyretic activity,50 effects on the cardiovascular system,51 effects on natriuresis,52 inflammatory response,53 anorexia, 54 and as already discussed several aspects of sexual function and behavior and others. During this time, we also made considerable efforts to develop selective agonists and antagonists for the MC1R, MC3R, MC4R, and MC5R receptors.55 Considerable progress has been made in our and other laboratories, and those discovered in our laboratory have proven to be very useful for demonstrating the involvement of melanocortin receptors in a number of biological functions which previously were unknown. This work will not be discussed here since despite the progress and the continuing need for such ligands, lack of support has become a problem though there is now considerable evidence that melanocortin receptors are involved in pain pathways especially in females56 and that the MC3R is involved in regulation of food intake.57
Conformation Constraint in Enkephalin and Other Neurotransmitters and the Biology of Pain
A long-term goal in our research pursued with biological colleagues over many years is the development of new ligands to help obtain an understanding of pain, and why it has been so difficult to obtain treatments for pain, especially prolonged and neuropathic pain, that do not lead to tolerance and major toxicities.
The discovery of the enkephalin in the 1970s58 led to a collaboration with Tom Burks and Hank Yamamura in the medical school that has continued to this day, though both unfortunately have passed away. After considering the problem through study of the biology of enkephalins (short half-lives in vivo and in vitro), we decided to constrain the conformational space of enkephalin by cyclization and by the use of d-amino acids where possible. Through a small series of cyclic analogues,59,60 we converted enkephalin to the cyclic pentapeptide analogue c-[d-Pen2, d-Pen5]enhephalin60 (Figure 2). We chose initially to use d-Pen2 in the 2 position of enkephalin and to make a cyclic disulfide-containing peptide because it was known that a d-amino acid in the 2-position of enkephalin was needed to maintain good binding affinity to the opioid receptors and because of the additional constraint the geminal dimethyl groups of Pen would bring in medium sized ring (13-membered ring). The choice of the 5-position for the other penicillamine residue was based on modeling studies suggesting it would stabilize a β-turn structure. This analogue drew much immediate attention because it was the first constrained, enzymatically stable, and highly δ opioid receptor selective analogue, and there was much interest in sorting out the physiological and pharmacological differences between the standard μ opioid ligands (e.g., morphine, fentanyl, etc.) from the biology of δ opioid ligands which were not known at that time. From a chemical design perspective, converting linear biologically active linear peptides into cyclic peptides drew interest from chemists involved in drug design,61,62 and now the cyclic approach has become a standard method in the design of the biologically active peptides and peptidomimetics. However, the major impact was the new insights it provide into the function of the δ opioid receptor both in terms of pain modulation and the origins of some of the μ opioid toxicities. Only a few of the early studies done in collaboration with our biological collaborators will be discussed here. However, hundreds of papers have been published since on many aspects of the biology of the δ opioid receptor and its functional relationship(s) to the μ and κ opioid receptors. Our collaborator Tom Burks immediately utilized DPDPE to establish that δ opioid receptors were involved in the analgesic effects of opioids, but the δ ligand did not inhibit intestinal motility (cause constipation) one of the major side effects of μ opioid ligands64 and in further studies were able to determine how μ, δ, and κ receptors mediated analgesia and gastrointestinal transit at spinal and supraspinal levels.65 The radiolabeled form of DPDPE60b was used to localize δ opioid receptors in the brain.66 Other studies determined the effects of δ ligands on drinking behavior;67 body temperature,68 inhibition of diarrhea without constipation,69 and many other insights about the properties of δ opioid receptors and ligands. Subsequent modification of DPDPE in χ space that led to complete selectivity (> 105) for the δ receptor will be discussed. Now I will turn to a pressing biological problem that still is unresolved today, finding opioid ligands with potent analgesia effects but without the major toxicities and tolerance of current opioids.
Figure 2.

Development of ligands for pain.
Biphalin is a bivalent analogue of enkephalin with a hydrazine bridge between the tetrapeptide chain first synthesized by Lipkowski and co-workers.70 It has high potency at both the μ and δ opioid receptors and is a potent analgesic. We became interested in this derivative because of its high potency at both μ and δ opioid receptors and because of the developing concept that multivalency could give synergistic effects in biological systems. Furthermore, since δ receptor selective ligands were found not to have the major toxicities of μ ligands, we proposed that “balanced” δ/μ ligands might have reduced toxicities and still have high potency in analgesia. An extensive evaluation by Frank Porreca and his students of biphalin showed the following: (1) given i.c.v., it was exceptionally potent in antinociception assays, 257 times that of morphine, and equipotent when given i.p.; (2) its antinociception was blocked by μ and δ opioid antagonists but not by k antagonists; (3) it had no inhibition of gastrointestinal propulsion; and (4) it caused little if any physical dependence.71 The insight that many of the toxic side effects of μ opioid ligands would be greatly reduced or eliminated by having a potent δ component led us to the realization that to utilize opioids for treatment of prolonged pain without the development of toxicities and undesirable effect such as dependence and tolerance, the ligand will need to have substantial affinity for δ receptors. Despite this knowledge, there still are no such drugs in clinical medicine. We now have done all the necessary preclinical biology and obtained tentative approval from the FDA to examine biphalin in clinical trials but have been unable to obtain funds for such studies either from industry or from the NIH.
Conversion of Somatostatin to a Potent μ Opioid Antagonist
Seven transmembrane G-protein-coupled receptors (GPCRs) are the largest class of proteins in the human genome. They are involved in virtually all aspects of intracellular communication and virtually all aspects of perception and behavior. It is well accepted that these receptors are evolutionarily related to one another and possess similar 3-D structures. But how about the ligands for these receptors; how are they related? Can a common template be found for them? We have suggested that β-turns and related structures may be such a template for many GPCR peptide ligands.
Somatostatin was originally isolated from the hypothalamus and was found to be an inhibitory hormone, and its biology attracted much attention as a potential drug by pharmaceutical companies. What attracted us was a rather obscure report72 that somatostatin (Figure 3) had weak (micromolar) opioid activity. We decided to investigate with Tom Burks and Hank Yamamura whether we could convert somatostatin into an opioid receptor selective ligand utilizing as a starting point truncated versions of somatostatin73 such as 2 (Figure 3) that had been designed for other purposes. In a short series of structure–activity relationship studies,74 we were able to obtain analogues such as 3 (Figure 3) that now had nanomolar affinity for opioid receptors and less than micromolar affinity for somatostatin receptors. In addition, these compounds were very highly selective for the μ opioid receptor, with little or no binding affinity for δ and κ opioid receptors,75,76 such as the two analogues 4a and 4b (Figure 3), which are known, respectively, as CTOP (4a) and CTAP (4b). The pharmacology of theses ligands was extensively examined, and the compounds were found to be highly stable to proteolytic breakdown and to be highly potent and prolonged acting μ opioid receptor antagonists. This led us to further examine in vitro and in vivo roles of μ opioid receptors in a variety of biological processes thought to be μ receptor mediated and activities previously unknown to involve μ opioid receptors.77-80 For example, they were critical in studies which established how opioid ligands could be used as antidiarrheals.81 A radiolabeled version also was prepared and that has become a standard μ opioid receptor ligand for binding studies82 and in the unlabeled form from many other biological studies. Conformational studies were made using NMR and demonstrated that these cyclic peptides did have β-turn structures.83
Figure 3.
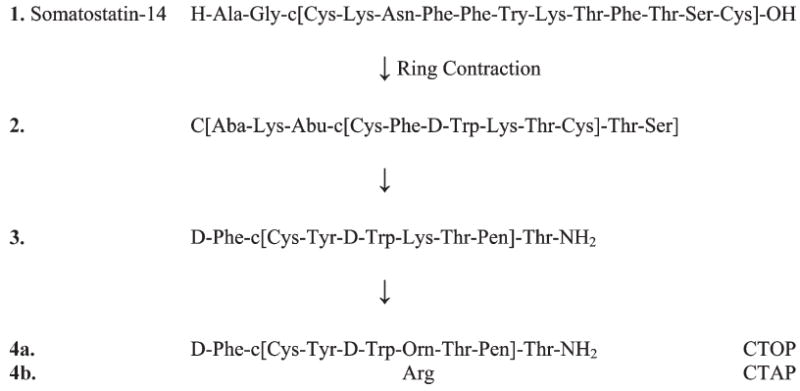
Conversion of somatostatin to a potent highly μ opioid receptor selective ligand.
Topographical Structural Consideration Peptide and Protein Design: χ Space, Asymmetric Synthesis, and Biological Activity
The protein-folding problem is a central scientific issue of modern biology. For the most part, peptide and protein chemists have focused their efforts on how peptide and protein backbone conformations organize themselves in folded proteins and how they get to the folded state. It is often considered that initial formation of these secondary structures in “nucleation sites” serves as an origin for protein folding. It occurred to us in the early 1980s based on our findings of ligand binding to carrier proteins2e,84,85 that the preferred side chain conformers gauche (−), gauche (+), and trans (Figure 4) might play as important a role as backbone conformation (phi (φ), psi (ψ) angles) in determining the affinity of peptide–protein interactions and the selectivity of ligand interactions when several receptor (enzyme, acceptor, etc.) subtypes were involved. This can be examined by crystallography if complexes can be crystallized or by NMR under certain conditions, but since we primarily were examining ligands for GPCRs, we sought a more universal approach. The approach we decided to take after considering the energy plot of chi (1) vs chi (2) for a typical α-amino acid such as tyrosine (Figure 5) was to impose conformation constraints primarily by covalent modification designed to introduce torsional barriers into standard α-amino acids in such a way as to conformationally bias side chains group to one of the three preferred gauche conformations, and to enhance torsional barriers sufficiently so that interconversion in χ space would have a significant energy barrier due to steric interactions or due to covalent bond formation. This has turned out to be a very fruitful scientific approach from several perspectives: (1) it led to development of new asymmetric synthetic organic methodology that can provide all the chiral isomers; (2) in structural organic chemistry, it provided new insights into conformation and topographical constraint while maintaining key structural elements necessary for peptide and protein folding and, most importantly, for ligand- receptor/acceptor binding interactions; and (3) from a biological perspective, it provided new and in some cases unexpected insights into the power of χ space and topography to modify and even enhance biological activity including affinity, selectivity, and efficacy. Aspects of this research has been reviewed,86,87 and though these reviews were not comprehensive they will be discussed only as necessary here. Rather we will concentrate on those findings with collaborators that have been particularly important in understanding biology and for furthering our insights into how chemistry can enhance biological understanding and lead to the development of new tools.
Figure 4.
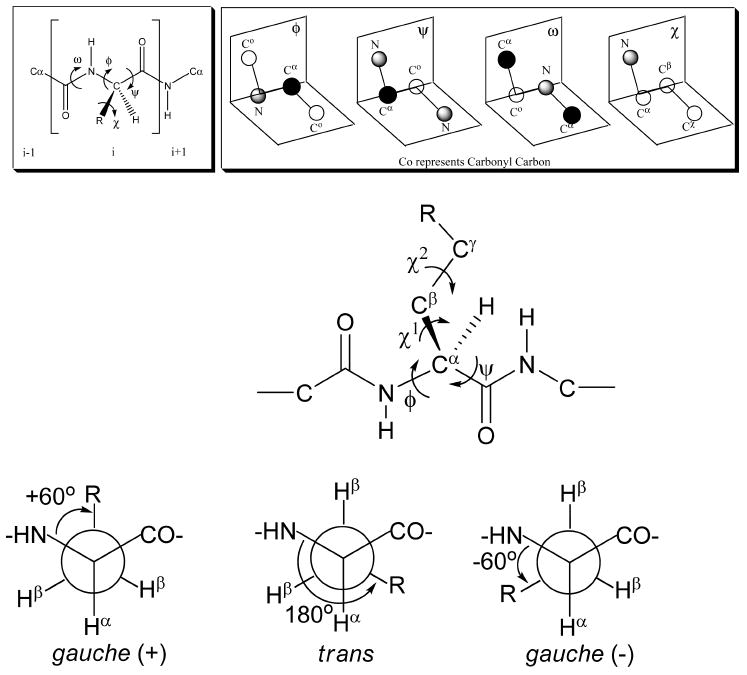
φ, ψ, ω, and χ torsional angles.
Figure 5.
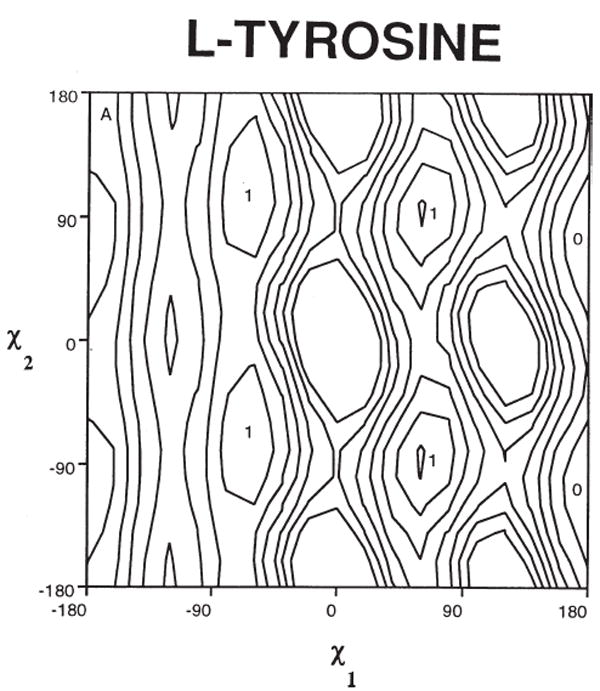
χ-1 vs χ-2 plot of tyrosine.
Though the peptide backbone and the N-terminal amino group or the C-terminal carboxylate or carboxamide can be critical moieties of the pharmacophore for a bioactive peptide, generally various side-chain functional groups constitute most of the pharmacophore which can organize in sequence or on separated residues and be brought together in 3D space in the binding process between the peptide ligand and its receptor/acceptor. In addition to the backbone (α-helix, β-turn etc), the topographical arrangements of the side chain functional groups that make up the pharmacophore is very important to their biological activity and receptor selectivity. For design in topographical χ space we concentrated on aromatic amino acids, since virtually all peptide hormones and neurotransmitters have one or more aromatic amino acid residues that are key pharmacophore elements. This has turned out to be very powerful because the dozen or so different peptide hormones and neurotransmitters we have uninvestigated over the years all have one or more key aromatic residues in their pharmacophore.
In Figure 6 are shown different structural templates which have been chosen for constraint in χ space for amino acids. In structure I, the major constraints to torsional energy barriers and specific gauche structures are steric effects or torsional strain effects. Figure 7 shows the χ1/χ2 plot of the four trimethyltyrosine structures with relative energies of the low energy gauche conformations (1 kcal energy contours). Clearly, the different gauche conformations have different low energy conformations. This suggests that if side-chain conformations are important for peptide–receptor interactions and information transduction, different conformations will lead to different activities. In II (Figure 6), the topographical structure is imposed by an additional covalent bond which actually precludes some torsional conformations in the six-membered nonaromatic ring. In structures of type III (Figure 6), constraint about the χ angles and preferred side-chain conformations are dictated primarily by steric considerations. One added advantage of this kind of structure is that chimeric amino acids can be prepared which contain structural elements from two amino acids. Such structures are particularly valuable in designing peptides with two different pharmacophores in the same structure (bivalent or multivalent ligands). Structures of type IV (Figure 6) are by their nature chimeric structures in the sense that the basic α-amino acid scaffold has the pyrolidine ring of proline or the pyrolidone ring of pyroglutamic acid (2-butyrolactam), and the R group can represent a specific functional group from many other amino acids found in peptides and proteins. We have developed robust asymmetric synthetic methods for all of these structures that allow one to obtain all of the enantiomeric and diastereomeric structures. Compounds of structure V are masked multivalent structures where the terminal olefin can serve as the starting point for multiple chemical reactions including ring-closing metathesis, aldehydes or ketones, other functional groups, starting materials for β-turn mimetics, etc. The overall goal is to make peptide backbone scaffolds as robust templates for synthesis of pharmacophore structures of great diversity. Nature has made the peptide scaffold a nontoxic scaffold to biological life, and we should learn from Nature. The inherent compatibility of the peptide scaffold with life is clear, and we should invest more effort to develop a robust nonpeptide synthetic chemistry around it. For all of the 1970s and 1980s I had an NSF grant to do that. However, when I got my best reviews in the early 1990s, the NSF cut off support for this research, and much remains undeveloped, though we continued to develop what we had begun. We will not discuss the synthetic methodologies we developed which generally can be readily scaled up. We will simply provide some key references for the asymmetry syntheses of the different kind of amino acids represented in Figure 6. Robust synthetic methods for compounds of structures I,89,90 structures II,91 structures III,92 structures IV,93-95 and structure V,96 have been developed. A few examples are discussed below to briefly illustrate some aspects of what we have developed.
Figure 6.
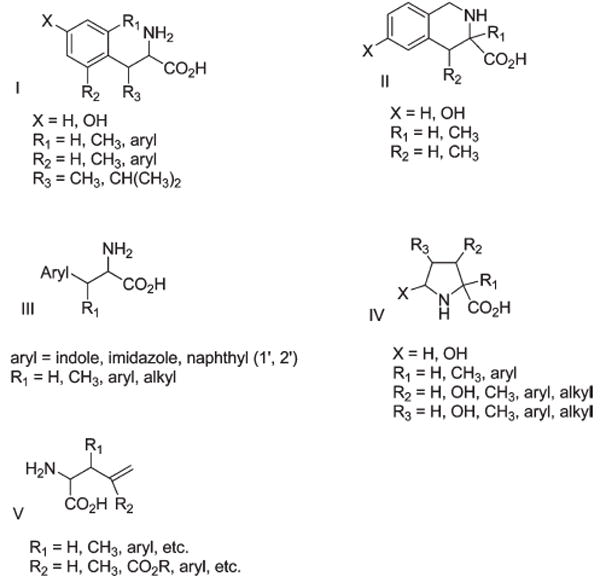
General structure of some χ-constrained aromatic amino acids.
Figure 7.
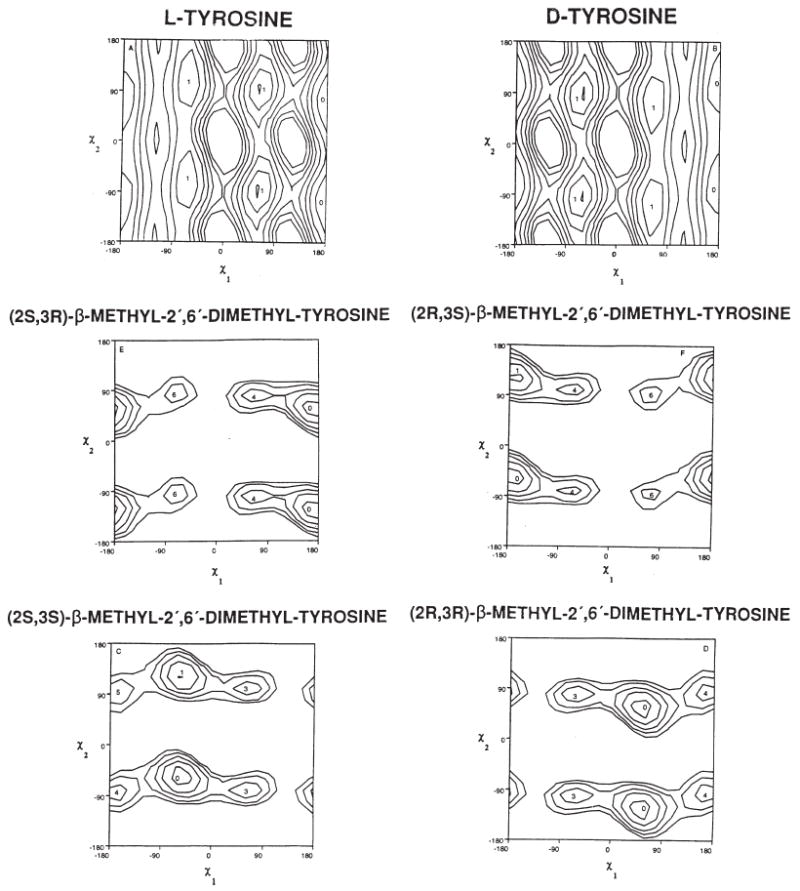
χ-1/χ-2 plot of l- and d-Tyr and for the four isomers of 2′,6′-dimethyl-β-methyltyrosine (TMT).
Figure 8 outlines the asymmetric synthesis of (2S,3R)-2′,6′-dimethyl-β-methyltyrosine. For a chiral auxiliary the classical benzene chiral auxiliary gave poor enantioselectivity, but we found that the phenyl derivative greatly improved enantioselectivity. Once this was established, the asymmetric bromination and the SN2 azide synthesis proceeded in high enantioselectivity, and subsequent reactions did not lead to racemization. Topographically modified analogues of tryptophan97 and histidine98 have been prepared using different but related asymmetric synthetic methods.
Figure 8.
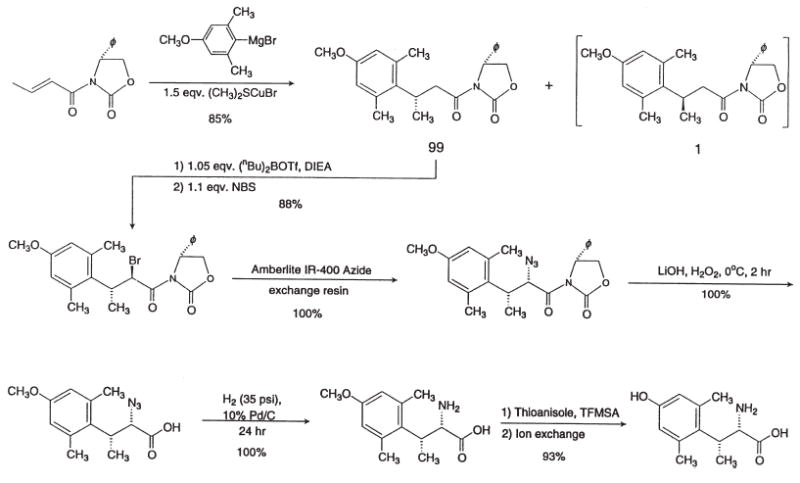
Asymmetric synthesis of (2S,3R)-2′,6′-dimethyl-β-methyltyrosine.
Figure 9 outlines the asymmetric synthesis of pyroglutamyl derivatives that can readily be converted to the corresponding proline analogues. This chemistry is highly stereoselective using simple starting materials that can readily be prepared in 100 g quantities or more as needed. And as shown in Figure 6, derivatives of IV with different stereo-chemistries and different R groups can be made using a varieties of chemistries including Michael-type chemistry, direct alkylation chemistry, and so forth. This chemistry opens up many new avenues for the design and synthesis of topographically novel peptides, peptidomimetics, and β-turn mimetics.
Figure 9.
Methods for the asymmetric synthesis of pyroglutamic acids analogues.
As a final example, we show new asymmetric synthetic methodology we have been developing to make all of the chiral isomers of structure V (Figure 6). As shown in Figure 10, the method lends itself to the introduction of a number functional groups that will provide a large chemical landscape for further development of novel topographical space in peptide design.
Figure 10.
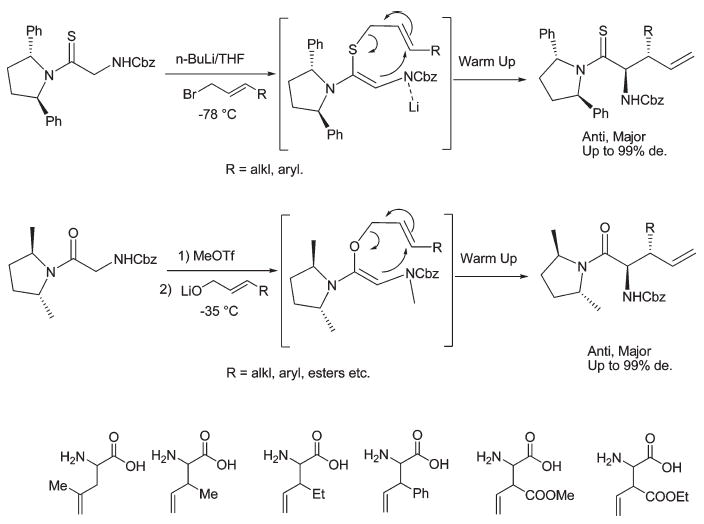
Asymmetric synthesis of β-functionalized γ,δ-unsaturated amino acids.
Application of χ (χ) Space Constraint to Biological Problems and Peptide Mimetic Design
A major question in Chemical Biology is the extent to which key pharmacophore elements in a peptide or protein structure utilize χ (χ) space (the side-chain conformations of amino acid residues in peptides) to modulate or even determine biological function. We have examined this question in several ways, and have found that in some cases, a specific preferred χ (χ) conformation can dramatically affect ligand–receptor interactions and the subsequent downstream biological effects, even behaviors, that are mediated by the particular ligand–receptor system.
For example, modification of enkephalin to the cyclic peptide c-[d-Pen2,d-Pen5]enkephalin (vide supra) gave a potent δ opioid receptor selective ligand with a well-defined conformational preference as determined by NMR studies in aqueous and DMSO solution,99 in its X-ray crystal structure,100 and by computational methods.99,101 However, the conformation preference of a key pharmacophore element, Tyr1, was still unclear, and we decided to examine this problem with our biological colleagues by using all 4-isomers of trimethyl- tyrosine (2S,3S)-, (2S,3R)-, (2R,3S)-, and (2R,3R)TMT1, incorporated into DPDPE. The binding affinity for the μ and δ receptors, and the biological activities in the classical mouse vas deference (MVD, δ receptors) and guinea pig ileum (GPI, μ receptor assays)102 are given in Table 2. Only the (2S,3R)TMT1 analogue has the δ opioid receptor binding affinity and receptor selectivity of DPDPE. This shows unambiguously the highly preferred χ1 torsional angle for binding to the δ receptor is the trans (180°) torsional angle in Tyr1, while the preferred torsional angle at the μ receptor for Tyr1 is −60°. Most interesting, the (2S,3R)-containing analogue is an antagonist at the μ receptor. Since all of the other conformational properties remain unchanged, these results demonstrate that a single constraint in χ space for a key pharmacophore residue can lead to potent agonist activity at one receptor subtype and antagonist activity at a second subtype receptor. To the best of our knowledge, this is the first time that a single change in torsional angle of a single pharmacophore element in a peptide could be demonstrated to be an agonist at one receptor subtype and an antagonist at another receptor subtype for a neurotransmitter. These results were confirmed in vivo where it was found as expected102b that the (2S,3S)TMT1 analogue is a potent analgesic while the (2S,3R)TMT1 analogue is a very weak analgesic due to antagonism at the μ receptor.
Table 2.
Binding Affinity and Biological Activity Properties of TMT1-DPDPE Derivatives
| peptide | vs [3H]CTOP (μ) | vs 3H[p-ClPhe4] DPDPE (δ) | GPI (μ) | MVD (δ) |
|---|---|---|---|---|
| DPDPE | 610 | 1.6 | 7300 | 4.1 |
| [(2S,3S)-TMT1]DPDPE | 720 | 211 | 293 | 170 |
| [(2S,3R)-TMT1]DPDPE | 4300 | 5.0 | 0% at 60 μM antagonist | 1.8 |
| [(2R,3R)-TMT1]DPDPE | 77000 | 3500 | 50000 | 2200 |
| [(2R,3S)-TMT1]DPDPE | 0% at 10 μM | 9% at 10 μM | 75% at 82 μM | 28% at 10 μM |
This three-dimensional knowledge provided the information to do ligand-based peptide mimetic design that is converting from a peptide scaffold directly to a nonpeptide scaffold by de novo design. This de novo design utilized modeling and computational chemistry considering several nonpeptide scaffolds and directly proposing the compounds to make. Thus, with a small library of a properly substituted 1,4-piperazine scaffold we were quickly able to obtain a nonpeptide ligand which could mimic the binding and receptor selectivity profile of the [TMT1]-DPDPE analogue of enkephalin103 and even mimic the peptide but not other nonpeptide ligands at a site specific mutated receptor.103b From these studies, we also were able to come up with a good 3D model for a ligand which would be a potent and selective δ opioid receptor ligand.
Use of χ (χ) Space To Develop an Inverse Agonist
As previously discussed, antagonists play a crucial role in understanding the chemical basis for biological activity in many systems. In the case of opioid ligands, Schiller and co-workers104 have shown that the dipeptide Tyr-Tic (Tic = tetrahydroisoquinoline-3-carboxylic acid) is a delta receptor antagonist (IC50 = 190 nM at the δ receptor). The Tic amino acid residue is a constrained α-amino acid with a well established side-chain conformation. We decided to investigate whether in the antagonist series for δ ligands the preferred Tyr χ angle would be the same as in the agonist series. Thus, we incorporated all four isomers of 2′,6′-dimethyl-β-methyltyrosine (TMT; (2S,3S; 2S,3R; 2R,3S; and 2R,2R) into the dipeptide to give four isomers of TMT-Tic.105 In this case, both the (2S,3R)-TMT-Tic-OH (IC50 = 9.3 nM) and the (2S,3S)-TMT-Tic-OH (IC50 = 124 nM) ligands were found to be potent δ-opioid receptor antagonists in the MVD assay. The most exciting results, however, came when we examined G-protein activation using the GTPγS assay with membranes from δ receptor transfected cell lines and found that the (2S,3R)-TMT-Tic-OH analogue was an inverse agonist at human δ opioid receptor.106 Inverse agonists are powerful potential drugs when one encounters constituitively active receptors in biological systems
In yet another example of the use of the χ-constrained amino acid TMT, incorporation of the four isomers of the O-methoxy derivative of TMT in oxytocin107 provided unique new insights into the bioactive topography of oxytocin antagonists.
Use of Chi (χ) Constraint To Explain Prolonged Biological Activity
As discussed above, NDP-α-MSH (MT-l) and the highly constrained cyclic α-MSH analogue MT-II show very prolonged biological activities in vitro and in vivo. As previously discussed, this effect on pigmentation is both cAMP and Ca2+ dependent. Interestingly, however, after several hours, the cAMP levels return to basal activity and yet prolonged skin darkening persists in vivo for many days. The biological explanation for this biology has been sought for many years (we have provided these peptides to many biological investigators worldwide, but they have not found a biological explanation). As a chemist, my basic hypothesis of such unexpected effects is that any change in biological activity must involve a change in structure. Structure–biological activity relationships indicated that the d-Phe7 substitution in α-MSH analogues was a key structural element for prolonged biological activity since l-Phe7 analogues do not have prolonged biological activity. Thus, we decided to use our χ constraint approach to see if we could discover a structural basis for biological activity prolongation in χ space. We used MT-II as our template because we thought, based on modeling, that its constrained 3D-conformational backbone structure would be maintained with the introduction of the four isomers of β-Me-Phe (all four isomers) in position 7108 and of β-Me-Trp (all four isomers) in position 9109 of MT-II. The β-Me-Phe7 analogues did provide some insights into the topographical requirements for prolonged biological activity at the melanocortin receptor,109 but the four β-Me-Trp-containing analogues were decisive, and we will limit our discussions here to the results with the 4 β-Me-Trp analogues. As shown in Table 3, the potencies of the four isomers vary over almost 3 orders of magnitude in the frog skin assay and over a factor of 30 in the human MC4R binding assay (comparable results were found in the second messenger cAMP assay;108 data not shown). The prolongation of biological activity in the frog skin is shown in Figure 11. The most exciting and important result is that the four isomers demonstrate the full spectrum of prolonged biological activity with the (2R,3R)-β-MeTrp9 analogue showing prolonged activity like MT-II, whereas the (2S,3S) compound showed no prolonged activity (like the hormone α-MSH) with the others of intermediate activity. Notice that prolonged activity and potency are not related to each other; that is, the order of potency and prolongation of bioactivity are different (Table 4). So what is the origin of the prolongation? Clearly, it is not a matter of potency but rather a matter of the topographical differences in the structure of the pharmacophore. This was confirmed by evaluation of the NMR conformations of the 4 β-MeTrp9-containing isomers. They had the same backbone conformation as MT-II but different preferred χ-1 rotamer structures.109 These results beautifully illustrate the critical significance of χ space in determining biological activity in peptide–protein interactions involving hormones and neurotransmitters and their receptors. In this case similar results at the frog and human receptors could be demonstrated. In general, one would expect this to be important in all aspects of peptide–protein, protein–protein, and protein–nucleic acid interactions and in both health and disease states. This is still an underdeveloped area of drug design, but its further development may have profound implications for treatment of disease. Further development in synthetic organic chemistry will be needed to maximize progress and to explore the significance of χ space in health and disease.
Table 3.
Comparative Biological Activities of the Four Isomers of Ac-Nle-c-[Asp-His-DPhe-Arg-βMeTrp-Lys]-Nle at the MC1Rs in Frogs and Humans
| peptide | frog skin MC1R |
hMC1R binding |
||
|---|---|---|---|---|
| EC50 (nM) | relative potency | IC50 (nM) | relative potency | |
| MT-II | 0.10 | 1.0 | 0.50 | 1.0 |
| [(2S,3S)-β-MeTrp9]-MT-II | 0.44 | 0.23 | 0.50 | 1.0 |
| [(2R,3S)-β-MeTrp9]-MT-II | 0.06 | 1.6 | 3.0 | 0.17 |
| [(2S,3R)-β-MeTrp9]-MT-II | 29 | 0.0035 | 15 | 0.030 |
| [(2R,3R)-β-MeTrp9]-MT-II | 0.30 | 0.30 | 2.0 | 0.25 |
Figure 11.
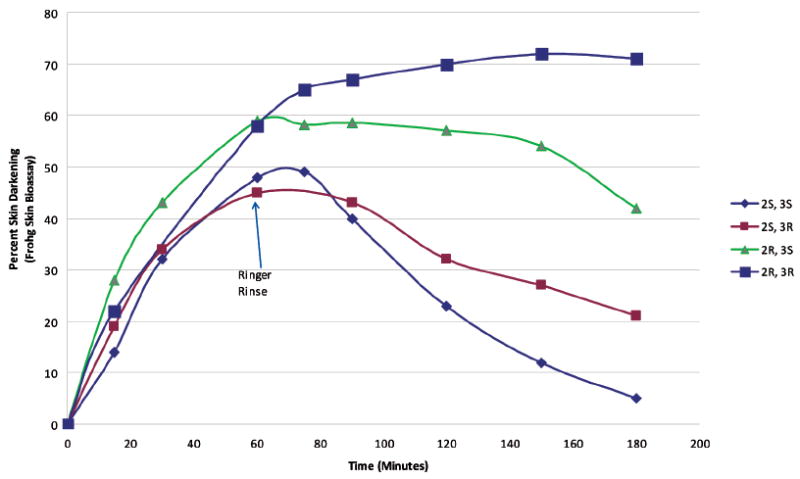
Measurement of the prolonged biological activity of the four isomers of [β-MeTrp9]-MT-II in the frog skin bioassay.108
Table 4.
Comparison of Potency and Prolonged Biological Activity of the Four Isomers of Ac-Nle-c-[Asp-His-DPhe-Arg-β-MeTrp-Lys]-NH2 at the Frog Skin and Human MC1R
| melanocortin receptor | relative potency | relative prolonged activity |
|---|---|---|
| frog skin (MC1R) | (2R,3S) > (2S,3S) > (2R,3R) > (2S,3R) | (2R,3R) > (2R,3S) > (2S,3R) > (2S,3S) |
| human MC1R | (2S,3S) > (2R,3R) > (2R,3S) > (2S,3R) | (2S,3S) > (2R,3R) > (2R,3S) > (2S,3R) |
Glucagon Antagonists and Multiple Signaling Pathways for GPCRs
I took my first sabbatical in 1975 at the National Institutes of Health to learn more about peptide hormone and neurotransmitter receptors and had the great fortune to spend a year in Martin Rodbell’s laboratory. Though I failed in my primary goal, to isolate in a purified soluble form the glucagon receptor (this still has not been done), I was fortunate to be there when Marty’s group was discovering the G-proteins and their functions and developing the binding assays and second messenger (cAMP) assay that made this possible. We took these new assays back to my laboratory and examined the SAR of glucagon and tried to discovery a glucagon antagonist and the role of glucagon in diabetes with an M.D., David Johnson. This is a very extensive research area which I will not discuss here except to make a few comments about the valuable insights that can be obtained by collaborating with a medical doctor. Early studies using semisynthetic methods of synthesis in collaboration with Marty Rodbell quickly established that the N-terminal residues in glucagon were vital for agonist biological activity while the C-terminal residues were critical for binding affinity.110 This realization led to the development of the first glucagon antagonist111 which was shown to block glycogenolysis in perfused rat liver112 and that it could lower blood glucose levels in diabetic animals which was done in collaboration with David Johnson.113 The availability of these antagonists and other analogues of glucagon eventually allowed us to establish with Miles Howslay that glucagon could activate two signaly transduction pathways. 114 This was one of the first demonstrations of this chemical phenomena for GPCRs and opened up a whole new line of research for investigating the mechanism of signaling by ligands and their G-protein-coupled receptors which is now a huge field. The significance is still far from being understood, especially in disease states such as diabetes, cancer, and others, but the chemical tools are available to do it now.
Multivalency: Drug Design for Disease
Efforts to treat our major degenerative diseases such as cancer, prolonged and neuropathic pain, mental diseases including addiction, and diabetes, cardiovascular disease, etc. have been largely unsuccessful and often are accompanied by significant toxicities. The standard drug design paradigm of identifying a target and developing a ligand (agonist, antagonist, etc.) for that target is often successful and has led to good drugs to treat symptoms and in some cases that are curative for a specific phenotype which often is just a small percentage of the patients with the disease. More recently, comparative genomic and proteomic studies of normal vs diseased tissue have demonstrated that, in many diseases, multiple changes in gene expression have occurred and that the treatment modalities can themselves enhance aspects of the disease. We have proposed that advantage can be taken of these findings in drug design for disease states.115,116 In this approach to drug design and development, ligands are developed that are multivalent, that target two or more receptors/acceptors that are critical for manifestation of the disease state, and that address the disease state relative to the normal state. Again, this requires close collaboration between chemists and biologists who are examining (determining) the key evolutionary changes (gene expression or modification) that are responsible for the disease state. We will briefly illustrate the approach we are taking with two examples: (1) the development of multivalent ligands that can treat neuropathic pain states such as allodynia and hyperalgesia in which potent opioid such as morphine are ineffective and (2) design of multivalent ligands that can detect cancer cells but not normal cells.
Design of Multivalent Ligands To Treat Neuropathic Pain without Development of Tolerance
Over 50 million people in the U.S. alone suffer from prolonged and neuropathic pain for which there is no adequate treatment. Considerable evidence has been developed during the past decade or so that the development of prolonged and neuropathic pain involves upregulation of neurotransmitters and their receptors in the ascending and descending pain pathways that are stimulating for neural transmission and thus can cause pain. These include neurotransmitters such as cholecystokinin, CGRP, substance P, and their receptors. Here we will concentrate on the upregulation of substance P and its receptor, the neurokinin-1 receptor (NK-1R), though we are investigating several others. Based on extensive biological studies, we hypothesized that design and synthesis of a multivalent ligand that had potent mixed δ and μ opioid agonist activities and potent antagonist activity at the NK-1R would have a number of highly desirable biological activities with few if any of the undesirable toxicities of current analgesic drugs. These desirable activities would include potent analgesic affects in both acute pain and in neuropathic pain states and without the development of tolerance. In Figure 12 we illustrate the design of a typical ligand with the specific example of a molecule we did design.117,118 Both adjacent and overlapping pharmacophores have been designed. In order to determine whether these multivalent ligands possess the necessary in vitro biological activity profile it is necessary to perform several biological activities as outlined in Table 5 for three of our lead compounds, done in collaboration with Frank Porreca, Hank Yamamura, and Josephine Lai. As can be seen, all three lead compounds have the same structure except for the C-termini. The initial compound 1 was modified from a C-terminal ester to a C-terminal amide (1→2) because it was found that the C-terminal ester has a very short half-life in serum (about 1 min), while the C-terminal amide had a half-life of over 4 h. The simple benzyl amide analogue 3 was prepared to increase aqueous solubility. As can be seen in Table 5,117,118 the designed ligands have very potent agonist activity at both the δ and μ receptors with good agonist activity in the GTPγS assay and were potent antagonists of the NK-1 receptor with picomolar binding in some cases. We have shown that all these ligands cross the blood–brain barrier (BBB) quite well. In order to explain the BBB penetrability, we examined the conformation of these three peptides in aqueous solution and in the presence of micelles using NMR. Interestingly, all three of these compounds have no discernable conformation in aqueous solution as expected. However, in the presence of micelles, they form highly stable conformations, the very large number of NOEs that were observed were used to establish that the compounds have helical like structures.118
Figure 12.

Design of a multivalent ligand for pain (Bzl = benzyl).
Table 5.
In vitro Biological Activity Profile for Novel Analgesic for Neuropathic Pain
| compd | binding assays |
second messenger [35S]GTPγS |
functional assays |
|||||
|---|---|---|---|---|---|---|---|---|
| hDOR vs [3H]DPDPE Ki (nM) | rMOR vs [3H]DAMGO Ki (nM) | hNK1 vs [3H]SP Ki | hDOR EC50 | hMOR EC50 | MVD (δ) IC50 (nM) | GPI (μ) IC50 (nM) | SP GPI ant. Ke | |
| H-Tyr-d-ala-Gly-Phe-Met-Pro-Leu-Trp-O-3′,5′Bzl(CF3)2 | 2.8 | 36 | 0.084 | 2.9 | 32 | 22 | 300 | 25 |
| H-Tyr-d-ala-Gly-Phe-Met-Pro-Leu-Trp-HN-3′-5′-Bzl(CF3)2 | 0.66 | 16 | 0.0065 | 8.6 | 7.0 | 15 | 490 | 10 |
| H-Trp-d-ala-Gly-Phe-Met-Pro-Leu-Trp-HN-Bzl | 0.64 | 1.8 | 3.2 | 2.6 | 21 | 4.8 | 61 | 9.9 |
How do these compounds behave in in vivo animal model for acute pain and for analgesic activity in the SNL rate model for neuropathic pain (allodynia and hyperalgesia)? Our biological collaborators, Drs. Todd Vanderah and Frank Porreca, have addressed these and other biological activities in vivo including an evaluation of toxicities and tolerance. A summary of the in vivo biological activities is given in Table 6 for compound 1. Although the results are preliminary they are exceptionally promising. These compounds have potent antinociceptive effects in both naïve animals and in animals which have neuropathic pain. For the latter animals, antinociceptive activities morphine has little or no efficacy. Most interesting and potentially important, these compounds do not lead to the development of tolerance in either naïve animal or in animals with neuropathic pain even after long-term use (10 or more days). Finally, these compounds do not appear to have any of the toxicities of current opioid-based analgesics, including no impairment of motor skills even at much higher doses than the analgesic doses. There is still much to learn about the biology of this new class of ligands, but this new approach in drug design appears to lead to unique biological activity profiles that directly address the neuropathic pain disease state. Whether it can reverse the changes in the expressed genome in ascending and descending pain pathways remains to be determined, but the lack of development of tolerance gives some hope.
Table 6.
Summary for in vivo Biological Activities from Multivalent Ligand H-Tyr-d-Ala-Gly-Phe-Met-Pro-Leu-Trp-0-3′5′-Bzl(CF3)2
|
Multivalent Ligands for the Detection and Treatment of Cancer
Despite enormous scientific efforts in the past 40 years, drugs that can treat cancer effectively and provide a cure are still rare. The primary approach, aside from the use of chemicals which are more toxic to cancer cells than normal cells, is to discover a major abnormality of the cancer cells vs the normal cells and to target that protein with a specific drug. Though there have been a number of notable successes using this approach, the successful outcomes usually involve a relative small subset of cancer patients even with the specific form of cancer (e.g., breast cancer). We have contributed to this area by our discoveries in the development in combinatorial chemistry in the one bead–one peptide (OBOP) or one bead–one compound (OBOC) approach 119 that allows one to utilize the power of solid-phase chemistry, especially solid-phase peptide chemistry, to synthesize large libraries of peptides, peptidomimetics, and various classes of other small organic compounds for which there is robust synthetic chemistry, to explore chemical space and discover those particular compounds that can be leads for the protein or nucleic acid targets that are developed. This led us (Syd Salmon, Kit Lam, Evan Hersh, Fahad Al-Obeidi, and myself) to found the first biotech company dedicated to the use of combinatorial chemistry in drug discovery (Selectide Corp., an important arm today of Sanofi-Aventis). Many other approaches to combinatorial chemistry have been developed, along with ancillary technologies such as high-throughput screening that are now part of most academic and industrial drug discovery efforts. This will not be discussed except to say that the fundamental ideas that came out of the development of combinatorial science were critical to the developments I will discuss below.
In collaboration with Bob Gillies,115,120 we have decided to take a new approach. Comparative genomics and proteomics of cancer vs normal cells have demonstrated that cancer cells that have 10-fold to 100-fold changes in the expressed genome can serve to distinguish cancer from normal cells.115 In this approach, we target cell surface proteins (receptors, enzymes, acceptors, etc.) that are up-regulated or newly expressed on cancer cell surfaces but not on normal cells. We hypothesized that we could target two or more cell surface proteins that would distinguish cancer from normal cells and get high selectivity. This also is done to obtain the synergies that are expected from multivalent vs monovalent interactions on a single cell surface. As a result of these synergies we would expect to see few false positives. In the design of these multivalent ligands, in addition to the ligands needed for molecular recognition, the design also must accommodate a unique agent for imaging (a wide variety of optical, magnetic and other imaging agents are possible) and also for treatment of the cancer (drug). Though the surface proteins are the target, it is realized that certain cell surface proteins (for example, GPCRs) are internalized on interaction with agonist ligands. This provides the opportunity in favorable cases to target both cell surface proteins and intracellular targets including organalles and the nucleus. In most of our studies to date, we have been developing the synthetic chemistry, molecular pharmacology, molecular biology, and related methods needed to make this a viable and robust approach with combinatorial possibilities for multivalency. As already discussed, in previous work we examined the advantages of multivalency using a water-soluble polymer scaffold, and though we obtained some exciting results in imaging cancer cells42-44 the methodology did not lend itself to in vivo imaging or drug delivery. Thus, we have developed new scaffolds120,121 which generally involve (-Pro-Gly-)n segments that are semi rigid and more flexible modified polyethyleneglycol (PEGO) units. Because cell surface receptors are relatively large (cross-sections of 20–40 Å or more) the ligands themselves must be separated by a distance of 25–75 Å. We have developed a very robust synthetic chemistry to do this (see Figure 13 for an example). This chemistry allows us to prepare homo- and heterobivalent ligands with addition sites that allow attachment of fluorophores and other imaging agents. Furthermore, we were able to demonstrate that these heterobivalent ligands with linker lengths estimated from modeling to be between 45 and 100 Å could cross-link receptors on cells containing receptors for the two ligands (a δ opioid receptor and a melanocortin receptor (MC4R) with synergies up to about 50-fold120a relative to cells that only had one of the receptors). These exciting findings demonstrate that our design works and that our ligands can cross-link two different proteins on a cell surface. Subsequent experiments (unpublished) have shown that similar results can be obtained with other heterobivalent ligands targeting these and other cell surface receptors. Furthermore, the contrast between cells containing two targets can be readily distinguished from cells containing only one receptor using bivalent ligand that also have a fluorescent probe attached. In this case the cells containing both receptors are completely “lit up” with the fluorescent probe, whereas the cells with one of the ligands can barely be visualized in the same time period. These results to date are very promising, and we are now examining the use of the heterobivalent ligands in in vivo studies with animal cancer models.
Figure 13.
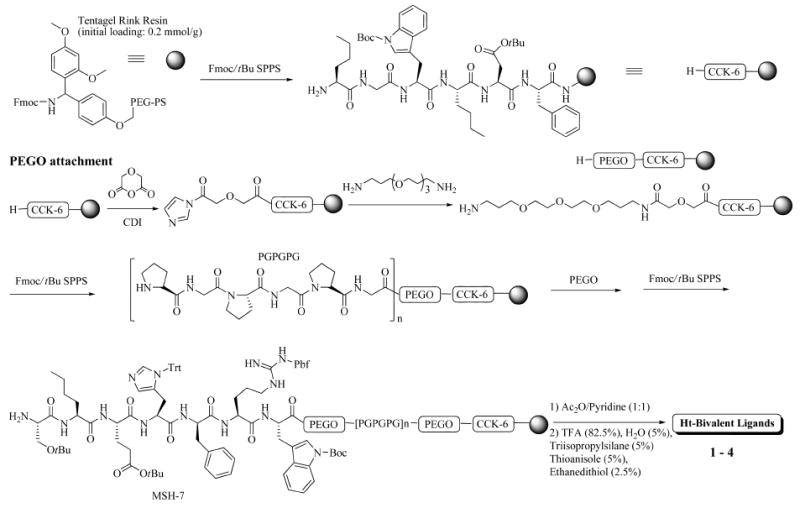
Synthetic scheme for the synthesis of a heterobivalent ligand.
From the above results, we believe that design of multimeric multivalent ligands can be a powerful tool for new approaches to drug development of disease states, and we are pursuing this approach vigorously with designed peptide and peptide mimetic ligands.
Concluding Remarks
Biology as a science is increasingly about chemistry and chemicals (genomes, proteomes, etc.), and understanding biology from evolution to disease requires chemistry. It seems self-evident that true collaboration between biologists and chemists is not only necessary but essential for scientific progress. However, old myths, habits, and institutional bias die hard, and not without considerable angst. Currently our universities and major government and private funding agencies are under severe financial stress. Despite the scientific problems and opportunities that are there for everyone to see, in the United States we are funding basic science at only about 50% the level of 30 or 40 years ago. This has created much stress on scientists and science and further retards the necessary reforms to promote and reward the collaborative science that is needed. Based on 45 years in the scientific trenches, I have found that collaboration is often not rewarded. Rejection does take its toll and significantly limits what we can accomplish. Nonetheless, I would not trade my scientific and academic career for anything. It has been such a joy to work with my students and biological collaborators and their students and to explore new territories of Nature that I could not have dreamt of. The joy of discovery or realization that different scientific paths were needed to understand or explain biology with chemistry has been fun and exciting.
I am especially grateful to my wife Pat who has been a physics Professor for over 40 years, and my children Tim, Steve, and Patrick whose patience, understanding, and love have given me endless support and joy. For my development as a scientist my mentors Professors A. William Johnson, A. T. Blomquist, Vincent du Vigneaud, and Carl (Speed) Marvel gave invaluable advice, criticism, support, and direction. Without them this journey would have never happened.
Acknowledgments
Over my 40 plus years I have had the good fortune of being blessed with over 70 outstanding graduate students and over 100 excellent postdocs, visiting scientists, and distinguished visitors, whose hard work, creativity, and dedication made this journey possible. Many of their names are given in the references provided in this Perspective. In addition, I have had collaborations with many leaders in several areas of biology and medicine and with their students, postdocs, and technicians. Without their enthusiasm and creativity much of this research would not have been possible. I am eternally grateful to them. I also want to thank the agencies, institutions, and companies who have supported our research over the years, including the National Institutes of Health for my entire career, the National Science Foundation for the first 20 plus years of my career, and for short periods of time, the Research Corporation, Abbott Laboratories, SmithKline, Merck, Ortho Pharmaceuticals, The State Department, Selectide Corporation, and the Arizona Biological Research Commission. Finally, and most importantly, I want to thank the University of Arizona and the Department of Chemistry and Biochemistry for giving me a wonderful home for 41 years and usually making me feel at home, even when I wandered into new unchartered territory. Over the years I have had wonderful Administrative Assistants who kept me organized and focused: special thanks to Lourdes Gallegos, Natasha Johnson, Cheryl McKinley, and Margie Colie.
References
- 1.Hruby VJ, Yamashiro D, du Vigneaud V. J Am Chem Soc. 1968;90:7106–7110. doi: 10.1021/ja01027a040. [DOI] [PubMed] [Google Scholar]
- 2.For those who might be interested in what an organic chemist could do at the interface of chemistry and biology at that time I list a few of our chemical accomplishments below and in ref 3 our contributions to biology. Barstow LE, Hruby VJ. J Org Chem. 1971;36:1305–1306.Hruby VJ, Groginsky CM. J Chromatogr. 1971;63:423–428. doi: 10.1016/s0021-9673(01)85665-x.Hruby VJ, Barstow LE. Macromol Synth. 1972;4:91–94.Hruby VJ, Brewster AI, Glasel JA. Proc Natl Acad Sci U S A. 1971;68:450–453. doi: 10.1073/pnas.68.2.450.Glasel JA, Hruby VJ, McKelvy JF, Spatola AF. J Mol Biol. 1973;79:555–575. doi: 10.1016/0022-2836(73)90406-3.Bower A, Sr, Hadley ME, Hruby VJ. Science. 1974;184:70–72. doi: 10.1126/science.184.4132.70.Hruby VJ. In: Chemistry and Biochemistry of Amino Acids, Peptides and Proteins. Weinstein B, editor. Vol. 3. Marcel Dekker; New York: 1974. pp. 1–188.Hruby VJ, Upson DA, Agarwal NS. J Org Chem. 1977;42:3552–3557. doi: 10.1021/jo00433a031..
- 3.Early biological work: Bower A, Sr, Hadley ME, Hruby VJ. Biochem Biophys Res Commun. 1971;45:1185–1191. doi: 10.1016/0006-291x(71)90143-4.Hruby VJ, Smith CW, Bower A, Sr, Hadley ME. Science. 1972;176:1331–1332. doi: 10.1126/science.176.4041.1331.Walter R, Kirchberger ML, Hruby VJ. Experientia. 1972;28:959–960. doi: 10.1007/BF01924972.Hruby VJ, Muscio FA, Groginsky CM, Gitu PM, Saba D, Chan WY. J Med Chem. 1973;16:624–629. doi: 10.1021/jm00264a010.Hadley ME, Bower A, Sr, Hruby VJ. Yale J Biol Med. 1973;46:602–616.Bower A, Sr, Hadley ME, Hruby VJ. Science. 1974;184:70–72. doi: 10.1126/science.184.4132.70.Hadley ME, Hruby VJ, Bower A., Sr Gen Comp Endrocrinol. 1975;26:24–35. doi: 10.1016/0016-6480(75)90212-9..
- 4.The instrument we designed later served as a prototype for commercial instruments produced by Vega Biotechnologies and was the first to have monitoring devices to assess synthetic progress. Hruby VJ, Barstow LE, Linhart T. Anal Chem. 1972;44:343–350. doi: 10.1021/ac60310a004..
- 5.Hruby VJ. Acc Chem Res. 2001;34:389–397. doi: 10.1021/ar990063q. [DOI] [PubMed] [Google Scholar]
- 6.Hruby VJ, Nikiforovich GV. In: Indian Acad Sci. Balaram P, Ramaseshan, editors. 1991. pp. 429–445. [Google Scholar]
- 7.For a review, see: Ramachandran GN, Sasisekharan U. Adv Protein Chem. 1968;23:283–248. doi: 10.1016/s0065-3233(08)60402-7..
- 8.(a) Meraldi J-P, Hruby VJ, Brewster AIR. Proc Nat Acad Sci U S A. 1977;74:1373–1377. doi: 10.1073/pnas.74.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blumenstein M, Hruby VJ, Yamamoto DM, Yang Y-CS. FEBS Lett. 1977;81:347–350. doi: 10.1016/0014-5793(77)80551-6. [DOI] [PubMed] [Google Scholar]; (c) Hruby VJ, Mosberg HI. In: Neurohypophyseal Peptide Hormones and Other Biologically Active Peptides. Schlesinger DH, editor. Elsevier; New York: 1981. pp. 227–237. [Google Scholar]; (d) Hruby VJ, Mosberg HI. Horm Antagonists. 1982:433–474. [Google Scholar]
- 9.Hruby VJ, Hadley ME. In: Proceedings of the XVIII Solvay Conference on Chemistry. Van Binst G, editor. 1986. pp. 269–289. [Google Scholar]
- 10.(a) Kramer TH, Wild KD, Toth G, Davis P, Hruby VJ, Burks TF, Porreca F. New Leads in Opioid Research. In: van Ree JM, Mulder AH, Wiegant VM, van Wimersma Greidanus TB, editors. Proceedings of the INRC Conference. Excerpta Medica; Amsterdam: 1990. pp. 222–224. [Google Scholar]; (b) Kramer TH, Davis P, Hruby VJ, Burks TF, Porreca F. J Pharmacol Exp Ther. 1993;266:577–584. [PubMed] [Google Scholar]; (c) Garcia-Sainz JA, Sanchez-Revilla L, Pelton JT, Trivedi DB, Hruby VJ. Biochem Biophys Acta. 1986;886:310–315. doi: 10.1016/0167-4889(86)90150-3. [DOI] [PubMed] [Google Scholar]
- 11.Horan PJ, Mattia A, Bilsky EJ, Weber SJ, Davis TP, Yamamura HI, Malatynska E, Applyard SM, Slaninová J, Misicka A, Lipkowski AW, Hruby VJ, Porreca F. J Pharmacol Exp Ther. 1993;265:1446–1454. [PubMed] [Google Scholar]
- 12.(a) Hruby VJ. In: Persp, Peptide Chem. Eberle A, Geiger R, Wieland T, editors. 1981. pp. 207–220. [Google Scholar]; (b) Hruby VJ. In: Top Mol Pharmacol. Burgen ASV, Roberts GCK, editors. Vol. 1. 1981. pp. 99–126. [Google Scholar]; (c) Hruby VJ, Knittel JJ, Mosberg HI, Rockway TW, Wilkes BC, Hadley ME. In: Peptides 1982. Blaha K, Malon P, editors. Walter de Gruyter; Berlin: 1983. pp. 19–30. [Google Scholar]; (d) Hruby VJ, Mosberg HI, Sawyer TK, Knittel JJ, Rockway TW, Ormberg J, Darman P, Chan WY, Hadley ME. Biopolymers. 1983;22:517–530. doi: 10.1002/bip.360220165. [DOI] [PubMed] [Google Scholar]
- 13.Wood SP, Tickle IJ, Treharne AM, Pitts JE, Mascarenhas Y, Li, Husain JY, Cooper S, Blundell TL, Hruby VJ, Wyssbrod HR, Buku A, Fishman AJ. Science. 1986;232:633–636. doi: 10.1126/science.3008332. [DOI] [PubMed] [Google Scholar]
- 14.Hruby VJ. Trends Pharmacol Sci. 1987;8:336–339. [Google Scholar]
- 15.Chan WY, Hruby VJ, Rockway TW, Hlavacek J. J Pharmacol Exp Therap. 1986;239:84–87. [PubMed] [Google Scholar]
- 16.For an example, see: Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. Proc Natl Acad Sci U S A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754..
- 17.Toniolo C. Int J Pept Protein Res. 1990;35:287–300. doi: 10.1111/j.1399-3011.1990.tb00052.x. [DOI] [PubMed] [Google Scholar]
- 18.(a) Burnett JB, Hadley ME, Heward CB, Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel ME. In: Phenotypic Expression in Pigment Cells. Seiji M, editor. University of Tokyo Press; Tokyo: 1981. pp. 331–338. [Google Scholar]; (b) Hadley ME, Anderson B, Heward CB, Sawyer TK, Hruby VJ. Science. 1981;213:1025–1027. doi: 10.1126/science.6973820. [DOI] [PubMed] [Google Scholar]
- 19.Hruby VJ, Sawyer TK, Yang Y-CS, Bregman MD, Hadley ME, Heward CB. J Med Chem. 1980;23:1432–1437. doi: 10.1021/jm00186a026. [DOI] [PubMed] [Google Scholar]
- 20.(a) Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, de Lauro Castrucci A-M, Hintz MF, Riehm JR, Rao RR. J Med Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]; (b) de Lauro Castrucci A-M, Hadley ME, Sawyer TK, Wilkes BC, Al-Obeidi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, Rao Hruby VJ. Gen Comp Endrocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 21.Haskell-Luevano C, Miwa H, Dickinson C, Hruby VJ, Yamada T, Gantz I. Biochem Biophys Res Commun. 1994;204:1137–1142. doi: 10.1006/bbrc.1994.2581. [DOI] [PubMed] [Google Scholar]
- 22.Sawyer TK, Hruby VJ, Darman PS, Hadley ME. Proc Nat Acad Sci U S A. 1982;79:1751–1755. doi: 10.1073/pnas.79.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Obeidi F, Hadley ME, Pettitt BM, Hruby V. J Am Chem Soc. 1989;111:3413–3416. [Google Scholar]
- 24.Al-Obeidi F, de Lauro Castrucci A-M, Hadley ME, Hruby VJ. J Med Chem. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- 25.(a) Hruby VJ. In: Progress in Brain Research. Chapter 18. Joosse J, Buijs RM, Tilders FJH, editors. Vol. 92. Elsevier Science Publishers; New York: 1992. pp. 215–224. [Google Scholar]; (b) Cowell SM, Balse-Srinivasan PM, Ahn J-M, Hruby VJ. In: Methods in Enzymology. Iyengar R, Hildebrandt JD, editors. Vol. 343. Academic Press; San Diego, CA: 2002. pp. 49–72. [DOI] [PubMed] [Google Scholar]
- 26.(a) Al-Obeidi F, Hruby VJ, Hadley ME, Sawyer TK, de Lauro Castrucci A-M. Int J Peptide Protein Res. 1990;35:228–234. doi: 10.1111/j.1399-3011.1990.tb00942.x. [DOI] [PubMed] [Google Scholar]; (b) Hadley ME, Hruby VJ, Jiang J, Sharma SD, Fink JL, Haskell-Luevano C, Bentley DL, Al-Obeidi F, Sawyer TK. Pigment Cell Res. 1996;9:213–234. doi: 10.1111/j.1600-0749.1996.tb00111.x. [DOI] [PubMed] [Google Scholar]
- 27.Hruby VJ, Lu D, Sharma SD, de L, Castrucci A, Kesterson RA, Al-Obeidi FA, Hadley ME, Cone RD. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 28.Hadley ME. Endocrinology. 4. Prentice Hall; Upper Saddle River, NY: 1996. [Google Scholar]
- 29.Hadley ME, Hruby VJ, Blanchard J, Dorr RT, Levine N, Dawson BV, Al-Obeidi F, Sawyer TK. In: Integration of Pharmaceutical Discovery and Development: Case Studies. Borchardt R, editor. Plenum Press; New York: 1998. pp. 575–595. [DOI] [PubMed] [Google Scholar]
- 30.(a) Sawyer TK, Yang Y-CS, Heward CB, Bregman MD, Fuller BB, Hadley ME, Hruby VJ. In: Peptides: Structure and Biological Function. Gross E, Meienhofer J, editors. Pierce Chemical Co; Rockford, IL: 1979. pp. 1017–1020. [Google Scholar]; (b) Bregman MD, Sawyer TK, Hadley ME, Hruby VJ. Arch Biochem Biophys. 1980;201:1–7. doi: 10.1016/0003-9861(80)90480-4. [DOI] [PubMed] [Google Scholar]; (c) de Lauro Castrucci A-M, Hadley ME, Hruby VJ. Gen Comp Endrocrinol. 1984;55:104–111. doi: 10.1016/0016-6480(84)90134-5. [DOI] [PubMed] [Google Scholar]
- 31.Upton JL, Woods R, Jessen GL, de L, Castrucci A-M, Hadley ME, Wilkes BC, Hruby VJ. Pigment Cell. 1985:139–143. [Google Scholar]
- 32.Marwan MM, Abdel Malek ZA, Kreutzfeld KL, Hadley ME, Wilkes BC, Hruby VJ, de Lauro Castrucci A-M. Mol Cell Endocrinol. 1985;41:171–177. doi: 10.1016/0303-7207(85)90020-6. [DOI] [PubMed] [Google Scholar]
- 33.Abdel Malek ZA, Kreutzfeld KL, Hadley ME, Bregman MD, Hruby VJ, Meyskens FL., Jr In vitro. 1986;22:75–81. doi: 10.1007/BF02623536. [DOI] [PubMed] [Google Scholar]
- 34.(a) Levine N, Lemus-Wilson A, Wood SH, Abdel-Malek ZA, Al-Obeidi F, Hruby VJ, Hadley MEJ. Invest Dermatol. 1987;89:269–273. doi: 10.1111/1523-1747.ep12471337. [DOI] [PubMed] [Google Scholar]; (b) Hadley ME, Wood SH, Jessen GL, Lemus-Wilson AL, Dawson BV, Levine N, Dorr RT, Hruby VJ. Life Sci. 1987;40:1889–1895. doi: 10.1016/0024-3205(87)90047-6. [DOI] [PubMed] [Google Scholar]
- 35.(a) Dorr RT, Lines L, Levine N, Brooks C, Xiang L, Hruby VJ, Hadley ME. Life Sci. 1996;58:1777–1784. doi: 10.1016/0024-3205(96)00160-9. [DOI] [PubMed] [Google Scholar]; (b) Dorr RT, Dvorakova K, Brooks C, Lines R, Levine N, Schram K, Miketova P, Hruby V, Alberts DS. Protochem Photobiol. 2000;72:526–532. doi: 10.1562/0031-8655(2000)072<0526:ieeati>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 36.de Lauro Castrucci A-M, Hadley ME, Lebl M, Zechel C, Hruby VJ. Regul Pept. 1989;24:27–35. doi: 10.1016/0167-0115(89)90208-5. [DOI] [PubMed] [Google Scholar]
- 37.(a) Wessells H, Fuciarelli K, Hansen J, Hadley ME, Hruby VJ, Dorr R, Levine NJ. Urology. 1998;160:389–393. [PubMed] [Google Scholar]; (b) Wessells H, Gralnek D, Dorr R, Hruby VJ, Hadley ME, Levine N. Urology. 2000;56:641–646. doi: 10.1016/s0090-4295(00)00680-4. [DOI] [PubMed] [Google Scholar]; (c) Wessells H, Hruby VJ, Hackett J, Han G, Balse-Srinivasan P, Vanderah TW. Ann N Y Acad Sci. 2003;994:90–95. doi: 10.1111/j.1749-6632.2003.tb03166.x. [DOI] [PubMed] [Google Scholar]
- 38.Wessells H, Hruby VJ, Hackett J, Han G, Balse-Srinivasan P, Vanderah TW. Neuroscience. 2003;118:755–762. doi: 10.1016/s0306-4522(02)00866-7. [DOI] [PubMed] [Google Scholar]
- 39.Wessells H, Levine N, Hadley ME, Dorr R, Hruby VJ. Int J Impotence Res. 2000;12:S74–S79. doi: 10.1038/sj.ijir.3900582. [DOI] [PubMed] [Google Scholar]
- 40.King SH, Mayorov AV, Balse-Srinivasan P, Hruby VJ, Vanderah TW, Wessells H. Curr Top Med Chem. 2007;7:1111–1119. [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma SD, Granberry ME, Jiang J, Leong SDL, Hadley ME, Hruby VJ. Bioconjugate Chem. 1994;5:591–601. doi: 10.1021/bc00030a015. [DOI] [PubMed] [Google Scholar]
- 42.Sharma SD, Jiang J, Hadley ME, Bentley DL, Hruby VJ. Proc Natl Acad Sci U S A. 1996;93:13715–13720. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Jiang J, Sharma SD, Hruby VJ, Fink JL, Hadley ME. Exp Dermatol. 1997;6:6–12. doi: 10.1111/j.1600-0625.1997.tb00139.x. [DOI] [PubMed] [Google Scholar]; (b) Jiang J, Sharma SD, Fink JL, Hadley ME, Hruby VJ. Exp Dermatol. 1996;5:325–333. doi: 10.1111/j.1600-0625.1996.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 44.(a) Jiang J, Sharma SD, Hruby VJ, Bentley DL, Fink JL, Hadley ME. Pigment Cell Res. 1996;9:240–247. doi: 10.1111/j.1600-0749.1996.tb00113.x. [DOI] [PubMed] [Google Scholar]; (b) Hadley ME, Hruby VJ, Jiang J, Sharma SD, Fink JL, Haskell-Luevano C, Bentley DL, Al-Obeidi F, Sawyer TK. Pigment Cell Res. 1996;9:213–234. doi: 10.1111/j.1600-0749.1996.tb00111.x. [DOI] [PubMed] [Google Scholar]
- 45.Cai M, Stankova M, Pond SJK, Mayorov AV, Perry JW, Yamamura HI, Trivedi D, Hruby VJ. J Am Chem Soc. 2004;126:7160–7161. doi: 10.1021/ja049473m. [DOI] [PubMed] [Google Scholar]; (b) Cai M, Varga EV, Stankova M, Mayorov A, Perry JW, Yamamura HI, Trivedi D, Hruby VJ. Chem Biol Drug Design. 2006;68:183–193. doi: 10.1111/j.1747-0285.2006.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.(a) Mamuren M, Chor SK, Whitesides GM. Angew Chem, Int Ed. 1998;37:2754. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (b) Gillies RJ, Hruby VJ. Expert Opin Ther Targets. 2003;7:137–139. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]; (c) Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Expt Opin Ther Targets. 2004;8:565–586. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 47.For extensive reviews see: Cone RD, editor. The Melanocortin Receptors. Humana Press; Totowa, NJ: 2000. p. 551..
- 48.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 49.(a) Zimanyr AIA, Pelleymounter MA. Curr Pharm Design. 2003;9:627–641. doi: 10.2174/1381612033391234. [DOI] [PubMed] [Google Scholar]; (b) Emerson PJ, Fisher MJ, Yan LZ, Mayer JP. Curr Top Med Chem. 2007:1121–1130. doi: 10.2174/156802607780906636. [DOI] [PubMed] [Google Scholar]
- 50.Huang Q-H, Entwistle ML, Alvaro JD, Duman RS, Hruby VJ, Tatro JB. J Neurosci. 1997;17:3343–3351. doi: 10.1523/JNEUROSCI.17-09-03343.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kunos G, Li S-J, Varga K, Archer P, Kesterson RA, Cone RD, Hruby VJ, Sharma SD. Fundam Clin Pharmacol. 1997;11(Suppl. 1):44s–48s. [Google Scholar]
- 52.Ni X-P, Kesterson RA, Sharma SD, Hruby VJ, Cone RD, Wiedemann E, Humphreys MH. Am J Physiol. 1998;274(43):R931–R938. doi: 10.1152/ajpregu.1998.274.4.R931. [DOI] [PubMed] [Google Scholar]
- 53.Huang Q-H, Hruby VJ, Tatro JB. Am J Physiol (Reg Integrat Compar Physiol) 1998;275:R524–R530. doi: 10.1152/ajpregu.1998.275.2.R524. [DOI] [PubMed] [Google Scholar]
- 54.Huang Q-H, Hruby VJ, Tatro JB. Am J Physiol. 1999;276:R864–R871. doi: 10.1152/ajpregu.1999.276.3.R864. [DOI] [PubMed] [Google Scholar]
- 55.Hruby VJ, Cai M, Grieco P, Han G, Kavarana M, Trivedi D. Ann N Y Acad Sci. 2003;994:12–20. doi: 10.1111/j.1749-6632.2003.tb03157.x. [DOI] [PubMed] [Google Scholar]
- 56.Mogil JS, Wilson SG, Chesler EJ, Rankin AL, Nemmani KVS, Lariviere WR, Groce MK, Wallace MR, Kaplan L, Staud R, Ness TJ, Glover TL, Stankova M, Mayorov A, Hruby VJ, Grisel JE, Fillingim RB. Proc Natl Acad Sci U S A. 2003;100:4867–4872. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.(a) Marks DL, Hruby V, Brookhart G, Cone RD. Peptides. 2006;27:259–264. doi: 10.1016/j.peptides.2005.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee M, Kim A, Conwell IM, Hruby V, Mayorov A, Cai M, Wardlaw SL. Peptides. 2008;29:440–447. doi: 10.1016/j.peptides.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lord JAH, Waterfield AA, Hughes J, Kosterlitz HW. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- 59.(a) Mosberg HI, Hurst R, Hruby VJ, Galligan JJ, Burks TF, Gee K, Yamamura HI. Biochem Biophys Res Commun. 1982;106:506–512. doi: 10.1016/0006-291x(82)91139-1. [DOI] [PubMed] [Google Scholar]; (b) Mosberg HI, Hurst R, Hruby VJ, Galligan JJ, Burks TF, Gee K, Yamamura HI. Life Sci. 1983;32:2565–2569. doi: 10.1016/0024-3205(83)90239-4. [DOI] [PubMed] [Google Scholar]
- 60.(a) Mosberg HI, Hurst R, Hruby VJ, Gee K, Yamamura HI, Galligan JJ, Burks TF. Proc Natl Acad Sci U S A. 1983;80:5871–5874. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Akiyama K, Gee KW, Mosberg HI, Hruby VJ, Yamamura HI. Proc Natl Acad Sci U S A. 1985;82:2543–2547. doi: 10.1073/pnas.82.8.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hruby VJ. Life Sci. 1982;31:189–199. doi: 10.1016/0024-3205(82)90578-1. [DOI] [PubMed] [Google Scholar]
- 62.Kessler H. Angew Chem, Int Ed. 1982;21:512–523. [Google Scholar]
- 63.Hruby VJ. Nat Rev Drug Discovery. 2002;1:847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 64.Galligan JJ, Mosberg HI, Hurst R, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1984;229:641–648. [PubMed] [Google Scholar]
- 65.Porreca F, Mosberg HI, Hurst R, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1984;230:341–348. [PubMed] [Google Scholar]
- 66.(a) Gulya K, Gehlert DR, Wamsley JK, Mosberg HI, Hruby VJ, Duckles SP, Yamamura HI. Eur J Pharmacol. 1985;111:285–286. doi: 10.1016/0014-2999(85)90770-8. [DOI] [PubMed] [Google Scholar]; (b) Gulya K, Gehlert D, Wamsley JK, Mosberg HI, Hruby VJ, Duckles SP, Yamamura HI. J Pharm Exp Ther. 1986;238:720–726. [PubMed] [Google Scholar]
- 67.Spencer RL, Deupress D, Hsiao S, Mosberg HI, Hruby VJ, Burks TF, Porreca F. Pharmacol Biochem Behav. 1986;25:77–82. doi: 10.1016/0091-3057(86)90233-9. [DOI] [PubMed] [Google Scholar]
- 68.Spencer RL, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1988;246:92–102. [PubMed] [Google Scholar]
- 69.Shook JE, Lemcke PK, Gehrig CA, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1989;249:83–90. [PubMed] [Google Scholar]
- 70.(a) Lipkowski AW, Konecka AM, Sroczynska I. Peptides. 1982;3:697–700. doi: 10.1016/0196-9781(82)90173-5. [DOI] [PubMed] [Google Scholar]; (b) Lipokowski AW, Konecka AM, Sroczynska I, Przeiwlocki R, Staha L, Tam SW. Life Sci. 1987;40:2283–2288. doi: 10.1016/0024-3205(87)90065-8. [DOI] [PubMed] [Google Scholar]
- 71.Horan PJ, Mattia A, Bilsky EJ, Weber SJ, Davis TP, Yamamura HI, Malatynska E, Applyard SM, Slaninová J, Misicka A, Lipkowski AW, Hruby VJ, Porreca F. J Pharmacol Exp Ther. 1993;265:1446–1454. [PubMed] [Google Scholar]
- 72.Rezek M, Havlicek V, Leybin L, LaBello FS, Frisien H. Can J Pharmacol. 1978;56:227–231. doi: 10.1139/y78-033. [DOI] [PubMed] [Google Scholar]
- 73.(a) Veber DF, Strachen RG, Birgstrand SJ, Holly FW, Homnick CF, Hirshmann R, Torchiana M, Saperstein R. J Am Chem Soc. 1976;98:2307–2369. [Google Scholar]; (b) Veber DF, Holly FW, Paleveda WJ, Nutt RF, Bergstrand SJ, Torchiana M, Glitzen MS, Saperstein R, Hirschmann R. Proc Natl Acad Sci U S A. 1979;75:2636–2640. doi: 10.1073/pnas.75.6.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.(a) Pelton JT, Gulya K, Hruby VJ, Duckles S, Yamamura HI. Peptides. 1985;6:159–163. doi: 10.1016/0196-9781(85)90026-9. [DOI] [PubMed] [Google Scholar]; (b) Pelton JT, Gulya K, Hruby VJ, Duckles S, Yamamura HI. Proc Natl Acad Sci U S A. 1985;82:236–239. doi: 10.1073/pnas.82.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kazmierski WM, Wire WS, Lui GK, Knapp RJ, Shook JE, Burks TF, Yamamura HI, Hruby VJ. J Med Chem. 1988;31:2170–2177. doi: 10.1021/jm00119a019. [DOI] [PubMed] [Google Scholar]; (b) Kazmierski WM, Hruby VJ. Tetrahedron. 1988;44:697–710. [Google Scholar]
- 76.Kazmierski WM, Yamamura HI, Hruby VJ. J Am Chem Soc. 1991;113:2275–2283. [Google Scholar]
- 77.(a) Gulya K, Pelton JT, Hruby VJ, Yamamura HI. Life Sci. 1986;38:2221–2230. doi: 10.1016/0024-3205(86)90574-6. [DOI] [PubMed] [Google Scholar]; (b) Shook JE, Pelton JT, Wire WS, Hirning LD, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1987;240:772–777. [PubMed] [Google Scholar]
- 78.(a) Gulya K, Lui GK, Pelton JT, Kazmierski WM, Hruby VJ, Yamamura HI. In: Progress in Opioid Research: Proc 1986 I N R C. Holaday JW, Law PY, Herz A, editors. NIDA Research Monograph 75; NIDA; Rockville, MD: 1987. pp. 209–212. [PubMed] [Google Scholar]; (b) Shook JE, Pelton JT, Lemcke PF, Porreca F, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1987;242:1–7. [PubMed] [Google Scholar]
- 79.(a) Shook JE, Pelton JT, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1987;243:492–500. [PubMed] [Google Scholar]; (b) Shook JE, Kazmierski WM, Pelton JT, Hruby VJ, Burks TF. The 49th Annual Committee on Problems in Drug Dependence. NIDA Research Monograph 81; NIDA; Rockville, MD: 1988. pp. 143–148. [Google Scholar]; (c) Mulder AH, Wardeh G, Kazmierski WM, Hruby VJ. Eur J Pharmacol. 1988;157:109–114. doi: 10.1016/0014-2999(88)90477-3. [DOI] [PubMed] [Google Scholar]; (d) Mulder AH, Wardeh G, Hogenboom F, Kazmierski WM, Hruby VJ, Schoffelmeer ANM. Eur J Pharmacol. 1991;205:1–6. doi: 10.1016/0014-2999(91)90761-e. [DOI] [PubMed] [Google Scholar]
- 80.Ayres EA, Villar R, Kramer TH, Kazmierski WM, Hruby VJ, Burks TF. Proc West Pharm Soc. 1988;31:41–43. [PubMed] [Google Scholar]
- 81.Shook JE, Lemcke PK, Gehrig CA, Hruby VJ, Burks TF. J Pharmacol Exp Ther. 1989;249:83–90. [PubMed] [Google Scholar]
- 82.Hawkins KN, Knapp RJ, Lui GK, Gulya K, Kazmierski WM, Wan Y-P, Pelton JT, Hruby VJ, Yamamura HI. J Pharmacol Exp Ther. 1989;248:73–81. [PubMed] [Google Scholar]
- 83.(a) Pelton JT, Cody WL, Hruby VJ, Gulya K, Hirning LD, Yamamura HI, Burks TF. In: Peptides: Structure and Function, Proceedings of the Ninth American Peptide Symposium. Deber CM, Hruby VJ, Kopple KD, editors. Pierce Chemical Co; Rockford, IL: 1985. pp. 623–626. [Google Scholar]; (b) Wynants C, Sugg EE, Hruby VJ, Van Binst G. Int J Peptide Protein Res. 1987;30:541–547. doi: 10.1111/j.1399-3011.1987.tb03363.x. [DOI] [PubMed] [Google Scholar]; (c) Pelton JT, Whalon M, Cody WL, Hruby VJ. Int J Peptide Protein Res. 1988;31:109–115. doi: 10.1111/j.1399-3011.1988.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 84.(a) Glasel JA, McKelvy JF, Hruby VJ, Spatola AF. Ann N Y Acad Sci. 1973;222:778–788. doi: 10.1111/j.1749-6632.1973.tb15304.x. [DOI] [PubMed] [Google Scholar]; (b) Glasel JA, Hruby VJ, McKelvy JF, Spatola AF. J Biol Chem. 1976;251:2929–2937. [PubMed] [Google Scholar]
- 85.(a) Blumenstein M, Hruby VJ. Biochem Biophys Res Commun. 1976;68:1052–1058. doi: 10.1016/0006-291x(76)90302-8. [DOI] [PubMed] [Google Scholar]; (b) Blumenstein M, Hruby VJ, Yamamoto DM, Yang Y-CS. FEBS Lett. 1977;81:347–350. doi: 10.1016/0014-5793(77)80551-6. [DOI] [PubMed] [Google Scholar]; (c) Blumenstein M, Hruby VJ. Biochemistry. 1977;16:5169–5177. doi: 10.1021/bi00643a002. [DOI] [PubMed] [Google Scholar]; (d) Blumenstein M, Hruby VJ, Yamamoto DM. Biochemistry. 1978;17:4971–4977. doi: 10.1021/bi00616a018. [DOI] [PubMed] [Google Scholar]; (e) Blumenstein M, Hruby VJ, Viswanatha V. Biochemistry. 1979;18:3552–3556. doi: 10.1021/bi00583a018. [DOI] [PubMed] [Google Scholar]; (f) Blumenstein M, Hruby VJ, Viswanatha V. Biochem Biophys Res Commun. 1980;94:431–437. doi: 10.1016/0006-291x(80)91249-8. [DOI] [PubMed] [Google Scholar]; (g) Blumenstein M, Hruby VJ, Viswanatha V. In: Biomolecular Stereodynamics. Sarma RH, editor. III. Academic Press; New York: 1981. pp. 353–368. [Google Scholar]; (h) Blumenstein M, Hruby VJ, Viswanatha V, Chaturvedi D. Biochemistry. 1984;23:2153–2161. doi: 10.1021/bi00305a008. [DOI] [PubMed] [Google Scholar]
- 86.Liao S, Hruby VJ. In: Methods in Molecular Medicine, Vol 23: Peptidomimetic Protocols. Kazmierski WM, editor. Humana Press Inc.; Totowa, NJ: 1999. pp. 175–187. [DOI] [PubMed] [Google Scholar]
- 87.(a) Hruby VJ, Li G, Haskell-Luevano C, Shenderovich MD. Biopolymers (Peptide Science) 1997;43:219–266. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; (b) Hruby VJ. Biopolymers. 1993;33:1073–1082. doi: 10.1002/bip.360330709. [DOI] [PubMed] [Google Scholar]
- 88.(a) Hruby VJ, Balse PM. Curr Med Chem. 2000;7:945–970. doi: 10.2174/0929867003374499. [DOI] [PubMed] [Google Scholar]; (b) Hruby VJ, Han G, Gitu PM. Synthesis of Peptides and Peptidomimetics. In: Goodman M, Felix A, Moroder L, Toniolo C, editors. Methods of Organic Chemistry (Houben-Weyl) E22c. Thieme Verlag; Stuttgart: 2002. pp. 5–51. [Google Scholar]
- 89.(a) Dharanipragada R, Nicolás E, Tóth G, Hruby VJ. Tetrahedron Lett. 1989;30(49):6841–6844. [Google Scholar]; (c) Nicolás E, Dharanipragada R, Tóth G, Hruby VJ. Tetrahedron Lett. 1989;30(49):6845–6848. [Google Scholar]; (c) Nicolás E, Russell KC, Hruby VJ. J Org Chem. 1993;58:766–770. [Google Scholar]; (d) Nicolas E, Russell KC, Knollenberg J, Hruby VJ. J Org Chem. 1993;58:7565–7571. [Google Scholar]; (e) Li G, Patel D, Hruby VJ. Tetrahedron Lett. 1994;35:2301–2304. [Google Scholar]; (f) Han Y, Liao S, Qiu W, Cai C, Hruby VJ. Tetrahedron Lett. 1997;38:5135–5138. [Google Scholar]
- 90.(a) Li G, Russell KC, Jarosinski MA, Hruby VJ. Tetrahedron Lett. 1993;34:2565–2568. [Google Scholar]; (b) Jiao D, Russell KC, Hruby VJ. Tetrahedron. 1993;49:3511–3520. [Google Scholar]; (c) Li G, Boteju LW, Patel D, Hruby VJ. Peptides: Chemistry Structure and Biology. In: Hodges RS, Smith JA, editors. Proc 13th Amer Pept Symp. ESCOM Publishers; Leiden: 1994. pp. 181–183. [Google Scholar]; (d) Qian X, Russell KC, Boteju LW, Hruby VJ. Tetrahedron. 1995;51:1033–1054. [Google Scholar]; (e) Haskell-Luevano C, Boteju LW, Hruby VJ. Letters Pept Sci. 1994;1:163–170. [Google Scholar]
- 91.(a) Valle G, Kazmierski WM, Crisma M, Bonora GM, Toniolo C, Hruby VJ. Int J Peptide Protein Res. 1992;40:222–232. [PubMed] [Google Scholar]; (b) Kazmierski WM, Hruby VJ. Tetrahedron Lett. 1991;32:5769–5772. [Google Scholar]; (c) Kazmierski WM, Urbanczyk-Lipkowski Z, Hruby VJ. J Org Chem. 1994;59:1789–1795. [Google Scholar]
- 92.(a) Liao S, Han Y, Qui W, Bruck M, Hruby VJ. Tetrahedron Lett. 1996;37:7917–7920. [Google Scholar]; (b) Lin J, Liao S, Han Y, Qiu W, Hruby VJ. Tetrahedron: Asymmetry. 1998;8:3213–3221. [Google Scholar]; (c) Liao S, Shenderovich MD, Lin J, Hruby VJ. Tetrahedron. 1997;53:16645–16662. [Google Scholar]; (d) Lin J, Liao S, Hruby VJ. Tetrahedron Lett. 1998;39:3117–3120. [Google Scholar]; (e) Wang W, Zhang J, Xiong C, Hruby VJ. Tetrahedron Lett. 2002;43:2137–2140. [Google Scholar]; (f) Wang W, Xiong C, Zhang J, Hruby VJ. Tetrahedron. 2002;58:3101–3110. [Google Scholar]; (g) Wang W, Cai M, Xiong C, Zhang J, Trivedi D, Hruby VJ. Tetrahedron. 2002;58:7365–7374. [Google Scholar]
- 93.(a) Soloshonok VA, Cai C, Hruby VJ, Van Meervelt L, Mischenko N. Tetrahedron. 1999;55:12031–12044. [Google Scholar]; (b) Soloshonok VA, Cai C, Hruby VJ, Van Meervelt L. Tetrahedron. 1999;55:12045–12058. [Google Scholar]; (c) Soloshonok VA, Cai C, Hruby VJ. Tetrahedron Lett. 2000;41:135–139. [Google Scholar]; (d) Soloshonok VA, Cai C, Hruby VJ. Org Lett. 2000;2:747–750. doi: 10.1021/ol990402f. [DOI] [PubMed] [Google Scholar]; (e) Tamaki M, Han G, Hruby VJ. J Org Chem. 2001;66:1038–1042. doi: 10.1021/jo005626m. [DOI] [PubMed] [Google Scholar]
- 94.(a) Soloshonok VA, Cai C, Hruby VJ. Tetrahedron: Asymmetry. 1999;10:4265–4269. [Google Scholar]; (b) Soloshonok VA, Cai C, Hruby VJ. Angew Chem, Int Ed. 2000;39:2172–2175. [PubMed] [Google Scholar]; (c) Soloshonok VA, Cai C, Hruby VJ, Van Meervelt L, Yamazaki T. J Org Chem. 2000;65:6688–6696. doi: 10.1021/jo0008791. [DOI] [PubMed] [Google Scholar]; (d) Cai C, Soloshonok VA, Hruby VJ. J Org Chem. 2001;66:1339–1350. doi: 10.1021/jo0014865. [DOI] [PubMed] [Google Scholar]
- 95.(a) Qiu W, Soloshonok VA, Cai C, Tang X, Hruby VJ. Tetrahedron. 2000;56:2577–2582. [Google Scholar]; (b) Tang X, Soloshonok VA, Hruby VJ. Tetrahedron: Asymmetry. 2000;11:2917–2925. [Google Scholar]; (c) Soloshonok VA, Tang X, Hruby VJ, Van Meervelt L. Org Lett. 2001;3:341–343. doi: 10.1021/ol000330o. [DOI] [PubMed] [Google Scholar]; (d) Soloshonok VA, Ueki H, Tiwari R, Cai C, Hruby VJ. J Org Chem. 2004;69:4984–4990. doi: 10.1021/jo0495438. [DOI] [PubMed] [Google Scholar]
- 96.(a) Gu X, Ndungu JM, Qiu W, Ying J, Carducci MD, Wooden H, Hruby VJ. Tetrahedron. 2004;60:8233–8243. [Google Scholar]; (b) Qu H, Gu X, Min BJ, Liu Z, Hruby VJ. Org Lett. 2006;8(19):4215–4218. doi: 10.1021/ol061414l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qu H, Gu X, Liu Z, Min BJ, Hruby VJ. Org Lett. 2007;9:3997–4000. doi: 10.1021/ol701704h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu Z, Qu H, Gu X, Min BJ, Nyberg J, Hruby VJ. Org Lett. 2008;10:4105–4108. doi: 10.1021/ol801657q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.(a) Boteju LW, Wegner K, Qian X, Hruby VJ. Tetrahedron. 1994;50:2391–2404. [Google Scholar]; (b) Han G, Lewis A, Hruby VJ. Tetrahedron Lett. 2001;42:4601–4603. [Google Scholar]; (c) Wang W, Xiong C, Yang J, Hruby VJ. Tetrahedron Lett. 2001;42:7717–7719. [Google Scholar]; (d) Xiong C, Wang W, Cai C, Hruby VJ. J Org Chem. 2002;67:1399–1402. doi: 10.1021/jo010860d. [DOI] [PubMed] [Google Scholar]
- 98.(a) Li G, Boteju LW, Patel D, Hruby VJ. Peptides: Chemistry Structure and Biology. In: Hodges RS, Smith JA, editors. Proc 13th Amer Peptide Symp. ESCOM Publishers; Leiden: 1994. pp. 181–183. [Google Scholar]; (b) Wang S, Tang X-J, Hruby VJ. Tetrahedron Lett. 2000;41:1307–1310. [Google Scholar]; (c) Li G, Patel D, Hruby VJ. Tetrahedron Asymmetry. 1993;4:2315–2318. [Google Scholar]
- 99.Hruby VJ, Kao L-F, Pettitt BM, Karplus M. J Am Chem Soc. 1988;110:3351–3359. [Google Scholar]
- 100.(a) Flippen-Anderson JL, Hruby VJ, Collins N, George C, Cudney B. J Am Chem Soc. 1994;116:7523–7531. [Google Scholar]; (b) Collins N, Flippen-Anderson JL, Haaseth RC, Deschamps JR, George C, Kövér K, Hruby VJ. J Am Chem Soc. 1996;118:2143–2152. [Google Scholar]
- 101.Hruby VJ, Pettitt BM. In: Computer Aided Drug Design. Perun TJ, Propst C, editors. Marcel Dekker, Inc.; New York: 1989. pp. 405–460. [Google Scholar]
- 102.(a) Qian X, Shenderovich MD, Kövér KE, Davis P, Horváth R, Zalewska T, Yamamura HI, Porreca F, Hruby VJ. J Am Chem Soc. 1996;118:7280–7290. [Google Scholar]; (b) Bilsky EJ, Qian X, Hruby VJ, Porreca Frank. J Pharmacol Exp Therap. 2000;293:151–158. [PubMed] [Google Scholar]
- 103.(a) Liao S, Shenderovich MD, Alfaro-Lopez J, Stropova D, Hosohata K, Davis P, Jernigan KA, Porreca F, Yamamura HI, Hruby VJ. Peptides: Frontiers of Peptide Science. In: Tam JP, Kaumaya PTP, editors. Proc In 15th Am Peptide Symp. ishers; Dordrecht, The Netherlands: 1999. pp. 214–215. [Google Scholar]; (b) Liao S, Alfaro-Lopez J, Shenderovich MD, Hosohata K, Lin J, Li X, Stropova D, Davis P, Jernigan KA, Porreca F, Yamamura HI, Hruby VJ. J Med Chem. 1998;41:4767–4776. doi: 10.1021/jm980374r. [DOI] [PubMed] [Google Scholar]; (c) Alfaro-Lopez J, Okayama T, Hosohata K, Davis P, Porreca F, Yamamura HI, Hruby VJ. J Med Chem. 1999;42:5359–5368. doi: 10.1021/jm990337f. [DOI] [PubMed] [Google Scholar]; (d) Shenderovich MD, Liao S, Qian X, Hruby VJ. Biopolymers. 2000;53:565–580. doi: 10.1002/(SICI)1097-0282(200006)53:7<565::AID-BIP4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 104.Schiller PW, Nguyen TM-D, Weltrowska G, Wilkes BC, Marsden BJ, Lemieux C, Chung NN. Proc Natl Acad Sci U S A. 1992;89:11871–11875. doi: 10.1073/pnas.89.24.11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liao S, Lin J, Shenderovich MD, Han Y, Hosohata K, Davis P, Qiu W, Porreca F, Yamamura HI, Hruby VJ. Bioorg Med Chem Lett. 1997;7:3049–3052. [Google Scholar]
- 106.(a) Hosohata K, Burkey TH, Alfaro-Lopez J, Hruby VJ, Roeske WR, Yamamura HI. Eur J Pharmacol. 1999;380:R9–R10. doi: 10.1016/s0014-2999(99)00516-6. [DOI] [PubMed] [Google Scholar]; (b) Hosohata K, Varga EV, Alfaro-Lopez J, Tang X, Vanderah TW, Porreca F, Hruby VJ, Roeske WR, Yamamura HI. J Pharmacol Exp Therap. 2003;304:683–688. doi: 10.1124/jpet.102.042929. [DOI] [PubMed] [Google Scholar]
- 107.Liao S, Shenderovich MD, Zhang Z, Maletinska L, Slaninova J, Hruby VJ. J Am Chem Soc. 1998;120:7393–7394. [Google Scholar]
- 108.Haskell-Luevano C, Boteju LW, Miwa H, Dickinson C, Gantz I, Yamada T, Hadley ME, Hruby VJ. J Med Chem. 1995;38:4720–4729. doi: 10.1021/jm00023a012. [DOI] [PubMed] [Google Scholar]
- 109.Haskell-Luevano C, Toth K, Boteju L, Job C, de L, Castrucci AM, Hadley ME, Hruby VJ. J Med Chem. 1997;40:2740–2749. doi: 10.1021/jm970018t. [DOI] [PubMed] [Google Scholar]
- 110.(a) Lin MC, Wright DE, Hruby VJ, Rodbell M. Biochemistry. 1975;14:1559–1563. doi: 10.1021/bi00679a002. [DOI] [PubMed] [Google Scholar]; (b) Hruby VJ, Wright DE, Lin MC, Rodbell M. Metabolism. 1976;25(Suppl. 1):1323–1325. doi: 10.1016/s0026-0495(76)80133-3. [DOI] [PubMed] [Google Scholar]; (c) Wright DE, Hruby VJ, Rodbell M. J Biol Chem. 1978;253:6338–6340. [PubMed] [Google Scholar]
- 111.(a) Bregman MD, Trivedi DB, Hruby VJ. J Biol Chem. 1980;255:11725–11733. [PubMed] [Google Scholar]; (b) Sawyer TK, Darman PS, Hruby VJ, Hadley ME. In: Peptides: Synthesis, Structure, Function. Rich DH, Gross E, editors. Pierce Chemical Co; Rockford, IL: 1981. pp. 387–390. [Google Scholar]
- 112.(a) Khan BA, Bregman MD, Nugent CA, Hruby VJ, Brendel K. Biochem Biophys Res Commun. 1980;93:729–736. doi: 10.1016/0006-291x(80)91138-9. [DOI] [PubMed] [Google Scholar]; (b) Hruby VJ, Mosberg HI. In: Peptides: Synthesis, Structure, Function. Rich DH, Gross E, editors. Pierce Chemical Co; Rockford, IL: 1981. pp. 375–378. [Google Scholar]
- 113.Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi DB. Science. 1982;215:1115–1116. doi: 10.1126/science.6278587. [DOI] [PubMed] [Google Scholar]
- 114.Wakelam MJ-O, Murphy GJ, Hruby VJ, Houslay MD. Nature. 1986;323:68–71. doi: 10.1038/323068a0. [DOI] [PubMed] [Google Scholar]
- 115.Gillies RJ, Hruby VJ. Expert Opin Ther Targets. 2003;7:137–139. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]
- 116.Hruby VJ, Porreca F, Yamamura HI, Tollin G, Agnes RS, Lee YS, Cai M, Alves I, Cowell S, Varga E, Davis P, Salamon Z, Roeske W, Vanderah T, Lai J. Am Assoc Pharm Sci J. 2006;8(3):E450–E460. doi: 10.1208/aapsj080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yamamoto T, Nair P, Davis P, Ma S-W, Navratilova E, Moye S, Tumati S, Lai J, Vanderah TW, Yamamura HI, Porreca F, Hruby VJ. J Med Chem. 2007;50:2779–2786. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamamoto T, Nair P, Jacobsen NE, Davis P, Ma S-W, Navratilova E, Moye S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. J Med Chem. 2008;51:6334–6347. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.(a) Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]; (b) Lebl M, Patek M, Kocis P, Krchnak V, Hruby VJ, Salmon S, Lam KS. Int J Peptide Protein Res. 1993;41:201–203. doi: 10.1111/j.1399-3011.1993.tb00132.x. [DOI] [PubMed] [Google Scholar]; (c) Hruby VJ. In: The Practice of Medicinal Chemistry. Chapter 9. Wermuth CG, editor. Academic Press; London: 1996. pp. 135–151. [Google Scholar]
- 120.(a) Vagner J, Xu L, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Angew Chem, Int Ed. 2008;47:1685–1688. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chem. 2006;17:1545–1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Josan JS, Vagner J, Handl H, Sankaranarayanan R, Gillies RJ, Hruby VJ. Int J Pept Res Ther. 2008;14(4):293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ying J, Köver KE, Gu X, Han G, Trivedi DB, Kavarana MJ, Hruby VJ. Biopolymers (Peptide Science) 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]