

Abstract
BACKGROUND AND PURPOSE
Ischemia-reperfusion injury plays an important role in the development of primary allograft failure after heart transplantation. Inhibition of the Na+/H+ exchanger is one of the most promising therapeutic strategies for treating ischemia-reperfusion injury. Here we have characterized the cardioprotective efficacy of zoniporide and the underlying mechanisms in a model of myocardial preservation using rat isolated working hearts.
EXPERIMENTAL APPROACH
Rat isolated hearts subjected to 6 h hypothermic (1–4°C) storage followed by 45 min reperfusion at 37°C were treated with zoniporide at different concentrations and timing. Recovery of cardiac function, levels of total and phosphorylated protein kinase B, extracellular signal-regulated kinase 1/2, glycogen synthase kinase-3β and STAT3 as well as cleaved caspase 3 were measured at the end of reperfusion. Lactate dehydrogenase release into coronary effluent before and post-storage was also measured.
KEY RESULTS
Zoniporide concentration-dependently improved recovery of cardiac function after reperfusion. The functional recovery induced by zoniporide was accompanied by up-regulation of p-extracellular signal-regulated kinase 1/2 and p-STAT3, and by reduction in lactate dehydrogenase release and cleaved caspase 3. There were no significant differences in any of the above indices when zoniporide was administered before, during or after ischemia. The STAT3 inhibitor, stattic, abolished zoniporide-induced improvements in functional recovery and up-regulation of p-STAT3 after reperfusion.
CONCLUSIONS AND IMPLICATIONS
Zoniporide is a potent cardioprotective agent and activation of STAT3 plays a critical role in the cardioprotective action of zoniporide. This agent shows promise as a supplement to storage solutions to improve preservation of donor hearts.
Keywords: zoniporide, ischemia-reperfusion injury, STAT3 pathway, sodium hydrogen exchange inhibitor, myocardial preservation
Introduction
Heart transplantation has become established as an effective therapy for patients with end-stage heart disease. Minimization of the ischemia-reperfusion injury incurred during organ procurement, storage and implantation is a key factor in improving short- and long-term outcomes of heart transplantation. This is achieved in current clinical practice by rapid, depolarizing arrest of the donor heart in a chilled specially formulated cardioplegic/storage solution such as St Thomas Solution (II) or Celsior followed by a period of static cold storage until the heart is reimplanted in the recipient (Hicks et al., 2006). However, these strategies are not without problems. The shift to more positive membrane potentials induced by hyperkalemic arrest leads to an initial increase in intracellular Na+ via a non-activating Na+ current that is exacerbated by activation of Na+/H+ exchange and inhibition of Na+/K+ ATPase under the conditions of ischemia and hypothermia. Importantly, these high intracellular Na+ levels cause an increase in intracellular Ca2+ via the Na+/Ca2+ exchanger operating in reverse mode (Fallouh et al., 2009). Two powerful approaches to minimize such high intracellular Ca2+ levels are the inhibition of the Na+/H+ exchanger (NHE) (Avkiran et al., 2001; Avkiran and Marber, 2002) and pharmacological activation of pro-survival signalling pathways (Murphy and Steenbergen, 2008).
Following the first report of the cardio-protective effects of NHE inhibition with amiloride (Karmazyn, 1988), numerous more specific and selective NHE inhibitors were developed (Karmazyn, 1999). Of those, HOE-642, more commonly known as cariporide, was the most effective and intensively studied and was shown to be cardioprotective in various animal models of myocardial ischemia-reperfusion (Karmazyn, 1999; Klein et al., 2000), in patients with acute myocardial infarction (Rupprecht et al., 2000) and in some high-risk patients undergoing coronary artery bypass grafting surgery (the GUARDIAN study) (Theroux et al., 2000).
In the setting of experimental cardiac arrest and preservation, cariporide was shown to improve post-ischemic cardiac function when added as a supplement to the St Thomas' cardioplegic solution or at reperfusion at 37°C, 28°C or 7.5°C (Shipolini et al., 1997). We have confirmed and extended these findings by showing that the inclusion of the combination of cariporide and glyceryl trinitrate in the arresting solution produced viable post-storage recovery of cardiac function in a rat isolated heart model after 10 h storage at 4°C (Gao et al., 2005). A role for pro-survival signalling was inferred from the inhibition of functional recovery by exposure of treated hearts to the adenosine triphosphate (ATP) sensitive potassium channel inhibitor, glibenclamide. These findings have recently been further verified in a translational model of clinical heart transplantation – a porcine orthotopic heart transplant model incorporating donor brain death. Here, donor hearts that were arrested and stored in Celsior supplemented with cariporide and glyceryl trinitrate could be successfully weaned from cardiopulmonary bypass after 14 h hypothermic storage (Hing et al., 2009).
An unexpectedly high fatal stroke rate in patients exposed to repeated high intravenous perioperative doses of cariporide (EXPEDITION trial) (Mentzer et al., 2008) has raised safety concerns regarding the clinical administration of cariporide and further development of the agent for clinical use has been prevented by the withdrawal of the compound from further testing in man. Zoniporide, a recently developed pyrazolylguanidine-based NHE inhibitor possesses greater potency and selectivity towards cardiac NHE (isoform 1), than cariporide (Marala et al., 2002). In line with the findings obtained with cariporide, zoniporide was shown to protect the myocardium during ischemia and reperfusion in several experimental settings including surgically relevant animal models of cardiopulmonary bypass (Knight et al., 2001; Clements-Jewery et al., 2004). An incidental finding during the pharmacological characterization of zoniporide was a significant binding affinity for µ and δ opioid receptors (Tracey et al., 2003). Such receptors have been shown to be responsible for transducing a number of the protective signalling pathways associated with the pre- and post-conditioning response (Zatta et al., 2008). However, no data on the cardioprotective effects of zoniporide in the setting of profound hypothermic storage as used in heart transplantation is as yet available.
Thus, the aims of the present study were to characterize the cardioprotective efficacy of zoniporide in an isolated working rat heart model of hypothermic myocardial preservation. We assessed specifically (i) the dose and timing of zoniporide administration that provides maximal recovery of cardiac function after 6 h hypothermic storage of the heart; (ii) the impact of zoniporide administration on myocardial damage and cell death as indicated by lactate dehydrogenase (LDH) release in the coronary effluent and by tissue levels of cleaved caspase 3; (iii) the levels of pro-survival signaling activity [extracellular signal-regulated kinase (ERK) 1/2, protein kinase B (Akt), STAT3 and glycogen synthase kinase (GSK)-3β phosphorylation], after zoniporide treatment; and (iv) to what extent activation of pro-survival signaling pathways played a role in zoniporide induced cardioprotection. Results from the current study indicated that zoniporide was a potent cardioprotective agent and activation of STAT3 pathway played a critical role in the cardioprotective action of zoniporide.
Methods
Animals
All animals received humane care in compliance with the guidelines set down by the National Health and Medical Research Council (Australia) and the ‘Guide for the Care and Use of Laboratory Animals’ (National Institutes of Health, Bethesda, MD) and all experimental procedures were approved by the Animal Ethics Committee of the Garvan Institute of Medical Research, Sydney, Australia (Animal Research Authority Ref Nos #03/24 and 06/25). Male Wistar rats weighing 320–380 g procured from the Animal Resource Centre (Canning Vale, Western Australia) were used in the present study.
Solution preparation
Perfusate
Krebs-Henseleit solution (37°C) was used for perfusion of the rat isolated hearts. Its composition was as follows: NaCl 118.0 mM; KCl 4.7 mM; MgSO4 1.2 mM; KH2PO4 1.2 mM; NaHCO3 25.0 mM; CaCl2 2.5 mM, glucose 11.0 mM. The components were dissolved in water purified through a MilliQ water purification system (Millipore, Australia). This solution was bubbled continuously with Carbogen (95% O2 and 5% CO2) at 37°C for an hour prior to the experiment. The resultant pH of the Krebs solution was between 7.3 and 7.4. The perfusate was filtered through an inline filter (5 µm pore size) during the course of pre-storage perfusion and post-storage reperfusion
Zoniporide
A fresh stock solution of 1 mM zoniporide was prepared on the day of the experiment and diluted as required into either Krebs buffer or Celsior to the desired concentration.
Stattic
Stattic was dissolved in 2 L of Krebs buffer to yield a final concentration of 10 µM. Stattic was added to the buffer during working mode perfusion.
Naloxone
Two ampoules (800 µg) of naloxone hydrochloride were added to 2 L of Krebs buffer (final concentration 1.1 µM naloxone). Naloxone was added to the buffer during working mode perfusion.
Langendorff and working rat heart preparations
A rat isolated working heart model of donor heart preservation previously described by us (Gao et al., 2005), was used in this study. Briefly, rats were anaesthetized with an intraperitoneal injection of ketamine (80 mg·kg−1) and xylazine (10 mg·kg−1). After bolus injection of 500 IU heparin into the renal vein, the heart was rapidly excised and arrested by immersion in ice-cold Krebs-Henseleit buffer. The aorta was cannulated, after which the heart was immediately perfused retrogradely in Langendorff mode with Krebs buffer at a hydrostatic pressure of 100 cm H2O. The warm ischemia time (i.e. between incision of diaphragm and cannulation of aorta) was kept to a minimum to prevent damage and/or preconditioning of the heart. The average warm ischemia time for all hearts was 3.08 ± 0.10 min. During Langendorff perfusion, a small incision was made in the left atrial appendage into which another cannula was inserted and tied off. This non-working preparation was run for 10 min and then converted to working mode by switching the supply of perfusate from the aorta to the left atrial cannula at a hydrostatic pressure of 15 cm H2O (pre-load). The working heart ejected perfusate via the aortic valve into the aortic cannula. The hydrostatic pressure in the aortic cannula was maintained at 100 cm H2O (after-load) throughout the working phase for all rat hearts.
Experimental protocol
Figure 1 illustrates the experimental timeline for the study. All hearts remained in working mode for 15 min prior to storage. After collection of baseline haemodynamic data at this time, the heart was arrested by infusion of ice-cold Celsior solution with or without supplements into the coronary circulation for 3 min from a reservoir 60 cm above the heart. All hearts were stored on ice (1–4°C) in 100 mL of the same solution for 6 h. At the conclusion of the storage period, hearts were remounted on the perfusion apparatus and reperfused in Langendorff mode for 15 min. After this initial stabilization, hearts were switched to working mode and functional measurements repeated at the end of 30 min. After completion of the functional measurements, rat heart tissue was rapidly collected by sectioning the ventricles horizontally into three parts. The apical section was discarded and the middle section was fixed in 4% paraformaldehyde for subsequent histological examination. The left ventricular free wall of the basal section was further dissected into 6–8 pieces and rapidly frozen in liquid nitrogen and then stored at −80°C for Western blot analysis. Coronary effluent was collected immediately after baseline pre-storage functional measurements were taken, then post-reperfusion at 1, 15 and 30 min after conversion to working mode then stored at −80°C until samples were batched for analysis.
Figure 1.
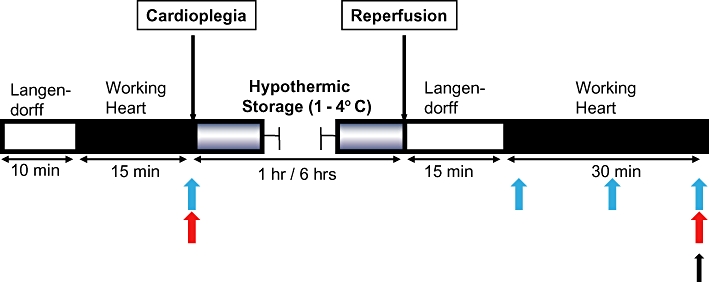
Experimental timeline. Blue arrows indicate times at which coronary effluent were collected; red arrows indicate times when measurement of cardiac functions were made; black arrow indicates the time point at which heart tissue was collected for Western blot and immunohistochemical analyses.
Functional measurements
Aortic pressure was monitored via a side arm of the aortic cannula with a pressure transducer (Ohmeda, Pty Ltd, Singapore). Aortic flow (AF) was measured by a flowmeter (Transonics Instruments Inc. Ithaca, NY, USA) in line with the aortic cannula. Aortic pressure and flow were recorded using PowerLab/4e (ADInstruments Pty Ltd, Sydney, Australia) and heart rate was derived from the AF trace. Coronary flow (CF) was measured by timed collection of the effluent draining from the apex of the heart. Cardiac output (CO) was derived from the sum of AF and CF. Measurements of functional indices made at 10 min after conversion to working mode during pre-storage perfusion were used as baseline. Any hearts having a baseline AF less than 35 mL·min−1, heart rate less than 200 beats·min−1, or CF less than 10 mL·min−1 were excluded from the experiment at this stage. At the end of the 30 min post-storage reperfusion, indices of cardiac function measured at baseline were then remeasured. Recovery of each parameter was expressed as a percentage of its pre-storage baseline value.
Experimental groups
The first series of experiments was designed to generate a concentration–response curve for the cardioprotective effect of zoniporide from which an optimum zoniporide concentration could be determined for further studies and to survey any direct effects of zoniporide on pre-storage cardiac function. Experimental groups were as follows (n = 6 hearts/group, cold storage time 6 h unless otherwise specified): Group 1: control group where hearts were not exposed to any supplement during the whole experimental procedure; Group 2: hearts were perfused with Krebs buffer containing 30 nM zoniporide prior to storage; Group 3: hearts perfused with Krebs buffer containing 100 nM zoniporide prior to storage; Group 4: hearts perfused with Krebs buffer containing 300 nM zoniporide prior to storage; Group 5: hearts perfused with Krebs buffer containing 1000 nM zoniporide prior to storage; Group 6: hearts perfused with Krebs buffer containing 3000 nM zoniporide prior to storage. The next experiments tested the effect of timing of the zonpioride supplement with the following: Group 7: hearts were arrested and stored in Celsior solution containing 1000 nM zoniporide; Group 8: hearts perfused with Krebs buffer containing 1000 nM zoniporide during post-storage reperfusion. The final experiments examined the modulation of the effect of zoniporide by the STAT3 inhibitor, stattic as follows: Group 9: hearts were perfused with Krebs buffer containing 10 µM stattic prior to storage; Group 10: hearts perfused with Krebs buffer containing 10 µM stattic during pre-storage perfusion followed by arrest and 6 h storage in Celsior solution containing 1000 nM zoniporide; Group 11: hearts perfused with Krebs buffer containing 1.1 µM naloxone during pre-storage perfusion followed by arrest and 6 h storage in Celsior solution containing 1000 nM zoniporide.
Assessment of cellular damage by LDH release into coronary effluent
Lactate dehydrogenase release into coronary effluent was measured with the TOX 7 assay kit (Sigma Aldrich, St Louis, MO, USA). Duplicate 75 µL aliquots were assayed according to the maker's instructions. The resultant absorbance of the tetrazolium product was measured at 492 nm in a 96-well plate reader. Values were normalized for CF at the time of collection.
Immunohistochemical detection of cleaved caspase 3
Rat hearts were fixed in 4% paraformaldehyde then embedded in paraffin blocks. Sections (5 µm thick) were cut and de-paraffinized in xylene then rehydrated through a series of decreasing concentrations of ethanol then phosphate-buffered saline. Antigens were retrieved by incubating sections in retrieval solution (pH 6.0, Novocastra, Newcastle Upon Tyne, UK) in a microwave oven for 4 min (‘high’ setting) then cooled for 20 min. Sections were then treated with 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity. Proteins were blocked using a Novolink Polymer Detection System (Novocastra, Newcastle Upon Tyne, UK). Sections were incubated in monoclonal antibody directed towards cleaved caspase 3 (Cell Signalling Technology, Danvers, MA, USA), diluted 1:50 at 4°C for 12 h followed by secondary antibody incubation. Primary antibody binding was detected with 3,3′-diaminobenzidine (DAB) and sections were counterstained with Harris haematoxylin, then dehydrated and coverslips applied. Slides were digitized with a Scanscope XT digital slide scanner (Aperio, Vista, CA, USA). Digital images were acquired using ImageScope programme (Aperio, version 10.0.36, Vista, CA, USA). Slides were viewed remotely using a desktop personal computer with the web-based ImageScope viewer. Ten representative images of each slide were acquired at 400× magnification for quantification of DAB staining, indication cleaved caspase 3, using Image J freeware (Version 1.41, National Institute of Mental Health, Bethesda, MA). This software incorporates a color deconvolution algorithm developed specifically for DAB, haematoxylin and eosin that allowed objective quantitation of staining intensity (Ruifrok and Johnston, 2001).
Western blot analysis
Samples of tissue (60 mg) from each heart were homogenized in ice-cold lysis buffer [150 mM NaCl, 50 mM Tris HCl, 1% Triton X-100, 1 mM sodium orthovanadate, 1 mM glycerophosphate, 5 mM dithiothreitol, 15% protease inhibitor cocktail (Roche complete ‘mini’; Roche Diagnostics, Australia), pH 7.4]. Samples were then centrifuged at 9400× g for 5 min at 4°C. Protein concentration of each lysate was measured using a Bradford assay kit (Pierce Biotechnology Inc, Rockford, IL, USA), with bovine serum albumin used as standard. Protein samples were boiled in sample loading buffer for 5 min before loading onto 8% SDS-polyacrylamide gels (30 µg per lane). After electrophoretic separation on a Protean III system (Bio – Rad Laboratories Pty. Ltd, Regents Park, Australia), proteins were transferred to polyvinylidene difluoride membrane (Millipore, Australia). Membranes were blocked for 1 h in Tris buffered saline (pH 7.4) which contained 1% BSA and 0.1% Tween −20. Membranes were probed overnight with rabbit polyclonal antibodies raised against total and phospho Akt (Ser 473), total and phospho ERK 1/2 (Thr 202/Tyr 204), total and phospho glycogen synthase kinase 3 beta (GSK-3β) (Ser 9) and total and phospho STAT3 (Tyr 705) or β-actin. The secondary antibody was a horseradish peroxidase-conjugated anti-rabbit IgG (GE Health Care Rydlemere, NSW, Australia). The protein bands were visualized using enhanced chemiluminescence (GE Health Care Rydlemere, NSW, Australia). Band intensities were quantified with Image J freeware (Version 1.41, National Institute of Mental Health, Bethesda, MA, USA) and normalized against a β-actin loading control.
Statistical analysis
All functional data and densitometric antibody intensities were expressed as mean ± SEM unless otherwise specified. Statistical analyses were performed on Statview for Windows, version 5.0.1 (SAS Institute Inc, Cary, NC, USA). Differences between groups were compared using one-way analysis of variance followed by Fisher's PLSD post hoc tests. P-values less than 0.05 were considered significant. EC50 value was obtained by nonlinear regression analysis, using Graph-Pad Prism software (version 5.0, La Jolla, CA, USA).
Materials
All chemicals unless otherwise stated were purchased from Sigma – Aldrich (St Louis, MO, USA) and were at least Analytical Reagent grade or equivalent. Ketamine was purchased from Parnell Laboratories (Alexandria, NSW, Australia), xylazine from Troy Laboratories (Smithfield, NSW, Australia) and heparin from Pfizer Pty Ltd (North Ryde, NSW, Australia). Celsior® solution, used for arresting and storing the heart (Menasche et al., 1994) was obtained from Genzyme (Naarden, the Netherlands). The formulation of Celsior is shown in online Table S1 for convenience. Naloxone (ampoules containing 400 µg naloxone hydrochloride in 1 mL of saline, pH 3.5) was sourced from Hospia, Australia Pty Ltd (Melbourne, Australia). Zoniporide was kindly provided by Pfizer Inc (Groton, CT, USA) under an independent external investigator agreement. All antibodies used for Western blot and immunochemistry assays were from Cell Signalling (Danvers, MA, USA). Channel, receptor and enzyme nomenclature in the present study conforms to that in Alexander et al.(2009).
Results
Baseline (pre-storage) cardiac function
Baseline cardiac functional parameters for experimental groups 1–11 are presented in Table 1. There were no statistical differences in any of the baseline cardiac functional parameters between all 11 groups. Importantly, neither zoniporide in the concentration range used (30–3000 nM) (Groups 2–6), 10 µM stattic (Groups 9 and 10) nor 1.1 µM naloxone (Group 11) had any direct effects on baseline cardiac function. Seven hearts were excluded from the study at this stage, because they failed to meet the functional entry criteria outlined in the Methods.
Table 1.
Baseline (pre-storage) values for indices of cardiac function
| Group number | Aortic flow (mL·min−1) | Coronary flow (mL·min−1) | Cardiac output (mL·min−1) | Heart rate (beats·min−1) | Number of hearts |
|---|---|---|---|---|---|
| 1 | 47 ± 2.1 | 19 ± 0.4 | 66 ± 2.3 | 259 ± 6.3 | 6 |
| 2 | 45 ± 2.6 | 18 ± 0.3 | 64 ± 2.4 | 227 ± 20.2 | 6 |
| 3 | 51 ± 4.5 | 17 ± 1.1 | 68 ± 5.5 | 238 ± 10.1 | 6 |
| 4 | 45 ± 2.4 | 18 ± 0.9 | 63 ± 2.8 | 254 ± 8.4 | 6 |
| 5 | 48 ± 3.2 | 19 ± 1.0 | 67 ± 3.7 | 246 ± 20.1 | 6 |
| 6 | 52 ± 2.4 | 19 ± 0.9 | 71 ± 2.7 | 232 ± 6.9 | 6 |
| 7 | 52 ± 4.2 | 18 ± 1.4 | 70 ± 5.4 | 227 ± 13.6 | 6 |
| 8 | 59 ± 2.1 | 18 ± 0.6 | 77 ± 2.2 | 239 ± 11.6 | 6 |
| 9 | 49 ± 1.5 | 20 ± 0.7 | 70 ± 1.2 | 239 ± 16.4 | 5 |
| 10 | 57 ± 4.1 | 20 ± 1.1 | 77 ± 5.2 | 246 ± 11.2 | 5 |
| 11 | 57 ± 3.8 | 19 ± 1.3 | 76 ± 3.4 | 238 ± 25.0 | 4 |
Effect of zoniporide on post-storage recovery of cardiac function
While recovery of control hearts not exposed to zoniporide was uniformly poor for all indices measured, the presence of zoniporide in the pre-storage perfusate resulted in a concentration-dependent increase in all parameters assessed after 6 h hypothermic storage (Figure 2A–D). Maximum recovery of all parameters was observed at 1000 nM zoniporide and increasing the zoniporide concentration to 3000 nM produced no significant increase in recovery over that observed at 1000 nM zoniporide. The calculated EC50 for zoniporide based on CO recovery was 176 nM. The optimum zoniporide concentration to achieve cardiac protection was deemed to be 1000 nM and this concentration was used in all further experiments.
Figure 2.
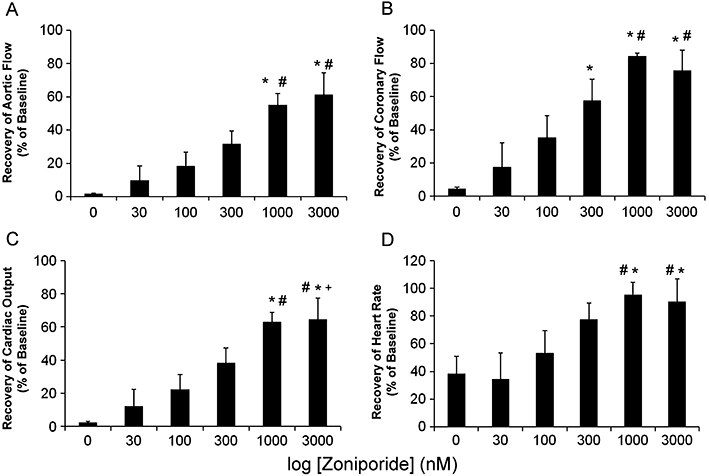
Cardioprotective efficacy of increasing concentrations of zoniporide applied during pre-storage perfusion indicated by recovery of aortic flow (A), coronary flow (B), cardiac output (C) and heart rate (D) after 45 min of reperfusion following 6 h hypothermic storage expressed as percentages of pre-storage baselines. Data are mean ± SEM (n = 6). *P < 0.05 versus Control; #P < 0.05 versus 30 nM; +P < 0.05 versus 100 nM. Calculated EC50 = 176 nM based on cardiac output recovery.
Effect of timing of zoniporide treatment
To examine the effect of timing of zoniporide exposure on post-storage recovery of cardiac function, recovery of hearts exposed to 1000 nM zoniporide added to the pre-storage perfusate was compared with recovery of hearts where 1000 nM zoniporide was added as a supplement to the arresting and storage solution or to the post-storage perfusate. Data in Figure 3 show that there were no significant differences between these three groups in post-storage recovery of any of the parameters measured.
Figure 3.
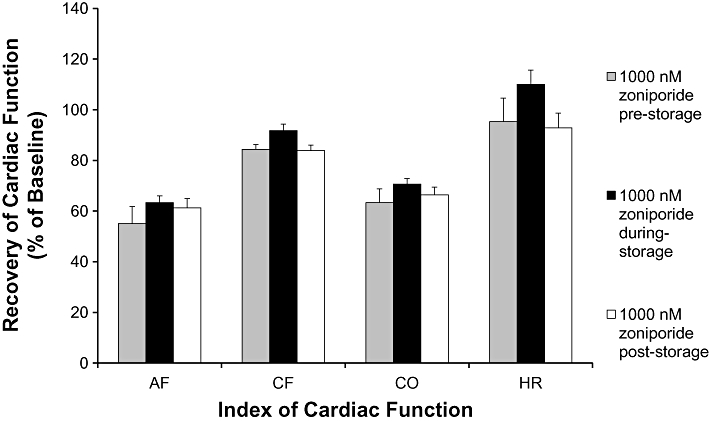
Effects of timing of zoniporide treatment at different phases of the experiment that is during pre-storage perfusion, during storage or at post-storage perfusion. Data are mean ± SEM of recoveries of aortic flow (AF), coronary flow (CF), cardiac output (CO) and heart rate (HR) after 45 min reperfusion expressed as percentages of pre-storage baselines (n = 6). No significant differences between recoveries of any of the functional indices between experimental groups. For recovery of Celsior alone (vehicle) control, refer to Figure 2.
Effect of zoniporide on indices of cardiac damage
Necrotic cellular damage
LDH levels in coronary effluent collected immediately prior to arrest and at intervals during reperfusion from hearts treated with 0–1000 nM zoniporide are shown in Figure 4A and B. Pre-arrest baseline LDH levels were minimal for all hearts with no significant differences between any of the groups. Reflecting the (non)recovery of cardiac contractile function observed in Figure 2, there was significant release of LDH from the control (untreated) hearts, reaching a 100-fold increase over basal levels 45 min after reperfusion (Figure 4A, P < 0.05, baseline vs. 45 min post-reperfusion). The presence of zoniporide produced a concentration-dependent decrease in coronary effluent levels of LDH (Figure 4A). In keeping with the functional results, the most significant reduction of LDH release was observed in the groups treated with 300 and 1000 nM zoniporide (Figure 4A, P < 0.01 vs. control). Indeed, LDH release in the 1000 nM zoniporide group 45 min after reperfusion was no different from its baseline (pre-ischemic) level. Consistent with independence of the timing of zoniporide treatment on functional improvement observed in Figure 3, there was no significant difference between coronary effluent levels of LDH from hearts exposed to 1000 nM zoniporide before, at arrest and during storage or at reperfusion (Figure 4B).
Figure 4.
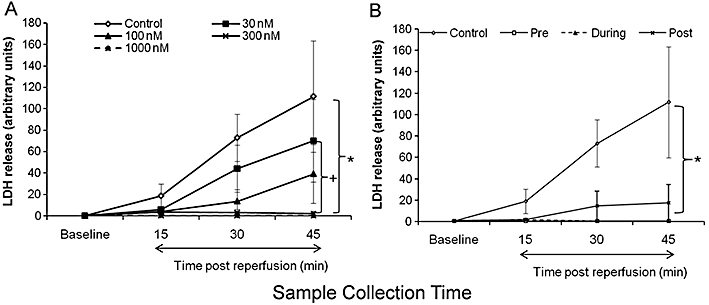
Effects of different concentrations and timing of zoniporide treatment on lactate dehydogenase (LDH) release. LDH levels in coronary effluents are expressed as absorbance at 492 nm divided by coronary flow then multiplied by 100. Data are mean ± SEM (n = 4–6). (A) Rat hearts treated with different doses of zoniporide during pre-storage perfusion. *P < 0.01 control versus 300 nM and 1000 nM, +P < 0.05 30 nM versus 1000 nM. (B) Rat hearts treated with 1000 nM zoniporide prior to cardioplegia (pre-storage), during storage (during-storage), and at reperfusion (post-storage). *P < 0.05 control versus all three treatment groups.
Apoptotic cellular damage
The presence of cleaved or activated caspase 3 is a definitive marker of apoptosis and a point of convergence for many pathological stimuli. It exists in cells as the inactive 35 kD procaspase that is cleaved to an active 17–20 kD peptide. Representative sections from hearts exposed to 0–1000 nM zoniporide collected at the end of 45 min reperfusion were stained with a primary monoclonal antibody directed towards cleaved caspase 3, that was visualized with brown staining (using the DAB reagent) (Figure 5B–F). Figure 5A, from a heart not exposed to any zoniporide was processed and stained with DAB in the absence of the primary (cleaved caspase 3) antibody to demonstrate the lack of non-specific binding of DAB. Control hearts showed extensive positive staining (Figure 5B) indicating increased accumulation of cleaved caspase 3 during post-storage reperfusion. Hearts exposed to 30 nM zoniporide during pre-storage perfusion also showed high levels of cleaved caspase 3 (Figure 5C). In hearts exposed to concentrations of zoniporide ≥100 nM during pre-storage perfusion, levels of cleaved caspase 3 were significantly reduced compared with control and 30 nM groups (Figure 5D–F). Quantitation of cleaved caspase 3-related DAB staining at higher power revealed that there were no significant differences in cleaved caspase3 levels between 100, 300 and 1000 nM groups (Figure 5G). We have demonstrated concordance of immunohistochemical detection of cleaved caspase 3 with the appearance of a protein band at 17 kD corresponding to cleaved caspase 3 by Western blotting in a previous study assessing the cardioprotective effects of the NHE inhibitor, cariporide in this model (Kwan et al., 2008).
Figure 5.
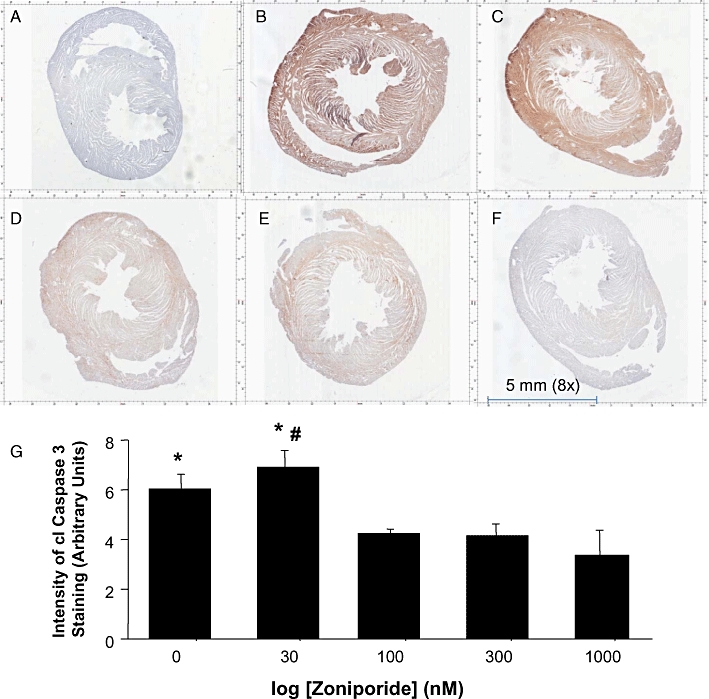
Effects of zoniporide applied during pre-storage perfusion on levels of cleaved caspase 3 after reperfusion of the heart. Shown here are representative photomicrographs of immunohistochemical staining of cleaved caspase 3 for (A) Negative control (No cleaved caspase 3 primary antibody; DAB only). Cleaved caspase 3 antibody present: (B) Control group (0 nM zoniporide); (C) 30 nM zoniporide; (D) 100 nM zoniporide; (E) 300 nM zoniporide; and (F) 1000 nM zoniporide. The dark brown color indicates positive staining of cleaved caspase 3. (G) Quantification of cleaved caspase 3 expressed as integrated density of positive staining acquired by Image J from ten randomized fields (400× magnification) of each heart section. Data are mean ± SEM. *P < 0.05 versus 1000 nM treatment; #P < 0.05 versus 100 and 300 nM.
Effects of zoniporide on pro-survival signalling
Figure 6 shows Western blots of a representative sample of hearts (6A–6D; upper panels) and quantified band intensities (lower panels), from experimental groups 1–5 (0–1000 nM zoniporide added to pre-storage perfusate), showing phosphorylation status of ERK 1/2 (Figure 6A), STAT3 (Figure 6B), Akt (Figure 6C) and GSK-3β (Figure 6D). The extent of phosphorylation of both ERK 1/2 and STAT3 was low in the absence or the presence of the lowest dose of zoniporide (30 nM), with a non-significant increase in phospho-ERK and phospho-STAT3 at 100 nM zoniporide. Phosphorylation of ERK was significantly increased at 300 nM zoniporide and was sustained at 1000 nM. The maximum extent of phosphorylation for STAT3 was observed at 300 nM zoniporide. There were no significant changes in the extent of phosphorylation of either Akt (Figure 6C), or GSK-3β (Figure 6D) with increasing zoniporide concentration at the time of sampling.
Figure 6.
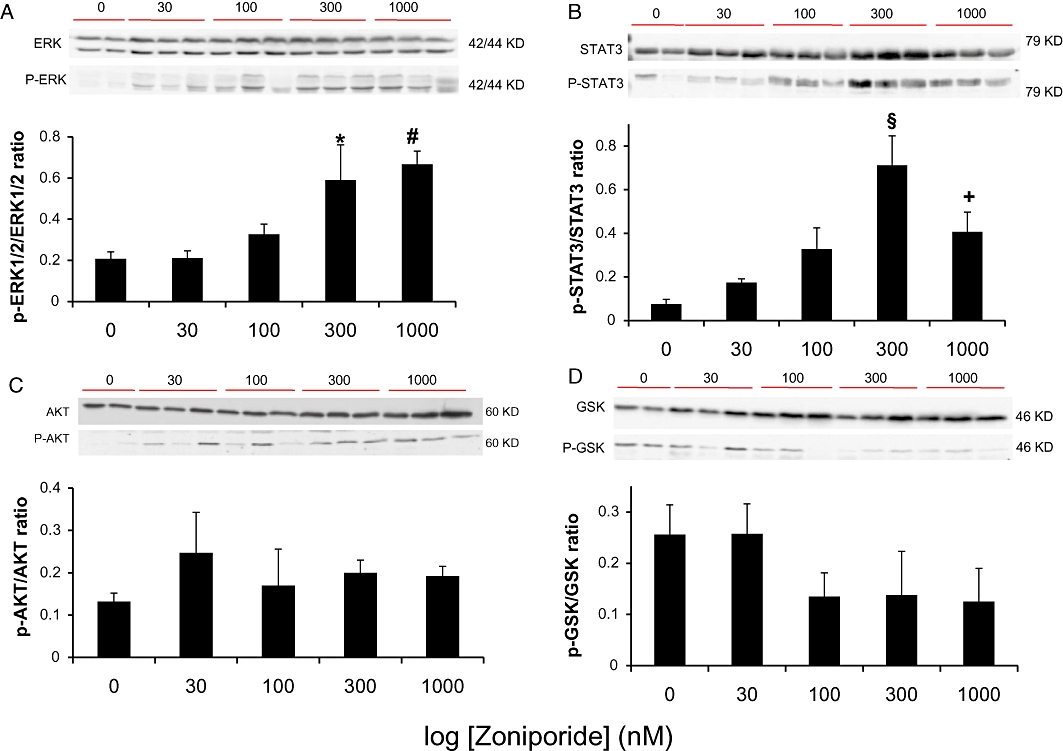
Effects of zoniporide applied during pre-storage perfusion on expressions of total and phospho-extracellular signal-regulated kinase (ERK) 1/2, STAT3, protein kinase B (Akt) and glycogen synthase kinase 3 beta (GSK-3β) after reperfusion of the heart. Representative Western blots of total and phospho protein levels and relative densitomertry expressed as ratios of phospho over total proteins are shown for (A) ERK 1/2 (B) STAT3 (C) Akt (D) GSK-3β. Data are mean ± SEM (n = 3–4). *P < 0.05 versus control and 30 nM; #P < 0.05 versus control, 30 nM and 100 nM. §P < 0.05 versus all other groups; +P < 0.05 versus control.
The extent of phosphorylation of these proteins was also compared in hearts exposed to 1000 nM zoniporide either before storage (Group 5), at arrest and during storage (Group 7) or at reperfusion (Group 8). There was no significant difference between the extent of phosphorylation of ERK 1/2 and STAT3 in hearts exposed to zoniporide before storage (above) compared to hearts exposed to zoniporide at arrest and during storage or at reperfusion (data not shown).
Effects of stattic, an inhibitor of STAT3 phosphorylation on the cardioprotective effect of zoniporide
As STAT3 phosphorylation was found to be increased in zoniporide treated hearts (Figure 6B) and activation of the STAT3 pathway has been recently shown to be another important pro-survival pathway implicated in protection against reperfusion injury (Lecour, 2009), we decided to investigate the potential cardioprotective role of STAT3 activation with the newly described specific inhibitor of activation and dimerization of STAT3, stattic (Schust et al., 2006). Stattic (at 100 µM) was first employed to good effect as a specific inhibitor of STAT3 in a murine isolated non-working heart model of myocardial ischemia-reperfusion injury (Goodman et al., 2008).
As there is no information on any possible direct effects of stattic on working heart models of reperfusion injury, we thought it prudent to exclude the possibility of any direct cardiotoxic effects of stattic in our model. To do this, the cold ischemic storage time was reduced to 1 h, a time after which hearts arrested and stored in celsior alone regain significant contractile function after reperfusion. The recovery of hearts arrested and stored in Celsior was compared to hearts pretreated with 10 µM stattic. The concentration of stattic used here was based on data from Schust et al. (2006). Post-storage functional recovery of these hearts is shown in Figure 7, where it can be seen that both groups of hearts recovered to an identical extent for all indices of cardiac function measured. Baseline (pre-storage) cardiac functional indices in hearts pretreated with 10 µM stattic are shown in Table 1 (Groups 9 and 10) were also not significantly different from any of the other groups. Thus, at this 10 µM concentration, stattic did not have any deleterious effect on baseline cardiac function or post-storage recovery.
Figure 7.
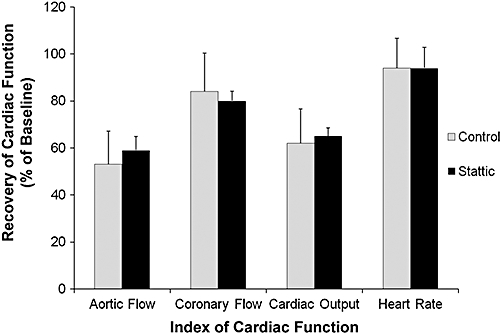
Comparison of cardiac functional recovery after 45 min reperfusion following 1 h hypothermic storage between hearts with Stattic or without (Control), during pre-storage perfusion. Data are mean ± SEM and expressed as percentages of pre-storage baselines. No significant differences between recoveries of any of the functional indices between experimental groups.
With this knowledge, we investigated the effect of stattic on the cardioprotective effect of zoniporide in hearts after 6 h of cold storage (Groups 9 and 10) (Figure 8A–C). Stattic pretreatment of hearts stored in Celsior supplemented with 1000 nM zoniporide abolished its protective effect for all indices of cardiac function examined. For simplicity, only the recovery of CO has been shown (Figure 8A). Pretreatment of zoniporide-stored hearts with 10 µM stattic completely abolished the protective effect of zonpioride and the release of LDH from the stattic + zoniporide treated hearts was also increased to levels not significantly different from control hearts in the absence of zoniporide (Figure 8B). In addition, Western blot analysis of total and phospho-STAT3 levels showed that the increased STAT3 phophorylation by zoniporide was abolished by pretreatment of the heart with stattic (Figure 8C, P < 0.05). Rat hearts pretreated with stattic followed by 6 h storage in Celsior solution did not differ from the control hearts in recovery of CO and LDH release (Figure 8A,B).
Figure 8.
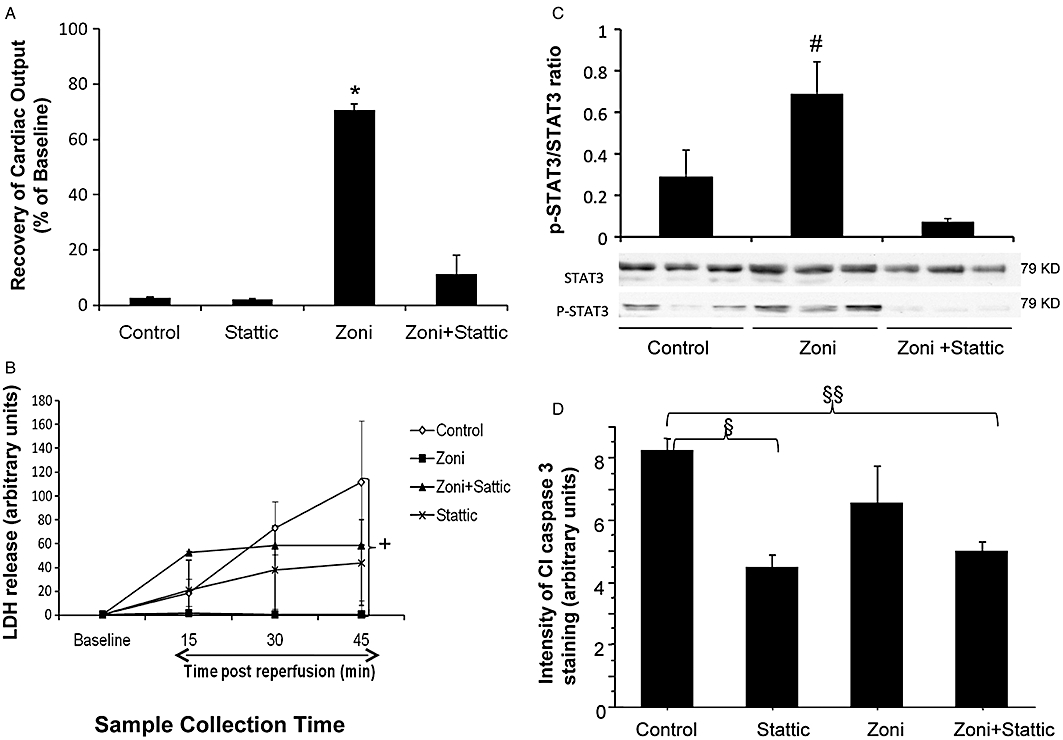
Effects of STAT3 inhibitor, stattic, on (A) Recovery of cardiac output after 6 h hypothermic storage; (B) lactate dehydogenase (LDH) release in coronary effluent; and (C) Levels of STAT3 phosphorylation; (D) Post-storage cleaved caspase 3 levels. Comparisons are between control group (hearts stored in Celsior solution without supplements), zoni group (hearts stored in Celsior solution supplemented with 1000 nM zoniporide), stattic group (hearts pretreated with 10 µM stattic then stored in Celsior solution without supplements), and zoni + stattic group (hearts pretreated with 10 µM stattic then stored in Celsior solution with 1000 nM zoniporide). Data are mean ± SEM (n = 5). *P < 0.005 versus all other groups; #P < 0.05 versus Control & zoniporide + static; +P < 0.05 control versus zoniporide; §P < 0.05 control versus stattic; §§P < 0.05 control versus zoniporide + stattic.
Figure 8D shows the effect of stattic on the tissue levels of cleaved (activated) caspase 3 staining in hearts after 6 h cold storage and 45 min reperfusion. Typical examples of immunohistochemical staining of cleaved caspase 3 in low power transverse sections of post-reperfused hearts are shown in the Supplementary Online Figure (Figure S1). Interestingly, exposure of the hearts to 10 µM stattic during pre-storage perfusion produced a significant decrease in cleaved caspase 3 levels compared to control hearts perfused with Krebs solution alone. This decrease was independent of whether or not the hearts were arrested and stored in 1000 nM zoniporide. Unlike hearts pretreated with 1000 nM zoniporide (Figure 5), the decrease in cleaved caspase 3 staining in hearts arrested and stored in 1000 nM zoniporide failed to reach significance (Figure 8D).
Effects of naloxone, a competitive inhibitor of µ, κ and δ opioid receptors on the cardioprotective effect of zoniporide
The possible involvement of opioid receptors in transducing the cardioprotective effect of zoniporide was tested by pretreating hearts to be arrested and stored in 1000 nM zoniporide with naloxone. The presence of naloxone did not significantly alter post-storage cardiac recovery of hearts arrested and stored in 1000 nM zoniporide (Table 2).
Table 2.
Effects of naloxone on post-storage recovery of cardiac function in hearts arrested and stored in 1000 nm zoniporide
| Treatment | Aortic flow | Coronary flow | Cardiac output | Heart rate |
|---|---|---|---|---|
| Zoniporide storage | 63 ± 2.6 | 92 ± 2.5 | 70 ± 2.1 | 110 ± 5.5 |
| Naloxone pretreatment/zoniporide storage | 60 ± 5.5 | 83 ± 4 | 65 ± 5 | 93 ± 4 |
Values shown in the Table are recovery values of each parameter expressed as % the pre-storage baseline value.
Discussion
The major findings from the present study showed that exposure of hearts to zoniporide resulted in: (i) a concentration- dependent recovery of post-storage cardiac function that was independent of the timing of zoniporide administration; (ii) a concentration- dependent decrease in both LDH release and tissue levels of cleaved caspase 3; (iii) an increase in ERK 1/2 and STAT3 phosphorylation at high zoniporide concentrations; and (iv) abolition of the protective effect of zoniporide by pretreating the hearts with the STAT3 inhibitor, stattic.
The present study demonstrates that zoniporide was a more potent cardioprotective agent than cariporide (HOE 642) in the isolated working heart model of donor heart preservation after 6 h hypothermic storage in aspartate-supplemented St Thomas' storage solution (Cropper et al., 2003). We have previously shown that the maximum cardioprotective concentration of cariporide in this model was 10 µM (Cropper et al., 2003), compared to 1 µM zoniporide in the present study (Figure 2). Further, maximal protection with cariporide, was not obtained until cariporide was present during pre-storage perfusion, arrest and storage and at reperfusion (Cropper et al., 2003), rather than during arrest and storage only, which is the case for zoniporide in the present study (Figure 3).
The principal mechanisms by which inhibitors of NHE-1 are thought to afford protective effects during myocardial ischemia and reperfusion are through the attenuation of intracellular Na+ and Ca2+ overload in cardiac myocytes and preservation of mitochondrial integrity (Karmazyn, 1999; Teshima et al., 2003). However, other important protective roles for NHE inhibitors have recently been identified including a reduction in inflammation (Buerke et al., 2008), a slow normalization of intracellular pH during the initial stages of reperfusion (Stromer et al., 2000), activation of mitochondrial ATP sensitive potassium channels (Miura et al., 2001), a decrease in mitochondrial permeability transition pore opening (Javadov et al., 2008) and decreased mitochondrial superoxide production (Garciarena et al., 2008).
There is also evidence that the NHE may be coupled to multiple intracellular signalling pathways (Xue et al., 2007), raising the possibility that multiple pathways may be involved in the prevention of ischemia reperfusion injury by NHE inhibitors. A number of studies suggest a central role of NHE in regulating the activity of mitogen-activated protein kinases, including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase and p38 MAPK, all believed to play a major role in the regulation of pivotal cellular processes such as cell survival/death balance (Rentsch et al., 2007; Pedersen et al., 2007a,b;). Also, recruitment of ezrin/radizin/moesin (ERM) and phosphoinositide 3-kinase-Akt has been proposed to be the mechanism by which NHE activation counteracted apoptotic cell death (Wu et al., 2004).
Effect of zoniporide on pro-survival signalling
Pro-survival signalling pathways involving ERK 1/2 and Akt and their down-stream target, GSK-3β have been shown to transduce cardiac protection against ischemia reperfusion injury by a number of pharmacological agents (Hausenloy and Yellon, 2007). Recently a growing body of work has indicated that the STAT3 pathway is also an important cardioprotective element, acting independently of ERK and Akt with both pathways postulated to target mitochondria through the permeability transition pore (Lacerda et al., 2009). In the present study, we examined the phosphorylation status ERK1/2, Akt, STAT3 and GSK-3β after 45 min reperfusion. We observed a dose-dependent increase in ERK1/2 phosphorylation that paralleled cardiac contractile recovery (Figure 2A–D) when rat hearts were perfused with increasing concentrations of zoniporide prior to 6 h storage (Figure 6A). Using the same experimental model we have also observed the same correlation between increased ERK1/2 phosphorylation and post-storage recovery of heart function when the structurally unrelated NHE inhibitor, cariporide, was employed (Kwan et al., 2008), suggesting that activation of ERK1/2 related signalling pathways is a class effect of NHE-1 inhibitors in this model. This observation is consistent with findings of Pedersen et al. (2007a) who showed that after osmotic cell shrinkage, ERK1/2 is inhibited in an NHE1-dependent and pH(i)-independent manner. Such shrinkage-related ERK1/2 inhibition was attenuated by NHE1 inhibitors.
There is increasing evidence that the Janus activated kinase-signal transducers and activators of transcription (JAK-STAT) signalling pathway also plays an important role in protection against ischemia reperfusion injury (Bolli et al., 2001; Butler et al., 2006; Gross et al., 2006; Lecour, 2009). The infarct-sparing and functional recovery observed after ischemic preconditioning (both immediate and delayed or ‘second window’) were associated with increased phosphorylation of STAT3 and could be abolished by prior treatment with the JAK2 inhibitor, AG490 (Hattori et al., 2001; Xuan et al., 2001). Pharmacological preconditioning, with growth factors and hormones, including insulin-like growth factor 1 (IGF1), erythropoietin and angiotensin II, has been shown to activate JAK-STAT family proteins through phosphorylation (Vignais et al., 1996; Negoro et al., 2000). Specifically, STAT3 has been shown to be essential in insulin and opioid-induced cardioprotection (Gross et al., 2006; Fuglesteg et al., 2008).
Post-conditioning of the hearts of young mice resulted in AG490-inhibitable STAT3 phosphorylation and infarct-sparing, while hearts from aged mice (>13 months) with lower levels of STAT3 as well as STAT3 knock-out mice were refractory to the post-conditioning stimulus (Boengler et al., 2008). In addition, the functional recovery of post-conditioned isolated (Langendorff) perfused hearts was associated with co-ordinated increases in the phosphorylation status of STAT3 and Akt (both inhibited by stattic (Goodman et al., 2008). Most recently, co-ordinated up-regulation of ERK 1/2 and STAT3 subsequent to activation of IL11 was observed as a result of pharmacological preconditioning of skeletal myoblast stem cells with diazoxide (Haider et al., 2010).
We have shown post-storage recovery of cardiac function was also associated with an increase in STAT3 phosphorylation in hearts exposed to zoniporide (Figure 6B). Interestingly, zoniporide was shown to bind to µ and δ opioid receptors at zoniporide concentrations employed in the present study (Tracey et al., 2003). Activation of these receptor subtypes has been implicated in the cardioprotective effects of pre- and post-conditioning ((Hausenloy and Yellon, 2007). Post-conditioning in a rabbit heart ischemia reperfusion model was recently found to be mediated through opioid receptor activation and JAK-STAT signalling (You et al., 2010), raising the possibility that opioid receptor activation may be responsible for the cardioprotective effects of zoniporide. However, this may not be the case in the present model, as pretreatment of hearts with the opioid antagonist, naloxone, failed to inhibit post-storage recovery of cardiac function after storage of these hearts in Celsior supplemented with 1000 nM zoniporide (Table 2).
To our knowledge, the present study is the first to demonstrate a critical role for the JAK-STAT pathway in the cardioprotective effects arising from inhibition of the NHE. We have shown that STAT3 phosphorylation was increased in rat hearts treated with zoniporide in concert with its cardioprotective effect. Previous studies have used the JAK inhibitor, AG-490 to infer a role for the JAK-STAT pathway, although reservations on the use of this agent have been expressed owing to the fact that it is not a specific inhibitor of STAT3 and it has inhibitory effects on other pro-survival pathways (Fuglesteg et al., 2008). In contrast, we have chosen to assess the cardioprotective role of STAT3 with a newly developed specific inhibitor of STAT3, stattic, which at the concentration employed in this study has been shown not to inhibit either ERK 1/2, Akt or STAT1 phosphorylation (Schust et al., 2006). We demonstrated that pre-storage exposure of hearts to this agent abolished the protective effect of zoniporide, as well as the ability of zoniporide to minimize cell damage measured by LDH release. In addition, zoniporide-associated STAT3 phosphorylation was also abolished (Figure 8A–C). Some studies have reported induction of apoptosis in cancer cell lines arising from inhibition of STAT3 (Turkson et al., 2005; Schust et al., 2006). Such a finding may compromise the interpretation of the inhibitory effects of stattic observed in Figure 8A. To rule out this likelihood, we have further demonstrated that recovery of cardiac function after 1 h of cold ischemic storage was not affected by the presence of 10 µM stattic (Figure 7).
Although no significant increases in the phosphorylation status of Akt and GSK-3β were shown here, the present findings do not necessarily rule out a role for them in mediating the cardioprotective effects of zoniporide. In the present study, we felt it important to demonstrate functional recovery in our hearts before we harvested the tissue, so tissue was taken 45 min after reperfusion. This was probably not the optimum time for assessment of pro-survival signalling. As has been emphasized recently, many phosphorylation events may be transient, reaching their maxima at shorter (less than 15 min post-reperfusion) rather than after longer times (Fan et al., 2009; 2010;).
Effect of zoniporide on indices of cell damage and cell death
Necrotic LDH release
Tritto and colleagues demonstrated that isolated rat hearts exposed to 7 µM cariporide during cardioplegia or at reperfusion had a threefold and 1.5-fold decrease in total LDH release, respectively, versus untreated control hearts (Tritto et al., 1998). In the present study, concentrations of zoniporide ≥ 300 nM abolished LDH release when given before or at arrest and during cold ischemic storage and a non-significant rise in LDH when zoniporide was given at reperfusion (Figure 4B).
Apoptosis
Previous studies have shown that pre-ischemic treatment of isolated rat hearts with the NHE inhibitor cariporide resulted in a decrease in TUNEL positive (apoptotic cells) after reperfusion (Chakrabarti et al., 1997). Here we measured cleaved caspase 3 as a marker of apoptosis and found significantly reduced levels of cleaved caspase 3 after reperfusion in rat hearts exposed to zoniporide concentrations above 100 nM, before arrest and storage (Figure 5A–G). In contrast, we observed a non-significant decrease in cleaved caspase 3 levels when 1000 nM zoniporide was present at arrest and during storage (Figure 8D). The decrease in the levels of cleaved caspase 3 may be secondary to ERK pathway activation. Such activation has previously been shown to inhibit pro-caspase 3 cleavage, although the mechanism of action of ERK was not elucidated at the time (Erhardt et al., 1999). Subsequently, it was shown that activated ERK was able to phosphorylate caspase 9 at Thr-125, a conserved MAP kinase consensus site. Phosphorylation at Thr-125 inhibited caspase 9 processing thus preventing pro-caspase 3 cleavage and activation (Allan et al., 2003).
An interesting incidental observation was the inhibition of caspase 3 cleavage when hearts were exposed to 10 µM stattic before arrest and storage (Figure 8D). As STAT3 activation and phosphorylation has been demonstrated to be associated with an anti-apoptotic phenotype (Negoro et al., 2000), a decrease in caspase 3 cleavage observed in the presence of profound inhibition of STAT3 phosphorylation (Figure 8C) may seem counterintuitive. However, this observation is concordant with that of Novosyadlyy et al. (2008), who failed to prevent IGF1 mediated protection from ER stress-associated apoptosis even after blocking Akt with wortmannin, ERK 1/2 with U1026 and STAT3 with stattic. Also, the ability of the ‘Michael acceptor’ properties of stattic to react with sulfhydryl residues on other proteins as inferred by McMurray (2006), in a commentary accompanying the first description of stattic also cannot be ruled out. A range of molecules with ‘Michael acceptor’ properties have been recently demonstrated to be potent inhibitors of caspases 3, 6 and 7 by reacting with active-site cysteine residues of these enzymes (see Chu et al., 2009).
Notwithstanding the observed decrease in caspase 3 cleavage, the presence of stattic significantly attenuated the post-storage functional recovery of hearts arrested and stored in zoniporide (Figure 8A), and increased LDH efflux in these hearts during reperfusion (Figure 8B). This finding is in line with a previous study that compared the mode of cell death in hearts subjected to injury prevented by the NHE inhibitor, cariporide, that found necrosis rather than apoptosis was the major mode of cell death (Otani et al., 2000).
In conclusion, results from our rat isolated heart model of donor heart preservation indicate that zoniporide is a potent cardioprotective agent that significantly improved donor heart functional recovery after 6 h hypothermic storage as well as reducing cardiomyocyte apoptosis and necrosis after reperfusion. The fact that maximal efficacy of the agent is achieved by adding it as a supplement to the arresting and storage solution has significant clinical implications for its use as zoniporide need only be added to the arresting and preservation solution, thus eliminating exposure of other donor organs to zoniporide and minimizing recipient exposure to the drug and any potential adverse effects it may cause (Mentzer et al., 2008).
Acknowledgments
This work has been supported bya grant from Australia National Heart Fundation given to Dr Mark Hicks (N° G 07S 3044). Authors also like to thank Dr Adam Hill for advice in use of Graph-Pad Prism software and Dr Andrew Jabbour for his help in use of Image J programme.
Glossary
Abbreviations
- AF
Aortic flow
- CF
Coronary flow
- CO
Cardiac output
- DAB
diaminobenzidine
- ERK
extracellular signal-regulated kinase
- GSK-3β
glycogen synthase kinase 3 beta
- JAK-STAT
janus activated kinase – signal transducers and activators of transcription
- LDH
lactate dehydrogenase
- NHE
sodium hydrogen exchanger
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Teaching Materials; Figs 1–8 as PowerPoint slide.
Figure S1 Typical low power photomocrographs of transverse sections of hearts showing the effect of pre-storage treatment with the STAT3 inhibitor, stattic, on the subsequent immunohistochemical deposition of cleaved caspase 3. Prestorage perfusate and storage compositions are as follows: a) Hearts perfused with Krebs solution then stored for 6 h in Celsior alone; b) Hearts perfused with Krebs solution containing 10 μM stattic then stored for 6 h in Celsior alone; c) Hearts perfused with Krebs solution, then stored for 6 h in Celsior containing 1000 nM zoniporide; d) Hearts perfused with Krebs solution containing 10 μM stattic then stored for 6 hours in Celsior containing 1000 nM zoniporide. Scale bar 5 mm; For quantitation of intensity of cleaved caspase 3 staining, refer to Figure 8D.
Table S1 Composition of Celsior arresting and preservation solution (Menasche et al., 1994)
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) Br J Pharmacol. (4th edn) 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003;5:647–654. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- Avkiran M, Marber MS. Na(+)/H(+) exchange inhibitors for cardioprotective therapy: progress, problems and prospects. J Am Coll Cardiol. 2002;39:747–753. doi: 10.1016/s0735-1097(02)01693-5. [DOI] [PubMed] [Google Scholar]
- Avkiran M, Gross G, Karmazyn M, Klein H, Murphy E, Ytrehus K. Na+/H+ exchange in ischemia, reperfusion and preconditioning. Cardiovasc Res. 2001;50:162–166. doi: 10.1016/s0008-6363(01)00228-0. [DOI] [PubMed] [Google Scholar]
- Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, et al. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res. 2008;102:131–135. doi: 10.1161/CIRCRESAHA.107.164699. [DOI] [PubMed] [Google Scholar]
- Bolli R, Dawn B, Xuan YT. Emerging role of the JAK-STAT pathway as a mechanism of protection against ischemia/reperfusion injury. J Mol Cell Cardiol. 2001;33:1893–1896. doi: 10.1006/jmcc.2001.1469. [DOI] [PubMed] [Google Scholar]
- Buerke U, Pruefer D, Carter JM, Russ M, Schlitt A, Prondzinsky R, et al. Sodium/hydrogen exchange inhibition with cariporide reduces leukocyte adhesion via P-selectin suppression during inflammation. Br J Pharmacol. 2008;153:1678–1685. doi: 10.1038/sj.bjp.0707647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KL, Huffman LC, Koch SE, Hahn HS, Gwathmey JK. STAT-3 activation is necessary for ischemic preconditioning in hypertrophied myocardium. Am J Physiol Heart Circ Physiol. 2006;291:H797–H803. doi: 10.1152/ajpheart.01334.2005. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Hoque AN, Karmazyn M. A rapid ischemia-induced apoptosis in isolated rat hearts and its attenuation by the sodium-hydrogen exchange inhibitor HOE 642 (cariporide) J Mol Cell Cardiol. 1997;29:3169–3174. doi: 10.1006/jmcc.1997.0561. [DOI] [PubMed] [Google Scholar]
- Chu W, Rothfuss J, Chu Y, Zhou D, Mach RH. Synthesis and in vitro evaluation of sulfonamide isatin Michael acceptors as small molecule inhibitors of caspase 6. J Med Chem. 2009;52:2188–2191. doi: 10.1021/jm900135r. [DOI] [PubMed] [Google Scholar]
- Clements-Jewery H, Sutherland FJ, Allen MC, Tracey WR, Avkiran M. Cardioprotective efficacy of zoniporide, a potent and selective inhibitor of Na+/H+ exchanger isoform 1, in an experimental model of cardiopulmonary bypass. Br J Pharmacol. 2004;142:57–66. doi: 10.1038/sj.bjp.0705749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cropper JR, Hicks M, Ryan JB, Macdonald PS. Cardioprotection by cariporide after prolonged hypothermic storage of the isolated working rat heart. J Heart Lung Transplant. 2003;22:929–936. doi: 10.1016/s1053-2498(02)00643-5. [DOI] [PubMed] [Google Scholar]
- Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol. 1999;19:5308–5315. doi: 10.1128/mcb.19.8.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallouh HB, Kentish JC, Chambers DJ. Targeting for cardioplegia: arresting agents and their safety. Curr Opin Pharmacol. 2009;9:220–226. doi: 10.1016/j.coph.2008.11.012. [DOI] [PubMed] [Google Scholar]
- Fan WJ, Genade S, Genis A, Huisamen B, Lochner A. Dexamethasone-induced cardioprotection: a role for the phosphatase MKP-1? Life Sci. 2009;84:838–846. doi: 10.1016/j.lfs.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Fan WJ, van Vuuren D, Genade S, Lochner A. Kinases and phosphatases in ischaemic preconditioning: a re-evaluation. Basic Res Cardiol. 2010;105:495–511. doi: 10.1007/s00395-010-0086-3. [DOI] [PubMed] [Google Scholar]
- Fuglesteg BN, Suleman N, Tiron C, Kanhema T, Lacerda L, Andreasen TV, et al. Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res Cardiol. 2008;103:444–453. doi: 10.1007/s00395-008-0728-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Hicks M, Macdonald PS. Improved preservation of the rat heart with celsior solution supplemented with cariporide plus glyceryl trinitrate. Am J Transplant. 2005;5:1820–1826. doi: 10.1111/j.1600-6143.2005.00967.x. [DOI] [PubMed] [Google Scholar]
- Garciarena CD, Caldiz CI, Correa MV, Schinella GR, Mosca SM, Chiappe de Cingolani GE, et al. Na+/H+ exchanger-1 inhibitors decrease myocardial superoxide production via direct mitochondrial action. J Appl Physiol. 2008;105:1706–1713. doi: 10.1152/japplphysiol.90616.2008. [DOI] [PubMed] [Google Scholar]
- Goodman MD, Koch SE, Fuller-Bicer GA, Butler KL. Regulating RISK: a role for JAK-STAT signaling in postconditioning? Am J Physiol Heart Circ Physiol. 2008;295:H1649–H1656. doi: 10.1152/ajpheart.00692.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross ER, Hsu AK, Gross GJ. The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK-3 beta. Am J Physiol Heart Circ Physiol. 2006;291:H827–H834. doi: 10.1152/ajpheart.00003.2006. [DOI] [PubMed] [Google Scholar]
- Haider KH, Idris NM, Kim HW, Ahmed RPH, Shujia J, Ashraf M. MicroRNA-21 is a key determinant in IL11/Stat3 anti-apoptotic signalling pathway in preconditioning of skeletal myoblasts. Cardiovasc Res. 2010;88:168–178. doi: 10.1093/cvr/cvq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori R, Maulik N, Otani H, Zhu L, Cordis G, Engelman RM, et al. Role of STAT3 in ischemic preconditioning. J Mol Cell Cardiol. 2001;33:1929–1936. doi: 10.1006/jmcc.2001.1456. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: united at reperfusion. Pharmacol Ther. 2007;116:173–191. doi: 10.1016/j.pharmthera.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Hicks M, Hing A, Gao L, Ryan J, Macdonald PS. Organ preservation. Methods Mol Biol. 2006;333:331–374. doi: 10.1385/1-59745-049-9:331. [DOI] [PubMed] [Google Scholar]
- Hing AJ, Watson A, Hicks M, Gao L, Faddy SC, McMahon AC, et al. Combining cariporide with glyceryl trinitrate optimizes cardiac preservation during porcine heart transplantation. Am J Transplant. 2009;9:2048–2056. doi: 10.1111/j.1600-6143.2009.02736.x. [DOI] [PubMed] [Google Scholar]
- Javadov S, Choi A, Rajapurohitam V, Zeidan A, Basnakian AG, Karmazyn M. NHE-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc Res. 2008;77:416–424. doi: 10.1093/cvr/cvm039. [DOI] [PubMed] [Google Scholar]
- Karmazyn M. Amiloride enhances postischemic ventricular recovery: possible role of Na+-H+ exchange. Am J Physiol. 1988;255:H608–H615. doi: 10.1152/ajpheart.1988.255.3.H608. [DOI] [PubMed] [Google Scholar]
- Karmazyn M. The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann N Y Acad Sci. 1999;874:326–334. doi: 10.1111/j.1749-6632.1999.tb09248.x. [DOI] [PubMed] [Google Scholar]
- Klein HH, Pich S, Bohle RM, Lindert-Heimberg S, Nebendahl K. Na(+)/H(+) exchange inhibitor cariporide attenuates cell injury predominantly during ischemia and not at onset of reperfusion in porcine hearts with low residual blood flow. Circulation. 2000;102:1977–1982. doi: 10.1161/01.cir.102.16.1977. [DOI] [PubMed] [Google Scholar]
- Knight DR, Smith AH, Flynn DM, MacAndrew JT, Ellery SS, Kong JX, et al. A novel sodium-hydrogen exchanger isoform-1 inhibitor, zoniporide, reduces ischemic myocardial injury in vitro and in vivo. J Pharmacol Exp Ther. 2001;297:254–259. [PubMed] [Google Scholar]
- Kwan J, Gao L, Macdonald PS, Hicks M. Protective effect of glyceryl trinitrate and cariporide in a model of donor heart preservation: activation of Erk1/2 pathway. Transplantation. 2008;86:391. [Google Scholar]
- Lacerda L, Somers S, Opie LH, Lecour S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc Res. 2009;84:201–208. doi: 10.1093/cvr/cvp274. [DOI] [PubMed] [Google Scholar]
- Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J Mol Cell Cardiol. 2009;47:32–40. doi: 10.1016/j.yjmcc.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Marala RB, Brown JA, Kong JX, Tracey WR, Knight DR, Wester RT, et al. Zoniporide: a potent and highly selective inhibitor of human Na(+)/H(+) exchanger-1. Eur J Pharmacol. 2002;451:37–41. doi: 10.1016/s0014-2999(02)02193-3. [DOI] [PubMed] [Google Scholar]
- McMurray JS. A new small-molecule Stat3 inhibitor. Chem Biol. 2006;13:1123–1124. doi: 10.1016/j.chembiol.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Menasche P, Termignon JL, Pradier F, Grousset C, Mouas C, Alberici G, et al. Experimental evaluation of Celsior, a new heart preservation solution. Eur J Cardiothorac Surg. 1994;8:207–213. doi: 10.1016/1010-7940(94)90117-1. [DOI] [PubMed] [Google Scholar]
- Mentzer RM, Jr, Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg. 2008;85:1261–1270. doi: 10.1016/j.athoracsur.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Miura T, Liu Y, Goto M, Tsuchida A, Miki T, Nakano A, et al. Mitochondrial ATP-sensitive K+ channels play a role in cardioprotection by Na+-H+ exchange inhibition against ischemia/reperfusion injury. J Am Coll Cardiol. 2001;37:957–963. doi: 10.1016/s0735-1097(00)01183-9. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negoro S, Kunisada K, Tone E, Funamoto M, Oh H, Kishimoto T, et al. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res. 2000;47:797–805. doi: 10.1016/s0008-6363(00)00138-3. [DOI] [PubMed] [Google Scholar]
- Novosyadlyy R, Kurshan N, Lann D, Vijayakumar A, Yakar S, LeRoith D. Insulin-like growth factor-1 protects cells from ER stress-induced apoptosis via enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ. 2008;15:1304–1317. doi: 10.1038/cdd.2008.52. [DOI] [PubMed] [Google Scholar]
- Otani H, Uchiyama T, Yamamura T, Nakao Y, Hattori R, Ninomiya H, et al. Effects of the Na+/H+ exchange inhibitor cariporide (HOE 642) on cardiac function and cardiomyocyte cell death in rat ischaemic-reperfused heart. Clin Exp Pharmacol Physiol. 2000;27:387–393. doi: 10.1046/j.1440-1681.2000.03248.x. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Darborg BV, Rasmussen M, Nylandsted J, Hoffmann EK. The Na+/H+ exchanger, NHE1, differentially regulates mitogen-activated protein kinase subfamilies after osmotic shrinkage in Ehrlich Lettre Ascites cells. Cell Physiol Biochem. 2007a;20:735–750. doi: 10.1159/000110434. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Darborg BV, Rentsch ML, Rasmussen M. Regulation of mitogen-activated protein kinase pathways by the plasma membrane Na+/H+ exchanger, NHE1. Arch Biochem Biophys. 2007b;462:195–201. doi: 10.1016/j.abb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Rentsch ML, Ossum CG, Hoffmann EK, Pedersen SF. Roles of Na+/H+ exchange in regulation of p38 mitogen-activated protein kinase activity and cell death after chemical anoxia in NIH3T3 fibroblasts. Pflugers Arch. 2007;454:649–662. doi: 10.1007/s00424-007-0233-3. [DOI] [PubMed] [Google Scholar]
- Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol. 2001;23:291–299. [PubMed] [Google Scholar]
- Rupprecht HJ, vom Dahl J, Terres W, Seyfarth KM, Richardt G, Schultheibeta HP, et al. Cardioprotective effects of the Na(+)/H(+) exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation. 2000;101:2902–2908. doi: 10.1161/01.cir.101.25.2902. [DOI] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Shipolini AR, Galinanes M, Edmondson SJ, Hearse DJ, Avkiran M. Na+/H+ exchanger inhibitor HOE-642 improves cardioplegic myocardial preservation under both normothermic and hypothermic conditions. Circulation. 1997;96:II-266–II-273. [PubMed] [Google Scholar]
- Stromer H, de Groot MC, Horn M, Faul C, Leupold A, Morgan JP, et al. Na(+)/H(+) exchange inhibition with HOE642 improves postischemic recovery due to attenuation of Ca(2+) overload and prolonged acidosis on reperfusion. Circulation. 2000;101:2749–2755. doi: 10.1161/01.cir.101.23.2749. [DOI] [PubMed] [Google Scholar]
- Teshima Y, Akao M, Jones SP, Marban E. Cariporide (HOE642), a selective Na+-H+ exchange inhibitor, inhibits the mitochondrial death pathway. Circulation. 2003;108:2275–2281. doi: 10.1161/01.CIR.0000093277.20968.C7. [DOI] [PubMed] [Google Scholar]
- Theroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, et al. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation. 2000;102:3032–3038. doi: 10.1161/01.cir.102.25.3032. [DOI] [PubMed] [Google Scholar]
- Tracey WR, Allen MC, Frazier DE, Fossa AA, Johnson CG, Marala RB, et al. Zoniporide: a potent and selective inhibitor of the human sodium-hydrogen exchanger isoform 1 (NHE-1) Cardiovasc Drug Rev. 2003;21:17–32. doi: 10.1111/j.1527-3466.2003.tb00103.x. [DOI] [PubMed] [Google Scholar]
- Tritto FP, Inserte J, Garcia-Dorado D, Ruiz-Meana M, Soler-Soler J. Sodium/hydrogen exchanger inhibition reduces myocardial reperfusion edema after normothermic cardioplegia. J Thorac Cardiovasc Surg. 1998;115:709–715. doi: 10.1016/S0022-5223(98)70337-X. [DOI] [PubMed] [Google Scholar]
- Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J Biol Chem. 2005;280:32979–32988. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- Vignais ML, Sadowski HB, Watling D, Rogers NC, Gilman M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol Cell Biol. 1996;16:1759–1769. doi: 10.1128/mcb.16.4.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KL, Khan S, Lakhe-Reddy S, Jarad G, Mukherjee A, Obejero-Paz CA, et al. The NHE1 Na+/H+ exchanger recruits ezrin/radixin/moesin proteins to regulate Akt-dependent cell survival. J Biol Chem. 2004;279:26280–26286. doi: 10.1074/jbc.M400814200. [DOI] [PubMed] [Google Scholar]
- Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci USA. 2001;98:9050–9055. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Zhou D, Yao H, Gavrialov O, McConnell MJ, Gelb BD, et al. Novel functional interaction between Na+/H+ exchanger 1 and tyrosine phosphatase SHP-2. Am J Physiol Regul Integr Comp Physiol. 2007;292:R2406–R2416. doi: 10.1152/ajpregu.00859.2006. [DOI] [PubMed] [Google Scholar]
- You L, Li L, Xu Q, Ren J, Zhang F. Postconditioning reduces infarct size and cardiac myocyte apoptosis via the opioid receptor and JAK-STAT signaling pathway. Mol Biol Rep. 2010 doi: 10.1007/s11033-010-0126-y. doi: 10.1007/s11033-010-0126-y. [DOI] [PubMed] [Google Scholar]
- Zatta AJ, Kin H, Yoshishige D, Jiang R, Wang N, Reeves JG, et al. Evidence that cardioprotection by postconditioning involves preservation of myocardial opioid content and selective opioid receptor activation. Am J Physiol Heart Circ Physiol. 2008;294:H1444–H1451. doi: 10.1152/ajpheart.01279.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.