

Abstract
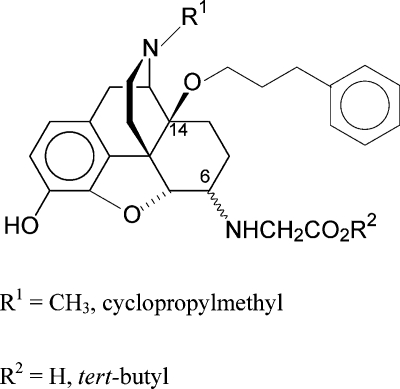
The synthesis and the effect of a combination of 6-glycine and 14-phenylpropoxy substitutions in N-methyl- and N-cycloproplymethylmorphinans on biological activities are described. Binding studies revealed that all new 14-phenylpropoxymorphinans (11−18) displayed high affinity to opioid receptors. Replacement of the 14-methoxy group with a phenylpropoxy group led to an enhancement in affinity to all three opioid receptor types, with most pronounced increases in δ and κ activities, hence resulting in a loss of μ receptor selectivity. All compounds (11−18) showed potent and long-lasting antinociceptive effects in the tail-flick test in rats after subcutaneous administration. For the N-methyl derivatives 13 and 14, analgesic potencies were in the range of their 14-methoxy analogues 9 and 10, respectively. Even derivatives 15−18 with an N-cyclopropylmethyl substituent acted as potent antinociceptive agents, being several fold more potent than morphine. Subcutaneous administration of compounds 13 and 14 produced significant and prolonged antinociceptive effects mediated through peripheral opioid mechanisms in carrageenan-induced inflammatory hyperalgesia in rats.
Introduction
The clinical management of pain, especially severe and chronic pain, is still a major challenge.1,2 Analgesic drugs such as opioids play a central role in pain control.2,3 Together with endogenous opioids, they modulate nociceptive transmission at different levels in the pain modulating pathways via interaction with opioid receptors.4−6 The efficacy of currently used opioid analgesics, including morphine, oxymorphone, oxycodone, and fentanyl, is frequently associated with the occurrence of undesired dose-limiting side effects.2,3 There is a continued search for opioids that are highly efficacious with reduced complications and improved patient compliance.
Work in our laboratory has been focused on the development of new opioid agonists and antagonists from the morphinan class of compounds.7−9 Introduction of a 14-methoxy group in oxymorphone resulted in 14-O-methyloxymorphone(10) (1; Figure 1), which not only increases affinity to opioid receptors while retaining the μ receptor selectivity but also markedly enhances the antinociceptive potency.10−12 Further work on 14-alkoxymorphinans led to the development of 14-methoxymetopon(13) (2; Figure 1). It was reported as a selective and high efficacy μ opioid receptor agonist showing potent centrally mediated antinociceptive effects and less pronounced typical opioid adverse actions.12−18 A derivative of the 14-alkoxymorphinan series of opioids, 14-phenylpropoxymetopon(19) (3; Figure 1), has been described as an extremely powerful analgesic with high affinity for all three opioid receptor types (μ, δ, and κ).(19) Similar results were provided on differently N-substituted 14-phenylpropoxymorphinan-6-ones (e.g., N-allyl and N-cyclopropylmethyl substituted morphinans 5 and 6).(20) It was established that the presence of a 14-phenylpropoxy group increases both the agonist potency and the affinity for all three opioid receptor types while concurrently diminishing the selectivity for any of the receptors. The two classical opioid antagonists naloxone (7) and naltrexone (8) were converted into highly active analgesic agents by introducing a phenylpropoxy group in position 14 (compounds 5 and 6, respectively).(20) Moreover, derivatives of the selective μ opioid receptor antagonist cyprodime having a phenylpropoxy group at C-14 have also acted as potent antinociceptives in different pain models in mice after subcutaneous (sc) administration.(21) Thus, the presence of this substituent directs to a profound alteration in the pharmacological profile of morphinan-6-ones.
Figure 1.

Structures of ligands related to 14-O-methyloxymorphone (1), naloxone (7), and naltrexone (8). CPM, cycloproplymethyl; Ph, phenyl.
Traditionally, analgesic effects of opioids have been associated with exclusive activation of opioid receptors in the central nervous system (CNSa). There is large evidence that intrinsic pain control can also occur at peripheral sites,6,22−24 which is supported by the identification of peripheral opioid receptors on sensory neurons.25,26 The contribution of the opioid system to peripheral pain control mechanisms gained considerable attention during the past years, leading to new directions in research focusing on exploration of the therapeutic potential of peripheral opioid receptors for superior management of pain.6,23,24,27,28 Targeting peripheral mechanisms can provide selective peripheral analgesia by avoiding the central complications associated with the use of opioids.6,23,27
Our research in the field of peripherally acting opioid antinociceptive agents has led us to obtain a series of 6-amino acid substituted derivatives (glycine, alanine, and phenylalanine) of 14-O-methyloxymorphone (1).(29) These compounds displayed high affinities at the μ opioid receptor and showed potent agonism.(30) A number of pharmacological studies reported that amino acid substitution in position 6 of 14-O-methyloxymorphone affords derivatives that produce potent antinociceptive actions in rodent models of acute nociception, inflammatory, visceral, and neuropathic pain.18,31−33 These antinociceptive effects were shown to be mediated by activating primarily peripheral opioid receptors. They showed markedly long-lasting antinociceptive actions compared to the conventional centrally acting μ opioids, fentanyl, morphine, the parent compound 1, and 14-methoxymetopon (2).8,31−33 The most potent compounds were the 6-glycine, 14-methoxy substituted derivatives 9 and 10, respectively (Figure 1). The 6β-glycine analogue 10 exhibited an antinociceptive potency comparable to fentanyl and was only 2-fold lower than that of 14-O-methyloxymorphone (1) after sc administration.(31)
In the present study, we have investigated the effect of a combination of 6-amino acid and 14-phenylpropoxy substitutions in N-methyl- and N-cycloproplymethylmorphinans on the biological profile represented by in vitro binding and antinociceptive activities after sc administration to rats. Structure−activity relationship (SAR) studies relating to the substitution pattern in positions 6 and 14 within this series were pursued. To this aim, 6-glycine derivatives having a 14-phenylpropoxy group (compounds 13, 14, 17, and 18) were synthesized and their in vitro and in vivo opioid activities were evaluated. We have also assessed the peripheral mechanisms of antinociceptive effects of derivative 13 and 14 after sc administration to rats with carrageenan-induced inflammatory pain. In addition, the pharmacological properties of the corresponding tert-butyl esters 11, 12, 15, and 16 were investigated in order to extend the SAR in the series of 14-alkoxymorphinans.
Chemistry
Reductive amination of the 14-phenylpropoxymorphinan-6-ones 4(21) and 6(20) was performed with glycine tert-butyl ester hydrochloride and NaCNBH3 in DMF/MeOH 10:1 at room temperature (Scheme 1). The diastereoisomers were separated by medium pressure liquid chromatography (MPLC) to obtain 11, 12, 15, and 16. Ester cleavage of the tert-butyl derivatives in dioxane/HCl generated the amino acids 13, 14, 17, and 18. Configuration assignments at C(6) are based on the coupling constants (J(5,6)) between H−C(5) and H−C(6). J(5,6) values for 6α-amino epimers are smaller (3.2−4.0 Hz) than for 6β-amino epimers (6.5−7.8 Hz).29,34,35 The results for compounds 11−18 agree with the earlier findings.
Scheme 1. Synthesis of 14-Phenylpropxymorphinans 11−18.

Reagents and conditions: (a) glycine-t-butylester hydrochloride, NaCNBH3, DMF/MeOH 10:1, RT; (b) separation of the diastereoisomers using column chromatography (silica gel); (c) 4 M HCl in dioxane, reflux.
Results and Discussion
In Vitro Opioid Receptor Binding Affinities
Binding affinities of the newly synthesized compounds 11−18 at opioid receptors were determined by in vitro competition binding assays using rat brain (μ, δ) and guinea pig brain (κ) membranes and employing [3H][d-Ala2,Me-Phe4,Gly-ol5]enkephalin ([3H]DAMGO, μ),(36) [3H][Ile5,6]deltorphin II (δ),(37) and [3H](5α,7α,8β-(−)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro(4−5)dec-8-yl]benzeneacetamide) ([3H]U69,593, κ)(38) as specific opioid radioligands.(30) The μ, δ, and κ opioid receptor binding affinities expressed as inhibition constants (Ki) are summarized in Table 1. The selectivity for the μ opioid receptor vs δ and κ receptors was defined by the ratio of the Ki values. For comparison purposes, the opioid binding affinity data for morphine, 3, the 6α- and 6β-glycine conjugates of 14-O-methyloxymorphone, 9 and 10, respectively, and the 14-phenylpropoxy analogue of naltrexone, compound 6, are included. All compounds 11−18 bound with high affinity at the μ opioid receptor (Ki = 0.16−1.40 nM), having considerably improved interaction with the μ site compared to morphine. As shown in Table 1, they also exhibit increased binding affinities at δ and κ receptors as indicated by the Ki values in the subnanomolar or low nanomolar range.
Table 1. Binding Affinities at μ, δ and κ Opioid Receptors.
|
Ki (nM)a |
selectivity ratio |
||||
|---|---|---|---|---|---|
| compd | μ receptor [3H]DAMGOb | δ receptor [3H][Ile5,6]deltorphin IIb | κ receptor [3H]U69,593c | δ/μ | κ/μ |
| morphined | 6.55 ± 0.74 | 217 ± 19 | 113 ± 9a | 33 | 17 |
| 3e | 0.20 ± 0.05 | 0.14 ± 0.02 | 0.54 ± 0.15 | 0.7 | 2.7 |
| 6f | 0.34 ± 0.06 | 0.48 ± 0.05 | 0.41 ± 0.09 | 1.4 | 1.2 |
| 9g | 0.89 ± 0.09 | 15.4 ± 1.4 | 43.2 ± 7.0a | 17 | 49 |
| 10g | 0.83 ± 0.02 | 7.86 ± 0.64 | 44.8 ± 0.1a | 10 | 54 |
| 11 | 1.40 ± 0.07 | 1.01 ± 0.06 | 1.26 ± 0.30 | 0.7 | 0.9 |
| 12 | 1.03 ± 0.25 | 0.92 ± 0.16 | 2.02 ± 0.68 | 0.9 | 2.0 |
| 13 | 0.19 ± 0.02 | 0.22 ± 0.02 | 0.73 ± 0.01 | 1.2 | 3.8 |
| 14 | 0.16 ± 0.02 | 0.19 ± 0.01 | 0.81 ± 0.03 | 1.2 | 5.1 |
| 15 | 1.38 ± 0.21 | 1.11 ± 0.07 | 1.49 ± 0.27 | 0.8 | 1.1 |
| 16 | 1.01 ± 0.21 | 0.40 ± 0.07 | 1.77 ± 0.37 | 0.4 | 1.8 |
| 17 | 0.27 ± 0.02 | 0.33 ± 0.10 | 0.64 ± 0.01 | 1.2 | 2.4 |
| 18 | 0.20 ± 0.04 | 0.35 ± 0.04 | 0.65 ± 0.01 | 1.7 | 3.2 |
To investigate the SAR for the novel 14-alkoxymorphinans, the following structural modifications were targeted: (i) replacement of the methoxy group with a phenylpropoxy group in position 14, (ii) substitution of the N-methyl with an N-cyclopropylmethyl group, (iii) modification of the substituent at position 6 (e.g., 6-glycine vs 6-glycine ester vs 6-keto), and (iv) α vs β orientation of the amino acid residue at C-6 of the morphinan skeleton. First, we have examined the result of the replacement of the 14-methoxy group in N-methyl substituted morphinans 9 and 10 with a phenylpropoxy group, leading to compounds 13 and 14, respectively, on the in vitro opioid binding profile. While the μ affinity was less affected, affinities at δ and κ receptors were increased significantly after introduction of a phenylpropoxy substituent (Table 1). Compared to analogues 9 and 10, affinities of the 14-phenylpropoxy substituted derivatives 13 and 14 were increased by about 7- and 5-fold at μ, 70- and 42-fold at δ, and 59- and 55-fold at κ receptors, respectively. On the basis of these data, it appears that a phenylpropoxy group in position 14 is not favorable for selective binding to μ, δ, or κ receptors by causing a notable loss in μ receptor selectivity, thus, corroborating and extending our previous findings in other series of 14-phenylpropoxy substituted morphinans.19,20 The present observations together with earlier reports10,11,19,20,39,40 provide additional evidence that interaction with opioid receptors is sensitive to the character and length of the substituent in position 14. It has been shown that different substitution patterns at C-14 in morphinan-6-ones give rise to compounds with improved or decreased selectivity for the μ receptor. An enhancement in the binding affinity at δ and κ receptors has been reported with other substituents at position 14 such as benzyloxy or naphthylmethoxy.(11) We have described that a 14-methoxy group results in higher selectivity for the μ receptor than 14-benzyloxy or 14-naphthylmethoxy substitution, indicating that small alkoxy groups are superior over arylalkoxy groups in this respect.(11) In contrast, replacement of the 14-methoxy with a phenylpropoxy group in cyprodime and other 4,5-oxygen bridge opened morphinan-6-ones was found to markedly increase binding affinities at μ but also at δ and κ receptors, however retaining μ selectivity.(21)
The effect on the opioid receptor binding profile upon different substitution at the nitrogen of the morphinan skeleton was also investigated. Replacement of the N-methyl group in 13 and 14 by a cyclopropylmethyl group resulted in analogues 17 and 18, respectively. The N-cyclopropylmethyl substitution was well-tolerated as indicated by the high affinity for all μ, δ, and κ receptors (Ki values in the subnanomolar range; Table 1). Further evidence for this observation comes from our previous works where nearly identical affinities for all opioid receptor types were noted for 3(19) and naltrexone derivative 6.(20) It appears that modification of the N-substituent, methyl vs cyclopropylmethyl, causes no major alterations in binding affinity and selectivity in this series of opioid compounds. In other series of morphinans which are unsubstituted or have a hydroxyl or a small alkoxy group at C-14, substitution of the N-methyl by a N-cyclopropylmethyl group leads to an increase in affinity at δ and κ opioid receptors but also to a raise in μ affinity, resulting in reduced μ receptor selectivity.11,41,42 A representative example is the comparison of binding affinities of oxymorphone (μ:δ:κ = 0.97:80.5:61.6)(11) with its N-cyclopropylmethyl analogue naltrexone (μ:δ:κ = 0.20:8.70:0.40).(42) On the other hand, it has been shown that replacement of the N-methyl group in 14-methoxymetopon (2) with a 2-phenylethyl group left the binding affinities at μ and δ opioid receptors essentially unchanged but affected interaction with the κ receptor.(11) In view of these results, the nature of the substituent at the nitrogen has a significant effect on both opioid receptor binding affinity and selectivity of the morphinan class of opioid compounds, depending on the character and length of the substituent in position 14.
The effect on opioid receptor binding affinities upon substitution of a keto group with a glycine residue in position 6 in the N-methyl and N-cyclopropylmethyl 14-phenylpropoxy substituted morphinans was examined. The 6-glycine conjugates 13 and 14 displayed similar binding affinities to all three opioid receptors as the 6-keto derivative 3. Moreover, in the N-cyclopropylmethyl series, in a direct comparison of the 14-phenylpropoxy analogue of naltrexone (6) with the 6-glycine derivatives 17 and 18, all compounds showed comparable affinities at μ, δ, and κ receptors (Table 1). According to the present results, the 6-glycine derivatives having a 14-phenylpropoxy group maintained the high affinity at the μ opioid receptor of the nonzwitterionic compounds 3 and 6, which is in agreement with a further SAR report in other series of 14-methoxymorphinans containing zwitterionic moieties such as amino acid residues.29,30 Thus, it is evident that replacement of the 6-keto group with a glycine residue does not have a detrimental effect on affinity at the μ receptor. Analogous observations were made for β-oxymorphamine(43) and β-naltrexamine(44) when having ionizable moieties at C-6. Also, it has been shown in 4,5-oxygen bridge opened N-methylmorphinans that a 6-cyano substituent in comparison to a 6-keto group has a minor impact on the ability to interact with opioid receptors.(45) These findings indicate that replacement of the 6-keto group with other substituents such as amino acids or a cyano group produces only minor changes in the opioid binding profile of morphinans.
The consequence of esterification of the glycine residue at C-6 on binding affinity and selectivity was also investigated. As shown in Table 1, the tert-butyl ester derivatives 11, 12, 15, and 16 exhibited a decrease in the affinity at μ, δ, and κ opioid receptors compared to the 6-glycine analogues 13, 14, 17, and 18, respectively. An exception was ester 16, which showed comparable δ receptor affinity to analogue 18 (0.40 vs 0.35 nM). However, all esters still display high interaction with opioid receptors as indicated by the Ki values in the low nanomolar range (1.01−1.40 nM at μ, 0.40−1.11 nM at δ, and 1.26−2.02 nM at κ receptors). On the other hand, the esters also showed to lack selectivity to a receptor type similar to the corresponding 6-glycine analogues.
When examining the impact of α/β orientation of the amino acid residue at position 6 on the in vitro opioid activity, it was noted that binding affinities and selectivities of α vs β epimers did not vary greatly in this series of newly synthesized 14-phenylpropoxymorphinans (Table 1).
Pharmacological Activities
The 6-glycine substituted 14-phenylpropoxymorphinans 11−18 were further evaluated in vivo for antinociceptive effects in rats after sc administration using the tail-flick test.(31) Their antinociceptive properties were compared to those of morphine and derivatives 9 and 10. The antinociceptive ED50 values are listed in Table 2. All compounds acted as potent antinociceptive agents with long-lasting action when administered sc, with peak ED50s of 26.8−349 nmol/kg. Peak antinociception occurred generally one hour after drug sc administration, and only derivatives 14 and 18 had a peak of action at two hours postinjection. Compared to morphine, the 14-phenylpropoxymorphinans of this series showed 17- to 226-fold increased analgesic potency, with compound 16 displaying the highest potency (Table 2). Antinociceptive potencies of the 6-glycine substituted 14-phenylpropoxy derivatives 13 and 14 were in the range of their 14-methoxy analogues 9 and 10, respectively (Table 2). It appears that a 14-phenylpropoxy substitution maintains the high analgesic activity, while it largely affects opioid binding affinities and μ receptor selectivity (Table 1).
Table 2. Antinociceptive Potencies in the Tail-Flick Test in the Rat after sc Administration.
| ED50 (nmol/kg, sc), 95% CLa time after drug administration |
|||||
|---|---|---|---|---|---|
| compd | 0.5 h | 1 h | 2 h | 3 h | relative potencyb morphine = 1 |
| morphine | 6053c | 7626 | 13285 | 1 | |
| (4037−9080) | (5084−11439) | (8856−19929) | |||
| 9d | 137 | 58.5c | 143 | 437 | 103 |
| (85.7−219) | (36.5−93.7) | (86.8−237) | (243−786) | ||
| 10d | 50.8 | 29.0c | 43.3 | 209 | |
| (32.7−78.7) | (18.8−44.9) | (27.9−67.1) | |||
| 11 | 63.5 | 58.3c | 84.0 | 148 | 104 |
| (15.4−262) | (18.0−188) | (31.4−255) | (28.4−775) | ||
| 12 | 164 | 92.3c | 124 | 189 | 66 |
| (78.6−341) | (39.5−215) | (53.0−292) | (69.8−514) | ||
| 13 | 127 | 81.1c | 87.9 | 116 | 75 |
| (65.0−246) | (46.4−139) | (43.9−176) | (48.5−278) | ||
| 14 | 180 | 143 | 130c | 192 | 47 |
| (122−247) | (85.8−238) | (73.4−231) | (90.0−414) | ||
| 15 | 365 | 348c | 363 | 427 | 17 |
| (195−687) | (212−570) | (215−615) | (205−692) | ||
| 16 | 33.8 | 26.8c | 34.4 | 56.0 | 226 |
| (18.8−60.5) | (14.1−51.0) | (17.7−66.8) | (21.9−143) | ||
| 17 | 1388 | 349c | 433 | 543 | 17 |
| (117−16526) | (113−1072) | (172−1084) | (180−1636) | ||
| 18 | 1561 | 183 | 147c | −e | 41 |
| (199−12223) | (17.7−1877) | (13.9−1554) | |||
Values in paranthesis are 95% confidence limits.
Relative potencies were calculated at the peak of action.
Peak of effect.
From ref (31).
−: no dose−response relationship.
Traditional and generally accepted SAR models have assigned critical importance in defining the pharmacological profile of agonist/antagonist action of morphinan-6-ones to the substitutent at the morphinan nitrogen.46−48 Substituents such as cyclopropylmethyl or allyl at the nitrogen have been commonly associated with an antagonist character in this series of opioids. On the other hand, there are a number of examples where substitution at C-14 affords potent analgesics independent of the substituent nature at the morphinan nitrogen.11,19−21,39,40 Even with a N-substituent that is linked with distinct antagonist character such as cyclopropylmethyl, antinociceptive agents can be obtained and examples include the clinically used buprenorphine,(49) nalfurafine (TRK-820) and its analogues,50,51 and 14-aminomorphinone derivatives.39,40 In the present study, the N-cyclopropylmethyl substituted compounds 17 and 18 and their corresponding tert-butyl esters 15 and 16 showed potent antinociceptive activities in rats after sc administration (Table 2). The in vivo pharmacology of derivatives 17 and 18 as antinociceptive agents was also supported by the functional data on the ligand-stimulated guanosine-5′-O-(3-[35S]thio)-triphosphate ([35S]GTPγS) binding to membranes from Chinese hamster ovary (CHO) stably transfected with human opioid receptors(21) (Table 3). Both compounds were highly potent partial agonists at the μ receptor and also at δ and κ receptors so that no significant selectivity for any receptor type was apparent. It is suggested that such compounds which interact with multiple receptor types may have an overall increased potency derived from the combined action at μ, δ, and κ opioid receptors.(52) Moreover, as found in this and our earlier studies11,19−21 and also by others,39,40,53−55 not necessarily the nature of the substituent at the nitrogen in morphine-like compounds but rather residues occupying a defined position in the vicinity to the morphinan nitrogen seem to be responsible for agonist/antagonist activity. Our SAR study also showed that replacement of the N-methyl group of compounds 13 and 14 with a cyclopropylmethyl group resulted in analogues 17 and 18, respectively, which retained the high antinociceptive potency after sc administration (Table 2). This holds also true when evaluating the N-methyl substituted 3(19) and naltrexone derivative 6,(20) which show comparable antinociceptive potencies in different analgesic tests in mice after sc administration.
Table 3. Stimulation of [35S]GTPγS Bindinga.
| μ receptor |
δ receptor |
κ receptor |
||||
|---|---|---|---|---|---|---|
| compd | EC50 (nM) | % stimb | EC50 (nM) | % stimc | EC50 (nM) | % stimd |
| 17 | 0.69 ± 0.10 | 34.7 ± 6.6 | 0.69 ± 0.29 | 26.1 ± 3.0 | 1.28 ± 0.35 | 28.2 ± 1.0 |
| 18 | 1.06 ± 0.28 | 24.2 ± 3.0 | 4.24 ± 0.47 | 34.7 ± 0.6 | 11.0 ± 1.4 | 30.4 ± 3.3 |
| morphine | 15.6 ± 0.5 | 93 ± 2.8 | 316 ± 5 | 103 ± 7 | 484 ± 213 | 62 ± 7 |
Membranes from CHO cells stably transfected with human μ, δ, or κ opioid receptors were incubated with varying concentrations of the compounds.
Compared to DAMGO.
Compared to DPDPE.
Compared to U69,593. Data represent mean ± SEM.
As shown in Table 2, esterification of the glycine residue in position 6 did not result in major alterations in antinociceptive activities and all tert-butyl esters were found to be greatly active displaying similar potencies to their corresponding glycine analogues. Notably, the N-cyclopropylmethyl tert-butyl ester 16 was about 6-fold more potent than derivative 18. In the tail-flick test, antinociceptive potencies did not significantly differ between the α and β epimers, which is in agreement with the in vitro biological data where also no major changes in opioid binding affinities were observed. Their ED50 ratios were ranging between 0.6 and 2.4 after sc administration, except for the pair 15 vs 16, where a 13-fold difference in the potency was calculated.
Opioids with hydrophilic groups such as amino acid residues attached to the C-6 position of the morphinan skeleton were targeted in an effort to obtain opioid compounds that would have potentially limited access to the CNS and thus to reduce the activation of central opioid receptors. We have reported on the potent antinociceptive action of the 6-glycine conjugate of 14-O-methyloxymorphone, compound 10, after systemic sc administration to rats with carrageenan-induced inflammatory hyperalgesia.(18) Further in vivo pharmacological studies were undertaken in the present study with the 14-phenylpropoxy derivative of 10, compound 14, and its corresponding 6α-glycine analogue 13 to assess their antinociceptive actions in a rat model of inflammatory pain. Subcutaneous administration of compounds 13 and 14, in a dose of 170 nmol/kg, produced a significant attenuation in pain-related behavior in the inflamed paw by increasing withdrawal latencies to mechanical stimulation having a long duration of action (up to 4 h) (Figure 2A). These observations are in line with the earlier pharmacological findings on potent and prolonged inhibitory effects of the 14-methoxy 6-glycine analogue 10 in rats with inflammatory hyperalgesia.(18) Moreover, its antinociceptive actions after sc administration was shown to be mediated through peripheral mechanisms not only in carrageenan-induced hindpaw inflammation(18) but also in other rodent pain models including tail-flick test,(31) formalin test,(31) and acetic acid-induced writhing.(32) The antinociceptive efficacy of derivatives 13 and 14 in tested sc doses in carrageenan-induced inflammatory pain was comparable to that reported for compound 10 after a sc dose of 100 μg/kg (i.e., 183 nmol/kg) and with markedly longer duration of action than morphine and 14-methoxymetopon (2).(18) To evaluate whether the antinociceptive effect of the 14-phenylpropoxy substituted derivatives 13 and 14 in rats with carrageenan-induced hyperalgesia is peripherally mediated, naloxone methiodide, the peripherally acting opioid antagonist, was sc coadministered with each opioid. As shown in Figure 2B, naloxone methiodide (426 nmol/kg) significantly reversed the antinociceptive effect of a 170 nmol/kg dose of compound 13 and 14 on mechanical hyperalgesia. Hence, replacement of the 14-methoxy group in compound 10 with a phenylpropoxy group gave rise to new derivatives showing peripherally mediated antinociceptive action in a rat model of inflammatory pain. Outcomes of these studies extend our earlier reports on 6-amino acid substituted derivatives (i.e., glycine, alanine and phenylalanine) of 14-O-methyloxymorphone (1)18,29−33 described as high affinity μ opioid receptor agonists that can induce potent antinociceptive effects via peripheral opioid receptor-specific mechanisms.
Figure 2.

(A) Antinociceptive effects of 6-glycine substituted 14-phenylpropoxymorphinans 13 and 14 after sc administration to rats with carrageenan-induced inflammatory hyperalgesia. (B) Antagonism by naloxone methiodide (NM) on the antinociceptive effect of compound 13 and 14 after sc coadministration on the inflamed paw withdrawal latencies to mechanical stimulation. Values are presented as % changes in withdrawal latencies of the inflamed paw from the pretreatment (pre-) values obtained at 3 h after carrageenan injection. Areas under the curves (AUC) of the respective time curves are represented. Data are shown as mean ± SEM *p < 0.05, **p < 0.01, and ***p < 0.001 vs vehicle-treated controls; #p < 0.05 vs agonist-treated animals.
Conclusions
We have described the synthesis and biological activities of a series of new 6-glycine substituted 14-phenylpropoxymorphinans which emerged as high affinity and potent opioid antinociceptive agents. Specific modifications in the substitution pattern, such as introduction of a 14-phenylpropoxy group and a glycine residue at position 6, led to an interesting alteration in opioid activity by influencing the biological profile of these compounds interacting with μ, δ, and κ opioid receptors. On the basis of the SAR that has emerged, it could be observed that there is a significant enhancement in affinity at all opioid receptor types upon introduction of a phenylpropoxy group at position 14 in 6-glycine substituted morphinans, with most pronounced increases in activity at δ and κ receptors, leading to a loss in μ receptor selectivity. In vitro binding affinities to all opioid receptors and selectivity were not appreciably affected upon substitution of the N-methyl with a cyclopropylmethyl group. Besides the increased affinity for opioid receptors, another attribute of the new 6-glycine substituted 14-phenylpropoxymorphinans is the potent and long-lasting antinociceptive effects in rats after sc administration, displaying considerably increased potency than morphine. For the N-methyl derivatives 13 and 14, antinociceptive potencies were in the range of their 14-methoxy analogues. Even derivatives 15−18 having a N-cyclopropylmethyl substituent acted as highly active antinociceptive agents, being several fold more potent than morphine. Subcutaneous administration of the N-methyl 6α- and 6β-glycine substituted 14-phenylpropoxymorphinans 13 and 14, respectively, significantly reduced mechanical hypersensitivity in rats with carrageenan-induced inflammatory pain, effects shown to be mediated via activation of peripheral opioid receptors. Such peripherally acting opioids may represent novel drug candidates for pain treatment.
Experimental Section
General Methods
The starting material thebaine was obtained from Tasmanian Alkaloids Ltd., Westbury, Tasmania, Australia. Melting points were determined on a Kofler melting point microscope and are uncorrected. 1H NMR spectra were obtained on a Varian Gemini 200 (200 MHz) spectrometer using tetramethylsilane (TMS) as internal standard for CDCl3 and 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) for D2O. Coupling constants (J) are given in Hz. IR spectra were taken on a Mattson Galaxy FTIR series 3000 in KBr pellets (in cm−1). Mass spectra were recorded on a Varian MAT 44 S apparatus. Elemental analysis was performed at the Institute of Physical Chemistry of the University of Vienna, Austria. For column chromatography (MPLC), Silica Gel 60 (0.040−0.063 mm, Fluka, Switzerland) was used. Thin-layer chromatography (TLC) was performed on silica gel plates Polygram SIL G/UV254 (Macherey-Nagel, Germany) with CH2Cl2/MeOH/NH4OH 90:9:1 as an eluent.
Radioligands [3H]DAMGO, [3H]U69,593, and [35S]GTPγS were purchased from PerkinElmer (Boston, MA, USA). [3H][Ile5,6]deltorphin II was obtained from the Institute of Isotopes Co. Ltd. (Budapest, Hungary). Naloxone hydrochloride, tris-(hydroxymethyl)-aminomethane (Tris), carrageenan (λ-carrageenan), naloxone methiodide, GTPγS, guanosine diphosphate (GDP), and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were obtained from Sigma Chemicals (St. Louis, MO, USA). All other chemicals were of analytical grade and obtained from standard commercial sources. Compounds 11, 12, 15, and 16 were used as bases and compounds 13, 14, 17, and 18 were used as hydrochloride salts. Purities of tested compounds were determined by elemental analysis and were ≥95%.
General Procedure for the synthesis of 13, 14, 17, and 18 (13 as example)
11 (0.18 g; 0.35 mmol) was dissolved in 5 mL of 4 M HCl in dioxane and stirred under reflux for 2 h. The colorless crystals were filtered off under nitrogen, washed with anhydrous diethyl ether, and dried.
[[4,5α-Epoxy-3-hydroxy-17-methyl-14β-[(3-phenylpropyl)oxy]morphinan-6α-yl]amino]acetic Acid Dihydrochloride (13·2HCl)
Colorless crystals (89%); mp > 207 °C (dec). IR (KBr) 1744 (C=O). 1H NMR (D2O) δ 7.47−7.35 (m, 5 arom H), 6.96 (d, J = 8.3, 1 arom H), 6.87 (d, J = 8.3, 1 arom H), 5.09 (d, J = 3.2, H-C(5)), 3.01 (s, CH3N). MS (ESI) m/z 479 [M + 1]+. Anal. (C28H34N2O5·2HCl·1.9H2O) C, H, N.
[[4,5α-Epoxy-3-hydroxy-17-methyl-14β-[(3-phenylpropyl)oxy]morphinan-6β-yl]amino]acetic Acid Dihydrochloride (14·2HCl)
Colorless crystals (93%). mp > 217 °C (dec). IR (KBr) 1740 (C=O). 1H NMR (D2O) δ 7.44 (s, 5 arom H), 6.94 (s, 2 arom H), 4.96 (d, J = 7.0, H-C(5)), 2.99 (s, CH3N). MS (ESI) m/z 479 [M + 1]+. Anal. (C28H34N2O5·2HCl·2H2O) C, H, N.
[[17-Cyclopropylmethyl-4,5α-epoxy-3-hydroxy-14β-[(3-phenylpropyl)oxy]morphinan-6α-yl]amino]acetic Acid Dihydrochloride (17·2HCl)
Colorless crystals (97%); mp > 218 °C (dec). IR (KBr) 1741 (C=O). 1H NMR (D2O) δ 7.29 (s, 5 arom H), 6.85 (d, J = 8.2, 1 arom H), 6.75 (d, J = 8.2, 1 arom H), 4.99 (d, J = 3.0, H-C(5)), 0.93 (m, CH-cp), 0.66 (m, CH2-cp), 0.36 (m, CH2-cp) (cp = cyclopropyl). MS (CI) m/z 519 [M + 1]+. Anal. (C31H38N2O5·2HCl·1.4H2O) C, H, N.
[[17-Cyclopropylmethyl-4,5α-epoxy-3-hydroxy-14β-[(3-phenylpropyl)oxy]morphinan-6β-yl]amino]acetic Acid Dihydrochloride (18·2HCl)
Colorless crystals (89%); mp > 223 °C (dec).IR (KBr) 1741 (C=O).1H NMR (D2O) δ 7.42 (s, 5 arom H), 6.94 (s, 2 arom H), 4.99 (d, J = 7.0 H-C(5)), 1.02 (m, CH-cp), 0.82−0.68 (m, CH2-cp), 0.46 (m, CH2-cp) (cp = cyclopropyl.MS (CI) m/z 519 [M + 1]+. Anal. (C31H38N2O5·2HCl·1.7H2O) C, H, N.
Opioid Receptor Binding Assays
Membranes were prepared from Sprague−Dawley rat or guinea pig brains, obtained from the Institut für Labortierkunde and Laborgenetik, Medizinische Universität Wien (Himberg, Austria), as previously described.(30) Binding experiments were performed in 50 mM Tris-HCl buffer (pH 7.4.) in a final volume of 1 mL containing 0.3−0.5 mg of protein and at least 10 concentrations of test compound as described.(18) Rat brain homogenates were incubated either with [3H]DAMGO (1 nM, 45 min, 35 °C) or [3H][Ile5,6]deltorphin II (0.5 nM, 45 min, 35 °C). Guinea pig brain homogenates were incubated with [3H]U69,593 (1 nM, 30 min, 30 °C). Reactions were terminated by rapid filtration through Whatman glass fiber filters using a Brandel M24R cell harvester, followed by washing with 50 mM Tris-HCl buffer (pH 7.4). Nonspecific binding was determined using 10 μM naloxone. The bound radioactivity was measured by liquid scintillation counting. Inhibition constant (Ki) values were calculated from competition binding curves using the nonlinear least-squares curve fitting by GraphPad Prism program. All experiments were performed in duplicate and repeated at least three times.
[35S]GTPγS Functional Assays
Functional assays were conducted on human opioid receptors stably transfected into CHO cells as described.(21) Aliquots of cell membranes (15 μg) in buffer A (20 mM HEPES, 10 mM MgCl2 and 100 mM NaCl, pH 7.4) were incubated with 0.05 nM [35S]GTPγS, 10 μM GDP, and test compounds, in a total volume of 1 mL, for 60 min at 25 °C. Nonspecific binding was determined using 10 μM GTPγS. Samples are filtered over glass fiber filters and counted as described for binding assays. Potency (EC50, nM) and percentage of stimulation (% stim) with respect to the standard agonists DAMGO (μ), DPDPE (δ), and U69,593 (κ) were calculated using GraphPad Prism program. The results are mean ± SEM from at least three determinations, each performed in triplicates.
In Vivo Assays. General Methods
Male Wistar rats (120−150 g) used in the tail-flick test were purchased from Charles River (Budapest, Hungary). Male Sprague−Dawley rats (250 g) used in carrageenan-induced inflammatory pain were purchased form B&K Universal Lab (Sollentuna, Sweden). Animals were maintained on a 12 h light/dark cycle with free access to food and water at all times except during testing. Experiments were carried out in accordance to the Declaration of Helsinki and the Guide for Care and Use of Laboratory Animals. The experimental protocols were approved by the Ethical Board of Semmelweis University, Budapest, and Ethics Committee for Animal Research (North Stockholm, Sweden).
Tail-Flick Test
The tail-flick test was performed according to the described procedure.(31) A beam light was focused on the tip of the tail and the latency required for the rat to remove its tail was determined before (basal latencies) and after drug administration, using an arbitrary cutoff time of twice the basal reaction time. Compounds were dissolved in distilled water and administered sc to rats in a volume of 5 mL/kg of body weight. Each drug dose or vehicle (distilled water) was tested in at least five animals. The effective dose 50% (ED50) values and 95% confidence limits were calculated according to the method of Litchfield and Wilcoxon.(56)
Carrageenan-Induced Hindpaw Inflammation
Unilateral inflammation was induced to nonanesthetized rats by injection of 100 μL of 1% carrageenan into the plantar surface of the right hindpaw as previously described.(18) All treatments were performed 3 h after carrageenan injection. Compounds were dissolved in physiological saline (0.9%) and administered sc in a volume of 100 μL. Control animals received the same volume of sc physiological saline solution. Each experimental group included four to seven animals. The Randall−Selitto test (Ugo Basile) was used to assess the hindpaw withdrawal latencies to mechanical stimulation as described.(18) A wedge-shaped, blunt piston with an area of 1 × 10 mm2 and a loading rate of 48 g/s was applied to the dorsal surface of the manually handled hindpaw, and the time required to initiate the struggle response was assessed and expressed in seconds. A cutoff time of 20 s was applied. Withdrawal latencies to mechanical stimulation were measured before injection of carrageenan (basal values), prior to administration of the drug 3 h postcarrageenan (pretreatment values), and after drug administration. The antinociceptive response is expressed as percentage (%) changes in hindpaw withdrawal latencies from the pretreatment value obtained at 3 h after carrageenan injection, and was calculated as 100 [(PX − P0)/P0], where P0 is the latency prior to drug administration at 3 h postcarrageenan and PX is the latency after drug administration at corresponding time points.
Statistical Analysis
Data are expressed as mean ± SEM. The areas under the curves (AUC) were calculated by the trapezoidal rule using NCSS software. Statistical analysis was carried out using one-way analysis of variances (ANOVA) with Dunnett’s post hoc test using SPSS software. A p value <0.05 was considered statistically significant.
Acknowledgments
We thank Tasmanian Alkaloids Pty. Ltd., Westbury, Tasmania, Australia, for the generous gift of thebaine. We acknowledge A. Kaletsch, I. Wachtl, and K. Parina for technical assistance. This work was supported by CAST, LISA, the Austrian Science Fund (FWF; P15481, P21350, and TRP 16-B18), the Hungarian grant OTKA-K 60999, and a contract from the National Institute on Drug Abuse (N01DA-1-8816).
Supporting Information Available
Additional experimental, spectroscopic data, elemental analysis results, and supplementary pharmacology figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
This paper is dedicated to Dr. Kenner C. Rice on the occasion of his 70th birthday.
Abbreviations: CHO, Chinese hamster ovary; CNS, central nervous system; DAMGO, [d-Ala2,Me-Phe4,Gly-ol5]enkephalin; DMF, N,N-dimethylformamide; DPDPE, [D-Pen2,D-Pen5]enkephalin; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; GDP, guanosine diphosphate; [35S]GTPγS, guanosine-5′-O-(3-[35S]thio)-triphosphate; MPLC, medium pressure liquid chromatography; SAR, structure−activity relationship; Tris, tris-(hydroxymethyl)-aminomethane; U69,593, 5α,7α,8β-(−)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro(4−5)dec-8-yl]benzeneacetamide.
Supplementary Material
References
- Breivik H.; Collett B.; Ventafridda V.; Cohen R.; Gallacher D. Survey of Chronic Pain in Europe: Prevalence, Impact on Daily Life, and Treatment. Eur. J. Pain 2006, 10, 287–333. [DOI] [PubMed] [Google Scholar]
- Marcus D. A.; Cope D. K.; Deodhar A.; Payne R.. Chronic Pain: An Atlas of Investigation and Management; Clinical Publishing: Oxford, 2009. [Google Scholar]
- Benyamin R.; Trescot A. M.; Datta S.; Buenaventura R.; Adlaka R.; Sehgal N.; Glaser S. E.; Vallejo R. Opioid Complications and Side Effects. Pain Physician 2008, 11, S105–S120. [PubMed] [Google Scholar]
- McNally G. P.; Akil H.. Opioid Peptides and Their Receptors: Overview and Function in Pain Modulation. In Neuropsychopharmacology: The Fifth Generation of Progress; Davis K. L., Charney D., Coyle J. T., Nemeroff C., Eds.; Lippincott Williams & Wilkins: Philadelphia, 2002; pp 34−46.
- Kieffer B. L.; Evans C. J. Opioid Receptors: From Binding Sites to Visible Molecules In Vivo. Neuropharmacology 2009, 56, 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C.; Clark J. D.; Oh U.; Vasko M. R.; Wilcox G. L.; Overland A. C.; Vanderah T. W.; Spencer R. H. Peripheral Mechanisms of Pain and Analgesia. Brain Res. Rev. 2009, 60, 90–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidhammer H. 14-Alkoxymorphinans: A Series of Highly Potent Opioid Agonists, Antagonists, and Partial Agonists. Curr. Top. Med. Chem. 1993, 1, 261–276. [Google Scholar]
- Schmidhammer H.Opioid Receptor Antagonists. In Progress in Medicinal Chemistry; Ellis G. P., Luscombe D. K., Oxford A. W., Eds.; Elsevier: Amsterdam, 1998; 35, 83−132. [PubMed] [Google Scholar]
- Schmidhammer H.; Spetea M.. Synthesis of 14-Alkoxymorphinan Derivatives and Their Pharmacological Actions. Top. Curr. Chem. 2011, 299, 63−91. [DOI] [PubMed] [Google Scholar]
- Schmidhammer H.; Aeppli L.; Atwell L.; Fritsch F.; Jacobson A. E.; Nebuchla M.; Sperk G. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 1. Highly Potent Opioid Agonists in the Series of (−)-14-Methoxy-N-methylmorphinan-6-ones. J. Med. Chem. 1984, 27, 1575–1579. [DOI] [PubMed] [Google Scholar]
- Lattanzi R.; Spetea M.; Schüllner F.; Rief S. B.; Krassnig R.; Negri L.; Schmidhammer H. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 22. Influence of the 14-alkoxy Group and the Substitution in Position 5 in 14-Alkoxymorphinan-6-ones on In Vitro and In Vivo Activities. J. Med. Chem. 2005, 48, 3372–3378. [DOI] [PubMed] [Google Scholar]
- Riba P.; Friedmann T.; Király K. P.; Al-Khrasani M.; Sobor M.; Asim M. F.; Spetea M.; Schmidhammer H.; Furst S. Novel Approach to Demonstrate High Efficacy of μ Opioids in the Rat Vas Deferens: A Simple Model of Predictive Value. Brain Res. Bull. 2010, 81, 178–184. [DOI] [PubMed] [Google Scholar]
- Schmidhammer H.; Schratz A.; Mitterdorfer J. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 8. 14-Methoxymetopon, an Extremely Potent Opioid Agonist. Helv. Chim. Acta 1990, 73, 1784–1787. [Google Scholar]
- Fürst S.; Búzás B.; Friedmann T.; Schmidhammer H.; Borsodi A. Highly Potent Novel Opioid Receptor Agonist in the 14-Alkoxymetopon Series. Eur. J. Pharmacol. 1993, 236, 209–215. [DOI] [PubMed] [Google Scholar]
- Freye E.; Schmidhammer H.; Latasch L. 14-Methoxymetopon, a Potent Opioid, Induces No Respiratory Depression, Less Sedation, and Less Bradycardia than Sufentanil in the Dog. Anesth. Analg. 2000, 90, 1359–1364. [DOI] [PubMed] [Google Scholar]
- King M. A.; Su W.; Nielan C. L.; Chang A. H.; Schütz J.; Schmidhammer H.; Pasternak G. W. 14-Methoxymetopon, a Very Potent μ-Opioid Analgesic with an Unusual Pharmacological Profile. Eur. J. Pharmacol. 2003, 459, 203–209. [DOI] [PubMed] [Google Scholar]
- Spetea M.; Tóth F.; Schütz J.; Ötvös F.; Tóth G.; Benyhe S.; Borsodi A.; Schmidhammer H. Binding Characteristics of [3H]14-Methoxymetopon, a High Affinity μ-Opioid Receptor Agonist. Eur. J. Neurosci. 2003, 18, 290–295. [DOI] [PubMed] [Google Scholar]
- Bileviciute-Ljungar I.; Spetea M.; Guo Y.; Schütz J.; Windisch P.; Schmidhammer H. Peripherally Mediated Antinociception of the μ-Opioid Receptor Agonist 2-[(4,5α-Epoxy-3-hydroxy-14β-methoxy-17-methylmorphinan-6β-yl)amino] acetic Acid (HS-731) after Subcutaneous and Oral Administration in Rats with Carrageenan-Induced Hindpaw Inflammation. J. Pharmacol. Exp. Ther. 2006, 317, 220–227. [DOI] [PubMed] [Google Scholar]
- Schütz J.; Spetea M.; Koch M.; Aceto M. D.; Harris L. S.; Coop A.; Schmidhammer H. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 20. 14-Phenylpropoxymetopon: An Extremely Powerful Analgesic. J. Med. Chem. 2003, 46, 4182–4187. [DOI] [PubMed] [Google Scholar]
- Greiner E.; Spetea M.; Krassnig R.; Schüllner F.; Aceto M.; Harris L. S.; Traynor J. R.; Woods J. H.; Coop A.; Schmidhammer H. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 18. N-Substituted 14-Phenylpropyloxymorphinan-6-ones with Unanticipated Agonist Properties: Extending the Scope of Common Structure−Activity Relationships. J. Med. Chem. 2003, 46, 1758–1763. [DOI] [PubMed] [Google Scholar]
- Spetea M.; Schüllner F.; Moisa R. C.; Berzetei-Gurske I. P.; Schraml B.; Dörfler C.; Aceto M. D.; Harris L. S.; Coop A.; Schmidhammer H. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 21. Novel 4-Alkoxy and 14-Phenylpropoxy Derivatives of the μ-Opioid Receptor Antagonist Cyprodime. J. Med. Chem. 2004, 47, 3242–3247. [DOI] [PubMed] [Google Scholar]
- Barber A.; Gottschlich R. Opioid Agonists and Antagonists: An Evaluation of Their Peripheral Actions in Inflammation. Med. Res. Rev. 1992, 12, 525–562. [DOI] [PubMed] [Google Scholar]
- Stein C.; Schäfer M.; Machelska H. Attacking Pain at its Source: New Perspectives on Opioids. Nature Med. 2003, 9, 1003–1008. [DOI] [PubMed] [Google Scholar]
- Kapitzke D.; Vetter I.; Cabot P. J. Endogenous Opioid Analgesia in Peripheral Tissues and the Clinical Implications for Pain Control. Ther. Clin. Risk Manag. 2005, 1, 279–297. [PMC free article] [PubMed] [Google Scholar]
- Stein C. Opioid Receptors on Peripheral Sensory Neurons. Adv. Exp. Med. Biol. 2003, 521, 69–76. [PubMed] [Google Scholar]
- Pol O.; Puig M. M. Expression of Opioid Receptors During Peripheral Inflammation. Curr. Top. Med. Chem. 2004, 4, 51–61. [DOI] [PubMed] [Google Scholar]
- DeHaven-Hudkins D. L.; Dolle R. E. Peripherally Restricted Agonists as Novel Analgesic Agents. Curr. Pharm. Des. 2004, 10, 743–757. [DOI] [PubMed] [Google Scholar]
- Smith H. S. Peripherally-Acting Opioids. Pain Physician 2008, 11, S121–S132. [PubMed] [Google Scholar]
- Schütz J.; Brandt W.; Spetea M.; Wurst K.; Wunder G.; Schmidhammer H. Synthesis of 6-amino Acid Substituted Derivatives of the Highly Potent Analgesic 14-O-Methyloxymorphone. Helv. Chim. Acta 2003, 86, 2142–2148. [Google Scholar]
- Spetea M.; Friedmann T.; Riba P.; Schütz J.; Wunder G.; Langer T.; Schmidhammer H.; Fürst S. In Vitro Opioid Activity Profiles of 6-Amino Acid Substituted Derivatives of 14-O-Methyloxymorphone. Eur. J. Pharmacol. 2004, 483, 301–308. [DOI] [PubMed] [Google Scholar]
- Fürst S.; Riba P.; Friedmann T.; Timar J.; Al-Khrasani M.; Obara I.; Makuch W.; Spetea M.; Schütz J.; Przewlocki R.; Przewlocka B.; Schmidhammer H. Peripheral versus Central Antinociceptive Actions of 6-Amino Acid-substituted Derivatives of 14-O-Methyloxymorphone in Acute and Inflammatory Pain in the Rat. J. Pharmacol. Exp. Ther. 2005, 312, 609–618. [DOI] [PubMed] [Google Scholar]
- Al-Khrasani M.; Spetea M.; Friedmann T.; Riba P.; Kiraly K.; Schmidhammer H.; Fürst S. DAMGO and 6β-Glycine Substituted 14-O-Methyloxymorphone but Not Morphine Show Peripheral, Preemptive Antinociception after Systemic Administration in a Mouse Visceral Pain Model and High Intrinsic Efficacy in the Isolated Rat Vas Deferens. Brain Res. Bull. 2007, 74, 369–375. [DOI] [PubMed] [Google Scholar]
- Obara I.; Makuch W.; Spetea M.; Schütz J.; Schmidhammer H.; Przewlocki R.; Przewlocka B. Local Peripheral Antinociceptive Effects of 14-O-Methyloxymorphone Derivatives in Inflammatory and Neuropathic Pain in the Rat. Eur. J. Pharmacol. 2007, 558, 60–67. [DOI] [PubMed] [Google Scholar]
- Jiang J. B.; Hanson R. N.; Portoghese P. S.; Takemori A. E. Stereochemical Studies on Medicinal Agents. 23. Synthesis and Biological Evaluation of 6-Amino Derivatives of Naloxone and Naltrexone. J. Med. Chem. 1977, 20, 1100–1102. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Portoghese P. S. Stereospecific Synthesis of the 6α- and 6β-Amino Derivatives of Naltrexone and Oxymorphone. J. Org. Chem. 1980, 45, 3366–3368. [Google Scholar]
- Handa B. K.; Lane A. C.; Lord J. A. H.; Morgan B. A.; Rance M. J.; Smith C. F. C. Analogues of β-LPH61−64 possessing selective agonist activity at μ-opiate receptors. Eur. J. Pharmacol. 1981, 70, 531–540. [DOI] [PubMed] [Google Scholar]
- Nevin S. T.; Kabasakal L.; Ötvös F.; Tóth G.; Borsodi A. Binding Characteristics of the Novel Highly Selective δ-Agonist, [3H]Ile5,6Deltorphin II. Neuropeptides 1994, 268, 261–265. [DOI] [PubMed] [Google Scholar]
- Lahti R. A.; Mickelson M. M.; McCall J. M.; Von Voightlander P. F. [3H]U69-593, a Highly Selective Ligand for the Opioid κ Receptor. Eur. J. Pharmacol. 1985, 109, 281–284. [DOI] [PubMed] [Google Scholar]
- Nieland N. P.; Moynihan H. A.; Carrington S.; Broadbear J.; Woods J. H.; Traynor J. R.; Husbands S. M.; Lewis J. W. Structural Determinants of Opioid Activity in Derivatives of 14-Aminomorphinones: Effect of Substitution in the Aromatic Ring of Cinnamoylaminomorphinones and Codeinones. J. Med. Chem. 2006, 49, 5333–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennison D.; Moynihan H.; Traynor J. R.; Lewis J. W.; Husbands S. M. Structural Determinants of Opioid Activity in Derivatives of 14-Aminomorphinones: Effects of Changes to the Chain Linking of the C14-Amino Group to the Aryl Ring. J. Med. Chem. 2006, 49, 6104–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobylecki R. J.; Carling R. W.; Lord J. A. H.; Smith C. F. C.; Lane A. C. Common Anionic Receptor Site Hypothesis: Its Relevance to the Action of Naloxone. J. Med. Chem. 1982, 25, 116–120. [DOI] [PubMed] [Google Scholar]
- Schüllner F.; Meditz R.; Krassnig R.; Morandell G.; Kalinin V. N.; Sandler E.; Spetea M.; White A.; Schmidhammer H.; Berzetei-Gurske I. P. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 19. Effect of 14-O-Benzylation on the Opioid Receptor Affinity and Antagonist Potency of Naltrexone. Helv. Chim. Acta 2003, 86, 2335–2341. [Google Scholar]
- Botros S.; Lipkowski A. W.; Larson D. L.; Stark A. P.; Takemori A. E.; Portoghese P. S. Opioid Agonist and Antagonist Activities of Peripherally Selective Derivatives of Naltrexamine and Oxymorphamine. J. Med. Chem. 1989, 32, 2068–2071. [DOI] [PubMed] [Google Scholar]
- Larson D. L.; Hua M.; Takemori A. K.; Portoghese P. S. Possible Contribution of a Glutathione Conjugate to the Long-duration Action of β-Funaltrexamine. J. Med. Chem. 1993, 36, 3669–3673. [DOI] [PubMed] [Google Scholar]
- Spetea M.; Greiner E.; Aceto M. D.; Harris L. S.; Coop A.; Schmidhammer H. Effect of a 6-Cyano Substituent in 14-Oxygenated N-Methylmorphinans on Opioid Receptor Binding and Antinociceptive Potency. J. Med. Chem. 2005, 48, 5052–5055. [DOI] [PubMed] [Google Scholar]
- Feinberg A. P.; Creese I.; Snyder S. H. The Opiate Receptor: A Model Explaining Structure−Activity Relationships of Opiate Agonists and Antagonists. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 4215–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casy A. F.; Parfitt R. T.. Opioid Analgesics: Chemistry and Receptors; Plenum Press: New York, 1986. [Google Scholar]
- Schmidhammer H.; Burkard W. P.; Eggstein-Aeppli L. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 4. Opioid Agonists and Partial Opioid Agonists in a Series of N-(Cyclobutylmethyl)-14-methoxymorphinan-6-ones. Helv. Chim. Acta 1989, 72, 1233–1240. [Google Scholar]
- Kintz P.; Marquet P.. Opioid Therapy of Drug Addiction; Humana Press: Totowa, NJ, 2002. [Google Scholar]
- Kawai K.; Hayakawa J.; Miyamoto T.; Imamura Y.; Yamane S.; Wakita H.; Fujii H.; Kawamura K.; Matsuura H.; Izumimoto N.; Kobayashi R.; Endo T.; Nagase H. Design, Synthesis, and Structure−Activity Relationship of Novel Opioid κ-Agonists. Bioorg. Med. Chem. 2008, 16, 9188–9201. [DOI] [PubMed] [Google Scholar]
- Nagase H.; Watanabe A.; Nemoto T.; Yamaotsu N.; Hayashida K.; Nakajima M.; Hasebe K.; Nakao K.; Mochizuki H.; Hirono S.; Fujii H. Drug Design and Synthesis of a Novel κ Opioid Receptor Agonist with an Oxabicyclo[2.2.2]Octane Skeleton and its Pharmacology. Bioorg. Med. Chem. Lett. 2010, 20, 121–124. [DOI] [PubMed] [Google Scholar]
- Lester P. A.; Traynor J. R. Comparison of the in Vitro Efficacy of μ, δ, κ, and ORL1 Receptor Agonists and Nonselective Opioid Agonists in Dog Brain Membranes. Brain Res. 2006, 1073−1074, 290–26. [DOI] [PubMed] [Google Scholar]
- Portoghese P. S.; Moe S. T.; Takemori A. E. A Selective δ1 Opioid Receptor Agonist Derived from Oxymorphone. Evidence for Separate Recognition Sites for δ1 Opioid Receptor Agonists and Antagonists. J. Med. Chem. 1993, 36, 2572–2574. [DOI] [PubMed] [Google Scholar]
- Portoghese P. S.; Sultana M.; Moe S. T.; Takemori A. E. Synthesis of Naltrexone-Derived δ-Opioid Antagonists. Role of Conformation of the δ Address Moiety. J. Med. Chem. 1994, 37, 579–585. [DOI] [PubMed] [Google Scholar]
- Brandt W. A Uniform Molecular Model of δ Opioid Agonist and Antagonist Pharmacophore Conformations. J. Comp.-Aided Mol. Des. 1998, 12, 615–621. [DOI] [PubMed] [Google Scholar]
- Litchfield J. T. Jr.; Wilcoxon F. A Simplified Method of Evaluating Dose−Effect Experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.