

Abstract
BACKGROUND AND PURPOSE
Severe acute pancreatitis (SAP) is characterized by trypsinogen activation, infiltration of leucocytes and tissue necrosis but the intracellular signalling mechanisms regulating organ injury in the pancreas remain elusive. Rho-kinase is a potent regulator of specific cellular processes effecting several pro-inflammatory activities. Herein, we examined the role of Rho-kinase signalling in acute pancreatitis.
EXPERIMENTAL APPROACH
Pancreatitis was induced by infusion of taurocholate into the pancreatic duct in C57BL/6 mice. Animals were treated with a Rho-kinase inhibitor Y-27632 (0.5–5 mg·kg−1) before induction of pancreatitis.
KEY RESULTS
Taurocholate infusion caused a clear-cut increase in blood amylase, pancreatic neutrophil infiltration, acinar cell necrosis and oedema formation in the pancreas. Levels of pancreatic myeloperoxidase (MPO), macrophage inflammatory protein-2 (MIP-2), trypsinogen activation peptide (TAP) and lung MPO were significantly increased, indicating local and systemic disease. Inhibition of Rho-kinase activity dose-dependently protected against pancreatitis. For example, 5 mg·kg−1 Y-27632 reduced acinar cell necrosis, leucocyte infiltration and pancreatic oedema by 90%, 89% and 58%, respectively, as well as tissue levels of MPO by 75% and MIP-2 by 84%. Moreover, Rho-kinase inhibition decreased lung MPO by 75% and blood amylase by 83%. Pancreatitis-induced TAP levels were reduced by 61% in Y-27632-treated mice. Inhibition of Rho-kinase abolished secretagogue-induced activation of trypsinogen in pancreatic acinar cells in vitro.
CONCLUSIONS AND IMPLICATIONS
Our novel data suggest that Rho-kinase signalling plays an important role in acute pancreatitis by regulating trypsinogen activation and subsequent CXC chemokine formation, neutrophil infiltration and tissue injury. Thus, these results indicate that Rho-kinase may constitute a novel target in the management of SAP.
Keywords: amylase, chemokines, inflammation, leucocytes, macrophage inflammatory protein-2, pancreas, trypsinogen
Introduction
The clinical course of acute pancreatitis includes a wide spectrum of presentations from simple and transient pain to development of local and systemic complications (Andersson et al., 2007). At present, there is no useful method to predict the severity and outcome of acute pancreatitis. Despite substantial investigative efforts, there is still no specific therapy available against acute pancreatitis and treatment is mainly limited to supportive care, which is partly related to an incomplete understanding of the underlying pathophysiology. In general, trypsinogen activation, inflammation and impaired microvascular perfusion have been implicated in the pathophysiology of pancreatitis (Wang et al., 2009; Zhang et al., 2009). Considering that trypsinogen activation seems to be an early and temporary process, inflammation in the pancreas persists longer and might be a more favourable target for specific therapeutic interventions (Regner et al., 2008). Tissue accumulation of leucocytes constitutes a hallmark of inflammation and numerous studies have documented a critical role of leucocyte recruitment in the pathophysiology of acute pancreatitis (Glasbrenner and Adler, 1993; Bhatia et al., 2000; Granger and Remick, 2005; Ryschich et al., 2009). Activation and tissue navigation of leucocytes are coordinated by secreted chemokines (Bacon and Oppenheim, 1998). The chemokine family is subdivided into two main groups (CC and CXC) based on structural properties. In the mouse, the CXC chemokine family includes macrophage inflammatory protein-2 (MIP-2), which is known to be a murine homologue of human growth-related oncogenic chemokines (Tekamp-Olson et al., 1990). MIP-2 is considered to predominately attract neutrophils and has been implicated as an important mediator of several severe conditions, such as endotoxaemia-induced lung and liver injury (Li et al., 2004; Mangalmurti et al., 2009), glomerulonephritis (Feng et al., 1995), bacterial meningitis (Klein et al., 2006) and hepatic ischaemia-reperfusion (Monson et al., 2007). Indeed, one previous study has shown that MIP-2 may also be an important regulator of neutrophil infiltration in the pancreas (Pastor et al., 2003). Although, the role of specific chemoattractants in leucocyte infiltration in the pancreas is relatively well described, the understanding of the signalling pathways orchestrating pro-inflammatory actions in the pancreas is limited.
Extracellular stress signals, such as ischaemia and infection, initiate intracellular signalling cascades that converge on specific transcription factors regulating gene expression of pro-inflammatory mediators. This signal transmission is largely regulated by intracellular kinases phosphorylating down-stream targets (Itoh et al., 1999). For example, small (∼21 kDa) guanosine triphosphatases of the Ras-homologus (Rho) family and one of their effectors, Rho-kinase, are known to act as molecular switches regulating numerous important cellular functions, such as cytoskeleton organization, cell adhesion, migration, reactive oxygen species formation and oncogenic transformation (Itoh et al., 1999; Alblas et al., 2001; Slotta et al., 2006). Notably, Rho-kinase inhibitors have been demonstrated to ameliorate reperfusion and endotoxaemic injury in the liver (Slotta et al., 2008) as well as protecting against tissue fibrosis (Kitamura et al., 2007), obstructive cholestasis (Laschke et al., 2008), cerebral and intestinal ischaemia (Shin et al., 2007; Santen et al., 2010) and pulmonary hypertension (Oka et al., 2008). However, the role of the Rho-kinase signalling in regulating trypsinogen activation, leucocyte recruitment and tissue injury in acute pancreatitis is not known.
Based on the above, we hypothesized that Rho-kinase signalling may play an important role in acute pancreatitis. We used a new experimental model of severe acute pancreatitis (SAP) in mice and interfered with Rho-kinase activity by administration of Y-27632, a specific Rho-kinase inhibitor.
Methods
Animals
All experiments were done in accordance with the legislation on the protection of animals and were approved by the Regional Ethical Committee for animal experimentation at Lund University, Sweden. Male C57BL/6 mice weighing 20–26 g (6–8 weeks) were maintained in a climate-controlled room at 22°C and exposed to a 12:12 h light-dark cycle. Animals were fed standard laboratory diet and given water ad libitum. Mice were anaesthetized by i.p. administration of 7.5 mg of ketamine hydrochloride (Hoffman-La Roche, Basel, Switzerland) and 2.5 mg of xylazine (Janssen Pharmaceutica, Beerse, Belgium) 100 g−1 body weight in 200 µL saline.
Experimental model of taurocholate-induced pancreatitis
The second part of duodenum and papilla of Vater was identified through a small (1–2 cm) upper midline incision. Traction sutures (7-0 prolene) were placed 1 cm from the papilla. A small puncture was made through the duodenal wall in parallel to the papilla of Vater with a 23G needle. A non-radiopaque polyethylene catheter (ID 0.28 mm) connected to a microinfusion pump (CMA/100, Carnegie Medicin, Stockholm, Sweden) was inserted through the punctured hole in the duodenum, via the papilla of Vater and 1 mm into the common bile duct. The common hepatic duct was identified at the liver hilum and clamped with a neurobulldog clamp. 10 µL of either 5% sodium taurocholate (Sigma-Aldrich, USA) or sodium chloride (0.9%) was infused for 5 min. Then the catheter was withdrawn and the common hepatic duct clamp was removed. The duodenal puncture closed by a purse-string suture (7-0 monofilament). The traction sutures were removed and abdomen was closed in two layers. Animals were allowed to wake up and given free access to food and water. Sham-operated animals underwent the same procedure without any infusion into the pancreas. Vehicle or the Rho-kinase inhibitor, Y-27632 [(R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide; Calbiochem, San Diego, USA], was given (0.5–5 mg·kg−1) i.p. 30 min prior to bile duct cannulation.
In separate experiments animals were treated with 5 mg·kg−1 Y-27632 2 h after taurocholate challenge. Animals were killed 24 h after the induction of pancreatitis. One group of mice received 5 mg·kg−1 Y-27632 alone without bile duct cannulation. Blood was collected from the tail vein for systemic leucocyte differential counts and determination of blood amylase levels. Blood samples were also collected from the inferior vena cava for flow cytometric studies of neutrophils. Pancreatic tissue was removed and kept in two pieces; one piece was snap-frozen in liquid nitrogen for biochemical analysis of myeloperoxidase (MPO), trypsinogen activating peptide (TAP) and MIP-2 and the other piece was fixed in formalin for later histological analysis.
Systemic leucocyte counts
Tail vein blood was mixed with Turks solution (0.2 mg gentian violet in 1 mL glacial acetic acid, 6.25% v/v) in a 1:20 dilution. Leucocytes were identified as monomorphonuclear (MNLs) and polymorphonuclear (PMNLs) cells in a Burker chamber.
Blood amylase
Amylase was quantified in blood with a commercially available assay (Reflotron®, Roche Diagnostics GmbH, Mannheim, Germany).
MPO assay
Frozen pancreatic tissue was preweighed and homogenized in 1 mL mixture (4:1) with phosphate-buffered saline and aprotinin 10 000 KIE mL−1 (Trasylol®, Bayer HealthCare AG, Leverkusen, Germany) for 1 min. The homogenate was centrifuged (15339×g, 10 min) and the supernatant was stored at −20°C and the pellet was used for MPO assay as previously described (Laschke et al., 2007). In brief, the pellet was mixed with 1 mL of 0.5% hexadecyltrimethylammonium bromide. Next, the sample was frozen for 24 h and then thawed, sonicated for 90 s, put in a water bath set at 60°C for 2 h, after which the MPO activity of the supernatant was measured. The enzyme activity was determined spectrophotometrically as the MPO-catalyzed change in absorbance in the redox reaction of H2O2 (450 nm, with a reference filter 540 nm, 25°C). Values are expressed as MPO units g−1 tissue.
Flow cytometry
For analysis of Mac-1 and CXCR2 expression on circulating neutrophils, blood was collected into syringes prefilled with 1:10 acid citrate dextrose at 24 h post taurocholate challenge. Immediately after collection, blood samples were incubated with an anti-CD16/CD32 antibody blocking Fcγ III/II receptors to reduce non-specific labelling for 10 min at room temperature and then incubated with FITC-conjugated anti-Mac-1 (clone M1/70, Integrin αM chain, rat IgG2b), APC-conjugated anti-Gr-1 (clone RB6-8C5, Rat IgG2b) and PerCP Cy5.5-conjugated anti-mouse CD182 (CXCR2) (clone TG11/CXCR2, rat IgG2a, Biolegend, San Diego, CA, USA) antibodies. Cells were fixed with 1% formaldehyde solution, erythrocytes were lysed using red blood cell lysing buffer (Sigma Chemical Co., St. Louis, MO, USA) and neutrophils were recovered following centrifugation. Flow-cytometric definition of neutrophils was based on Gr-1+ cells in the neutrophil population of cells based on forward and side scatter characteristics on a FACSort flow cytometer (Becton Dickinson, Mountain View, CA, USA). A viable gate was used to exclude dead and fragmented cells.
MIP-2 levels
Tissue levels of MIP-2 were determined in stored supernatant from homogenized pancreatic tissue by using double-antibody Quantikine enzyme linked immunosorbent assay kits (R & D Systems Europe, Abingdon, UK) using recombinant murine MIP-2 as standard. The minimal detectable protein concentration is less than 0.5 pg·mL−1.
Histology
Pancreas samples were fixed in 4% formaldehyde phosphate buffer overnight and then dehydrated and paraffin embedded. Six micrometer sections were stained (haematoxylin and eosin) and examined by light microscopy. The severity of pancreatitis was evaluated in a blinded manner by use of a preexisting scoring system including oedema, acinar cell necrosis and neutrophil infiltrate on a 0 (absent) to 4 (extensive) scale, as previously described in detail (Schmidt et al., 1992).
Radioimmunoassay
RIA was performed as described previously (Lindkvist et al., 2008). A 0.1 M Tris HCl buffer (pH 7.5) containing 0.15 M NaCl, 0.005 M EDTA and 2 g·L−1 bovine serum albumin (Sigma, St Louis, USA) was used as assay buffer. Samples of 100 µL diluted in assay buffer were incubated (16 h, 4°C) with 200 µL of [125 I]-Tyr-TAP ( = 20 000 counts min−1) in assay buffer and 200 µL of antiserum diluted 1/750 in assay buffer. Parallel incubations with the synthetic activation peptide TAP diluted in assay buffer in a series of concentrations from 0.078 to 20 nM, were used as standards in the assays. Free and bound radioactivities were separated by means of a second step antibody precipitation; 100 µL of a cellulose coupled anti-mouse IgG suspension (Sc-Cel® IDA, Boldon, England) was added to the samples. After 30 min of incubation, 1 mL of water was added and tubes were centrifuged (704×g, 5 min, room temperature). The supernatant was decanted and radioactivity of the precipitate was counted in a γ-spectrophotometer (1 min).
Tryspsinogen activation in isolated acinar cells
Pancreatic acini cells were prepared by collagenase digestion and gentle shearing as described previously (Saluja et al., 1999). Cells were suspended in HEPES-Ringer buffer (pH 7.4) saturated with O2 and passed through a 150 µm cell strainer (Partec, England). Isolated acinar cells (1 × 107 cells per well) were preincubated with vehicle or Y-27632 (100 nM, 10 min) and stimulated with 100 nM cerulein (37°C, 30 min). The buffer was then discarded and the cells were washed twice with buffer (pH 6.5) containing 250 mM sucrose, 5 mM 3-(morpholino) propanesulphonic acid (MOPS) and 1 mM MgSO4. The cells were next homogenized in cold (4°C) MOPS buffer using a potter Elvejham-type glass homogenizer. The resulting homogenate was centrifuged (56×g, 5 min), and the supernatant was used for assay. Trypsin activity was measured flourometrically using Boc-Glu-Ala-Arg-MCA as the substrate as described previously (Kawabata et al., 1988). For this purpose, a 200 µL aliquot of the acinar cell homogenate was added to a cuvette containing assay buffer (50 mM Tris, 150 mM NaCl, 1 mM CaCl2 and 0.1% BSA, pH 8.0). The reaction was initiated by the addition of substrate, and the fluorescence emitted at 440 nm in response to excitation at 380 nm was monitored. Trypsin levels (pg·mL−1) were calculated using a standard curve generated by assaying purified trypsin. Viability of the pancreatic acinar cells was higher than 95% as determined by trypan blue dye exclusion.
Statistics
Data are presented as mean values ± SEM. Statistical evaluations were performed using Kruskal-Wallis one-way analysis of variance on ranks followed by multiple comparisons versus control group (Dunnett's method). P < 0.05 was considered significant, and n represents the number of animals.
Results
Rho-kinase activity regulates tissue damage in pancreatitis
To study the role of Rho-kinase, we first examined blood amylase levels as an indicator of tissue damage in SAP. It was found that retrograde infusion of sodium taurocholate into the pancreatic duct enhanced blood amylase levels by nearly 17-fold (Figure 1, P < 0.05 vs. sham, n = 5−7). Administration of the Rho-kinase inhibitor Y-27632 reduced taurocholate-provoked levels of blood amylase from 834.4 ± 117.3 µKat·L−1 down to 141.2 ± 28.5 µKat·L−1, corresponding to an 83% reduction (Figure 1, P < 0.05 vs. vehicle + taurocholate, n = 5–7). Morphological examination revealed that pancreas tissue from control animals had a normal microstructure (Figure 2, n = 5–7), whereas taurocholate challenge caused severe destruction of the pancreatic tissue structure characterized by extensive acinar cell necrosis, oedema and massive infiltration of neutrophils (Figure 2, n = 5–7). It was observed that Rho-kinase inhibition protected against taurocholate-induced destruction of the tissue structure (Figure 2, n = 5–7). For example, inhibition of Rho-kinase activity decreased taurocholate-induced acinar cell necrosis by 90% and oedema by 58% in the pancreas (Figure 3A and B, P < 0.05 vs. vehicle + taurocholate, n = 5–7). Indeed, the number of circulating MNL and neutrophils increased in SAP, indicating systemic activation in this model (Table 1). Rho-kinase inhibition reversed systemic changes in leucocyte differential counts towards baseline levels in controls (Table 1). Notably, administration of 5 mg·kg−1 Y-27632 (n = 6) after induction of pancreatitis had no effect on taurocholate-induced acinar cell necrosis, oedema or infiltration of neutrophils in pancreas (not shown).
Figure 1.
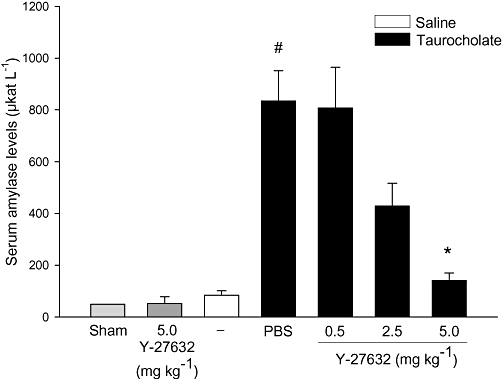
Blood amylase (µKat·L−1) in sham and control animals infused with saline alone into the pancreatic duct. Animals were treated with PBS or the Rho-kinase inhibitor Y-27632 (0.5–5.0 mg·kg−1) before infusion with sodium taurocholate into the pancreatic duct. One group of mice was given 5 mg·kg−1 Y-27632 alone without bile duct cannulation. Blood samples were obtained after 24 h. Data represent means ± SEM and n = 5–7. #P < 0.05 versus sham and *P < 0.05 versus PBS + taurocholate.
Figure 2.
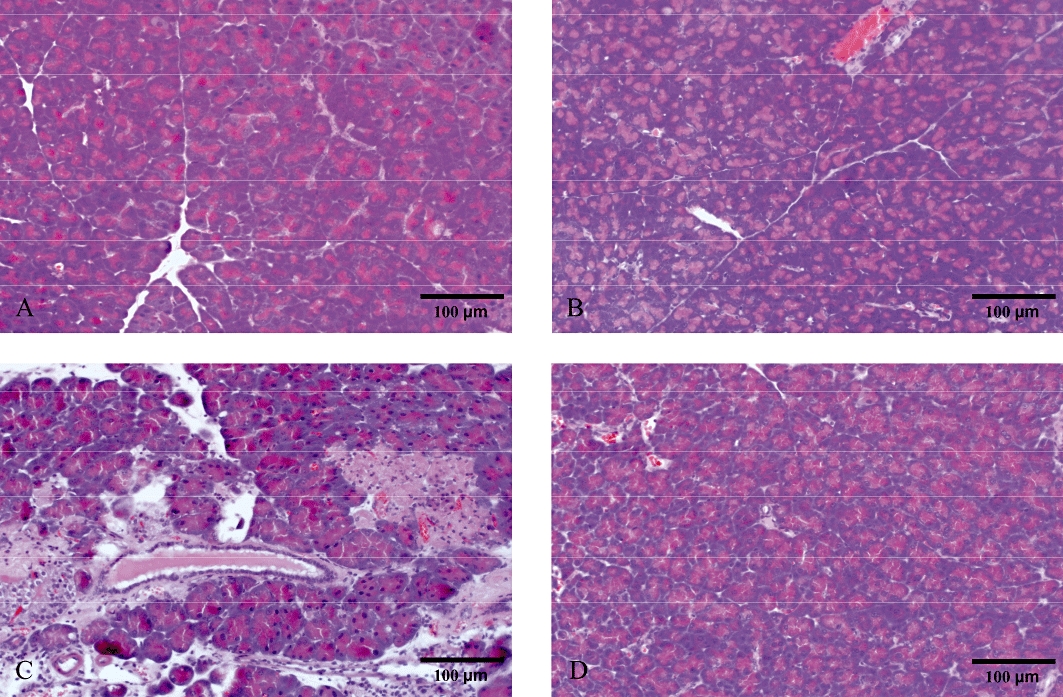
Representative haematoxylin and eosin stained sections of the pancreas. (A) Sham animals and (B) control animals infused with saline alone into the pancreatic duct. Taurocholate-exposed mice were pretreated with (C) PBS or (D) 5 mg·kg−1 of the Rho-kinase inhibitor Y-27632. Samples were harvested 24 h later. Bars represent 100 µm.
Figure 3.
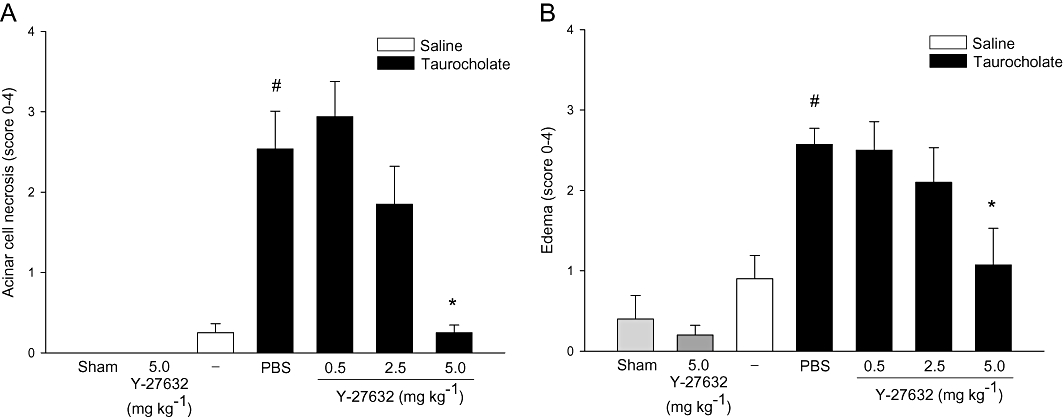
Rho-kinase regulates taurocholate-induced tissue damage in the pancreas. (A) Acinar cell necrosis and (B) oedema formation in sham, control (saline alone into the pancreatic duct) and taurocholate-exposed mice pretreated with PBS or the Rho-kinase inhibitor Y-27632 (0.5–5 mg·kg−1). One group of mice was given 5 mg·kg−1 Y-27632 alone without bile duct cannulation. Samples were obtained after 24 h. Data represent means ± SEM and n = 5–7. #P < 0.05 versus sham and *P < 0.05 versus PBS + taurocholate.
Table 1.
Systemic leucocyte differential counts
| PMNL | MNL | Total | |
|---|---|---|---|
| Sham | 0.8 ± 0.1 | 4.6 ± 0.2 | 5.4 ± 0.2 |
| Y-27632 5 mg·kg−1 | 0.9 ± 0.2 | 4.3 ± 0.2 | 5.2 ± 0.4 |
| Saline control | 0.9 ± 0.1 | 4.6 ± 0.1 | 5.5 ± 0.2 |
| PBS + pancreatitis | 1.7 ± 0.2# | 6.9 ± 0.6# | 7.6 ± 0.7# |
| Y-27632 0.5 mg·kg−1+ pancreatitis | 1.7 ± 0.3 | 6.1 ± 0.1 | 7.8 ± 0.4 |
| Y-27632 2.5 mg·kg−1+ pancreatitis | 1 ± 0.1* | 4.9 ± 0.1* | 5.9 ± 0.2* |
| Y-27632 5 mg·kg−1+ pancreatitis | 0.9 ± 0.1* | 5.3 ± 0.1* | 6.2 ± 0.2* |
Blood was collected from sham, saline control, taurocholate-treated animals receiving PBS or the Rho-kinase inhibitor Y-27632 (0.5–5 mg·kg−1). One group of mice received 5 mg·kg−1 Y-27632 alone without bile duct cannulation. Cells were identified as monomorphonuclear leucocytes (MNL) and polymorphonuclear leucocytes (PMNL). Data represents mean ± SEM, 106 cells mL−1 and n = 5–7.
P < 0.05 versus sham
P < 0.05 versus PBS + taurocholate.
Rho-kinase activity controls neutrophil recruitment in pancreatitis
Pancreatic levels of MPO were used as a marker of inflammatory cell infiltration. Peak levels of MPO were observed 24 h after taurocholate challenge (not shown) and this time-point was used for subsequent studies of neutrophil infiltration in the pancreas. It was found that challenge with taurocholate enhanced pancreatic levels of MPO by seven-fold (Figure 4A, P < 0.05 vs. sham, n = 5–7). Inhibition of Rho-kinase signalling decreased taurocholate-induced MPO levels in the pancreas by 73% (Figure 4A, P < 0.05 vs. vehicle + taurocholate, n = 5–7). Moreover, histological analysis of pancreatic tissue showed that taurocholate challenge provoked a clear-cut enhancement in extravascular neutrophils (Figure 4B, P < 0.05 vs. sham, n = 5–7). Notably, administration of 5 mg·kg−1 Y-27632 reduced taurocholate-provoked infiltration of neutrophils in the pancreas by 88% (Figure 4B, P < 0.05 vs. vehicle + taurocholate, n = 5–7). Neutrophil chemotaxis is known to be coordinated by MIP-2 (Pastor et al., 2003). Herein, we observed that MIP-2 levels were low but detectable in normal pancreas and that challenge with taurocholate markedly increased MIP-2 levels in the pancreas up to 22.1 ± 5.3 pg·mg−1 tissue (Figure 4C). Notably, Rho-kinase inhibition greatly decreased MIP-2 levels in the inflamed pancreas (Figure 4C). In addition, we noted that Mac-1 expression was increased on the surface of circulating neutrophils in mice with pancreatitis (Figure 5A), indicating systemic activation in this experimental model. Inhibition of Rho-kinase signalling markedly reduced neutrophil expression of Mac-1 in pancreatitis (Figure 5A). In contrast, the expression of chemokine receptor CXCR2 on neutrophils decreased after taurocholate challenge and administration of Y-27632 had no effect on CXCR2 expression on neutrophils in animals with pancreatitis (Figure 5B). As part of the systemic response to SAP, activated neutrophils accumulate in the lung. Indeed, we observed that MPO levels in the lung were significantly increased in animals with pancreatitis. Pretreatment with Y-27632 decreased pulmonary levels of MPO by 75% in mice challenged with taurocholate (Figure 5C). In contrast, treatment with 5 mg·kg−1 Y-27632 (n = 6) had no effect on MPO levels in the pancreas or the lung when given after taurocholate challenge (not shown).
Figure 4.
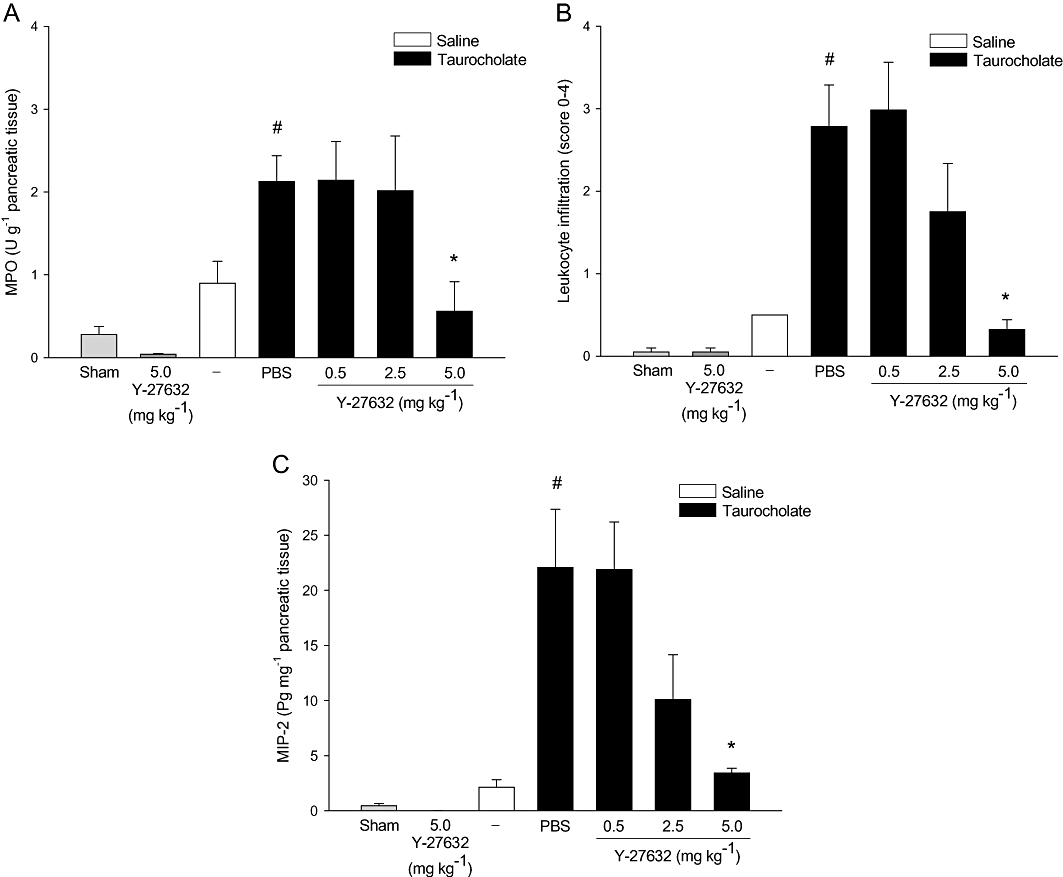
Rho-kinase regulates taurocholate-induced neutrophil accumulation in the pancreas. (A) MPO levels, (B) extravascular neutrophils, (C) MIP-2 levels in the pancreas in sham, control (saline alone into the pancreatic duct) and taurocholate-exposed mice pretreated with PBS or the Rho-kinase inhibitor Y-27632 (0.5–5 mg·kg−1). One group of mice was given 5 mg·kg−1 Y-27632 alone without bile duct cannulation. Samples were obtained after 24 h. Data represent means ± SEM and n = 5–7. #P < 0.05 versus sham and *P < 0.05 versus PBS + taurocholate.
Figure 5.
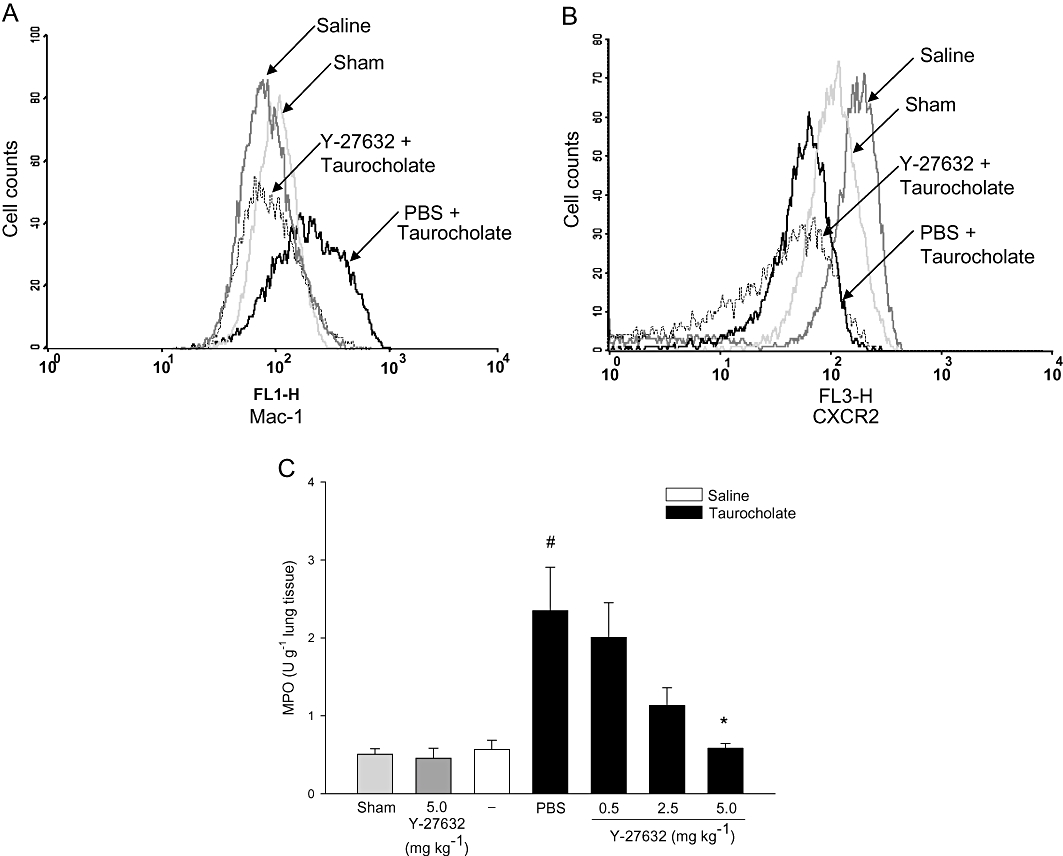
A representative sample of (A) Mac-1, (B) CXCR2 expression on neutrophils and (C) lung MPO levels in sham, control (saline alone into the pancreatic duct) and taurocholate-exposed mice pretreated with PBS or the Rho-kinase inhibitor Y-27632 (0.5–5 mg·kg−1). One group of mice was given 5 mg·kg−1 Y-27632 alone without bile duct cannulation. Samples were obtained after 24 h. Data represent means ± SEM and n = 5–7. #P < 0.05 versus sham and *P < 0.05 versus PBS + taurocholate.
Rho-kinase activity regulates trypsinogen activation in pancreatitis
TAP is a cleavage product from trypsinogen and TAP is a useful marker of trypsinogen activation (Gudgeon et al., 1990; Hartwig et al., 1999). Herein, it was found that taurocholate challenge increased TAP levels in the pancreas by 3-fold (Figure 6, P < 0.05 vs. sham, n = 5–7), suggesting that trypsinogen is indeed activated in this model of pancreatitis. Interestingly, we observed that administration of the Rho-kinase inhibitor significantly reduced taurocholate-induced TAP levels from 357.2 ± 28.2 down to 146.8 ± 56.8 µg·g−1 tissue, corresponding to a 61% reduction in trypsinogen activation (Figure 6, P < 0.05 vs. vehicle + taurocholate, n = 5–7).
Figure 6.
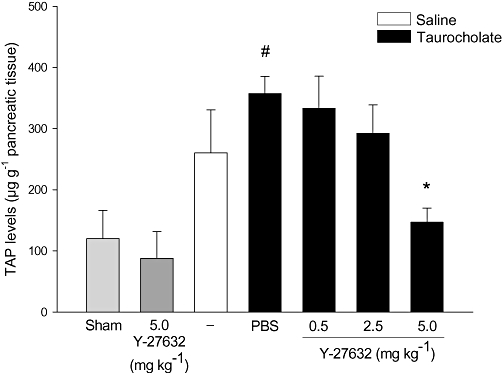
Rho-kinase regulates taurocholate-induced activation of trypsinogen. Levels of TAP in the pancreas in sham, control (saline alone into the pancreatic duct) and taurocholate-exposed mice pretreated with PBS or the Rho-kinase inhibitor Y-27632 (0.5–5 mg·kg−1). One group of mice was given 5 mg·kg−1 Y-27632 alone without bile duct cannulation. Samples were obtained after 24 h. Data represent means ± SEM and n = 5–7. #P < 0.05 versus sham and *P < 0.05 versus PBS + taurocholate.
Rho-kinase activity regulates activation of trypsinogen in acinar cells in vitro
We next determined whether Rho-kinase regulates trypsinogen activation in pancreatic acinar cells in vitro. For this purpose, we isolated acinar cells from the pancreas of mice and incubated the cells with cerulein as described previously (Saluja et al., 1999). It was found that cerulein stimulation increased trypsinogen activation by two-fold compared with unstimulated cells (Figure 7, P < 0.05 vs. control, n = 5). Notably, preincubation of the acinar cells with Y-27632 decreased secretagogue-induced activation of trypsinogen by 69% (Figure 7, P < 0.05 vs. vehicle + cerulein, n = 5).
Figure 7.
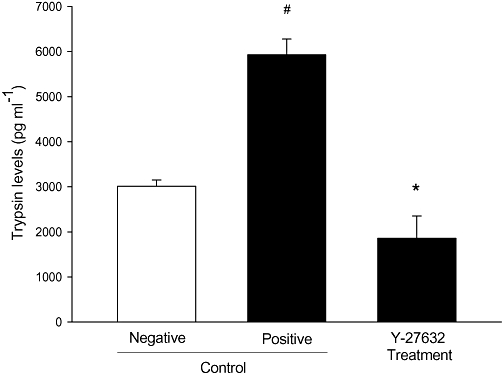
Rho-kinase regulates activation of trypsinogen in acinar cells. Acinar cell activation of trypsinogen was measured in (negative) control cells and cerulein-exposed acinar cell homogenate pretreated with vehicle (positive control) or the Rho-kinase inhibitor Y-27632 (100 nM). Trypsinogen activation was determined by measuring enzymatic activity of trypsin fluorometrically by using Boc-Gln-Ala-Arg-MCA as the substrate as described in detail in Methods. Trypsin levels (pg·mL−1) were calculated using a standard curve generated by assaying purified trypsin. Data represent means ± SEM and n = 5. #P < 0.05 versus control and *P < 0.05 versus positive control.
Discussion and conclusions
Signalling mechanisms regulating pathophysiological processes in acute pancreatitis are incompletely understood. The present study reveals that Rho-kinase signalling plays an integral part in the pathophysiology of SAP. In fact, inhibition of Rho-kinase activity markedly reduced acinar cell necrosis and blood amylase levels in acute pancreatitis. Our findings show that inhibition of Rho-kinase activity abolishes trypsinogen activation in the pancreas, which helps to explain the attenuated inflammatory response and tissue damage in SAP. These novel findings indicate that targeting Rho-kinase activity may be a useful approach to protect against SAP.
Rho-kinase activity is generally considered to regulate cytoskeletal dynamics, including cell contraction and vesicular trafficking, but there is increasing evidence also implicating Rho-kinase signalling in numerous features of inflammatory reactions, such as leucocyte migration, phagocytosis and cytokine formation (Riento and Ridley, 2003; Bokoch, 2005). In the present study, we observed that administration of Y-27632, a specific Rho-kinase inhibitor, markedly decreased tissue damage in SAP. For example, inhibition of Rho-kinase signalling reduced the taurocholate-induced increase in blood amylase by 83% and acinar cell necrosis by more than 90%, indicating that Rho-kinase indeed controls a substantial part of the tissue injury in pancreatitis. It should be mentioned that a previous study reported that Y-27632 increased secretion of amylase from acinar cells in cerulein-induced pancreatitis (Kusama et al., 2003). That observation may seem to be in contrast to our present findings but the reduction of blood amylase in our present work is more likely to be related to the protective effect of Y-27632 against taurocholate-induced cell injury in the pancreas. Nonetheless, our present data constitutes the first evidence in the literature that inhibition of the Rho-kinase signalling pathway protects against SAP. Thus, our data add SAP to the growing list of conditions, which may be ameliorated by interference with Rho-kinase signalling, including ischaemia-reperfusion (Bao et al., 2004; Shiotani et al., 2004), endotoxaemia (Thorlacius et al., 2006), septic lung injury (Tasaka et al., 2005), tissue fibrosis (Bourgier et al., 2005) and obstructive cholestasis (Laschke et al., 2009). In this context, it is interesting to note that the present findings show that inhibition of Rho-kinase attenuates TAP levels in animals with pancreatitis, suggesting that Rho-kinase is involved in the conversion of trypsinogen to active trypsin. This caused us to ask whether Rho-kinase may regulate trypsinogen activation in acinar cells in the pancreas. Interestingly, we found that Y-27632 abolished secretagogue-induced activation of trypsinogen in vitro, suggesting that Rho-kinase indeed regulates trypsinogen activation in acinar cells. Considering that trypsin formation is an early and important component in the pathophysiology of AP (Mithöfer et al., 1998), it is tempting to speculate that trypsinogen activation is a main target of the Rho-kinase inhibitor in AP. However, the relationship between trypsin activity on one hand and the inflammatory response on the other hand in SAP is not clearly delineated. It may be that both develop in parallel and potentiate each other or there may be a sequential relationship with one preceding the other. Nonetheless, we also observed that administration of Y-27632 after taurocholate challenge had no significant effect on inflammatory parameters or tissue damage in the pancreas, supporting the notion above that trypsinogen activation in acinar cells rather than secondary chemokine formation and neutrophil activation may be the main protective mechanism exerted by the Rho-kinase inhibitor. In this context, it should be noted that targeting Rho-kinase activity may have a limited influence on the treatment of patients with on-going pancreatitis considering that Rho-kinase-regulated activation of trypsinogen is an early feature in AP and that delayed treatment with the Rho-kinase inhibitor did not ameliorate tissue damage in the inflamed pancreas. However, it is possible that high-risk patients undergoing endoscopic retrograde cholangiopancreatography may benefit from prophylactic administration of Rho-kinase inhibitors.
Activation and extravascular accumulation of leucocytes are key components in the inflammatory response following injury and infection, but in certain instances, leucocytes may cause organ damage, including graft rejection and sepsis (Carlos and Harlan, 1994). In fact, numerous studies have documented that leucocyte recruitment constitutes a rate-limiting step in pancreatitis by demonstrating markedly attenuated tissue destruction in neutrophil-depleted animals (Kyriakides et al., 2001). Herein, we observed that taurocholate challenge increased MPO activity and the number of extravascular neutrophils in the pancreas. Administration of Y-27632 greatly decreased both MPO levels (73%) and extravascular neutrophils (88%) in the pancreas, suggesting that Rho-kinase activity is a potent regulator of neutrophil trafficking in pancreatitis. Specific adhesion molecules regulate the recruitment process of leucocytes to extravascular sites of inflammation (Kelly et al., 2007). Although the detailed role of specific adhesion molecules in supporting leucocyte recruitment in the pancreas is relatively unclear, numerous studies have shown that Mac-1 is a dominating molecule in mediating tissue infiltration of neutrophils (Asaduzzaman et al., 2008; Lee et al., 2009; Rahman et al., 2009). In the present study, we found that taurocholate challenge upregulated Mac-1 expression on neutrophils. Interestingly, administration of Y-27632 markedly reduced surface levels of Mac-1 on neutrophils, indicating that Rho-kinase signalling contributes to neutrophil expression of Mac-1 in pancreatitis. Moreover, this inhibitory effect on Mac-1 expression may also help explain the inhibitory action of Y-27632 on neutrophil accumulation in SAP observed herein. In addition, systemic complications of SAP include pulmonary accumulation of neutrophils (Sharif et al., 2009). Indeed, we observed that lung MPO activity was clearly increased in taurocholate-treated animals. Notably, inhibition of Rho-kinase function clearly attenuated pulmonary MPO levels, indicating that Y-27632 protects against systemic activation and infiltration of neutrophils in the lung. Considered together, these findings suggest that Rho-kinase signalling regulates both local and distant organ accumulation of neutrophils in acute pancreatitis.
It is generally held that secreted chemokines are fundamental regulators of leucocyte activation and tissue navigation. CXC chemokines, such as MIP-2, are particularly potent activators of neutrophils. It was, therefore, of great interest to examine local formation of MIP-2 in the pancreas in this study. We observed that taurocholate caused a clear-cut increase in MIP-2 formation in the pancreas. It is interesting to note that inhibition of Rho-kinase activity reduced taurocholate-induced expression of MIP-2 by 84%. Indeed, this marked attenuation of MIP-2 formation may account for the inhibitory effect of Y-27632 on neutrophil expression of Mac-1 as well as on the infiltration of neutrophils in the pancreas and lung. However, these findings do not exclude the possibility that Rho-kinase signalling also directly regulates Mac-1 expression and migratory function in neutrophils. For example, a previous study has reported that Rho-kinase can coordinate chemoattractant-induced leucocyte migration in vitro (Satoh et al., 2001). Moreover, it is valuable to note that these findings do not exclude a potential role of other protein kinases; p38 mitogen-activated protein kinase signalling has also been shown to play a role in pancreatitis (Chen et al., 2007). In general, MIP-2 effects are mediated through binding to the CXC chemokine receptor 2 (CXCR2), which is the high affinity receptor on murine neutrophils for MIP-2 (Cacalano et al., 1994; Jones et al., 1997). Herein, we observed that taurocholate challenge decreased CXCR2 expression on neutrophils, which is in line with other models of systemic inflammation such as sepsis (Rios-Santos et al., 2007) and trauma (Quaid et al., 1999). The reason behind the discrepancy between decreased expression of CXCR2 on the one hand and increased tissue migration of neutrophils on the other is not known but may be related to a relative accumulation of neutrophils with low levels of CXCR2 in the circulation after vascular extravasation of neutrophils with higher expression of CXCR2. Alternatively, there may be a time-dependent component, that is, neutrophils exit the circulation prior to the downregulation of CXCR2 in the remaining population of neutrophils in the blood. Nonetheless, we observed that administration of Y-27632 had no effect on CXCR2 expression on neutrophils in pancreatitis, suggesting that pancreatitis-associated CXCR2 shedding from the surface of neutrophils is not related to Rho-kinase activity.
Taken together, our novel data show that inhibition of Rho-kinase signalling ameliorates tissue damage in pancreatitis. Indeed, our findings show that Rho-kinase regulates trypsinogen activation in pancreatitis and that interference with Rho-kinase activity decreased MIP-2 formation, neutrophil activation (Mac-1 expression) and recruitment in the pancreas. Thus, our results not only elucidate important signalling mechanisms in pancreatitis but also suggest that targeting Rho-kinase activity may be a useful approach to protect against pathological tissue damage in SAP.
Acknowledgments
This work was supported by grants from the Swedish Medical Research Council (2009-4872), Crafoordska stiftelsen, Einar och Inga Nilssons stiftelse, Harald och Greta Jaenssons stiftelse, Greta och Johan Kocks stiftelser, Fröken Agnes Nilssons stiftelse, Franke och Margareta Bergqvists stiftelse för främjande av cancerforskning, Magnus Bergvalls stiftelse, Mossfelts stiftelse, Nanna Svartz stiftelse, Ruth och Richard Julins stiftelse, Svenska Läkaresällskapet, Allmäna sjukhusets i Malmö stiftelse för bekämpande av cancer, MAS fonder, Malmö University Hospital and Lund University. D Awla and A Abdulla are supported by a fellowship from the Kurdistan regional government and the Nanakali Group.
Glossary
Abbreviations
- i.p.
intraperitoneal
- MIP-2
macrophage inflammatory protein-2
- MNL
monomorphonuclear leucocytes
- MPO
myeloperoxidase
- MOPS
3-(morpholino) propanesulphonic acid
- PBS
phosphate-buffered saline
- PMNL
polymorphonuclear leucocytes
- RIA
radioimmunoassay
- SAP
severe acute pancreatitis
- TAP
trypsinogen activating peptide
Conflict of interest
The authors state no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alblas J, Ulfman L, Hordijk P, Koenderman L. Activation of Rhoa and ROCK are essential for detachment of migrating leucocytes. Mol Biol Cell. 2001;12:2137–2145. doi: 10.1091/mbc.12.7.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Andersson B, Andersson E, Axelsson J, Eckerwall G, Tingstedt B. Acute pancreatitis – from cellular signalling to complicated clinical course. HPB (Oxford) 2007;9:414–420. doi: 10.1080/13651820701713766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaduzzaman M, Zhang S, Lavasani S, Wang Y, Thorlacius H. LFA-1 and MAC-1 mediate pulmonary recruitment of neutrophils and tissue damage in abdominal sepsis. Shock. 2008;30:254–259. doi: 10.1097/shk.0b013e318162c567. [DOI] [PubMed] [Google Scholar]
- Bacon KB, Oppenheim JJ. Chemokines in disease models and pathogenesis. Cytokine Growth Factor Rev. 1998;9:167–173. doi: 10.1016/s1359-6101(98)00005-7. [DOI] [PubMed] [Google Scholar]
- Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, et al. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548–558. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Brady M, Shokuhi S, Christmas S, Neoptolemos JP, Slavin J. Inflammatory mediators in acute pancreatitis. J Pathol. 2000;190:117–125. doi: 10.1002/(SICI)1096-9896(200002)190:2<117::AID-PATH494>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005;15:163–171. doi: 10.1016/j.tcb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Bourgier C, Haydont V, Milliat F, Francois A, Holler V, Lasser P, et al. Inhibition of Rho kinase modulates radiation induced fibrogenic phenotype in intestinal smooth muscle cells through alteration of the cytoskeleton and connective tissue growth factor expression. Gut. 2005;54:336–343. doi: 10.1136/gut.2004.051169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, et al. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- Carlos TM, Harlan JM. Leucocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- Chen P, Zhang Y, Qiao M, Yuan Y. Activated protein C, an anticoagulant polypeptide, ameliorates severe acute pancreatitis via regulation of mitogen-activated protein kinases. J Gastroenterol. 2007;42:887–896. doi: 10.1007/s00535-007-2104-2. [DOI] [PubMed] [Google Scholar]
- Feng L, Xia Y, Yoshimura T, Wilson CB. Modulation of neutrophil influx in glomerulonephritis in the rat with anti-macrophage inflammatory protein-2 (MIP-2) antibody. J Clin Invest. 1995;95:1009–1017. doi: 10.1172/JCI117745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasbrenner B, Adler G. Pathophysiology of acute pancreatitis. Hepatogastroenterology. 1993;40:517–521. [PubMed] [Google Scholar]
- Granger J, Remick D. Acute pancreatitis: models, markers, and mediators. Shock. 2005;24(Suppl. 1):45–51. doi: 10.1097/01.shk.0000191413.94461.b0. [DOI] [PubMed] [Google Scholar]
- Gudgeon AM, Heath DI, Hurley P, Jehanli A, Patel G, Wilson C, et al. Trypsinogen activation peptides assay in the early prediction of severity of acute pancreatitis. Lancet. 1990;335:4–8. doi: 10.1016/0140-6736(90)90135-r. [DOI] [PubMed] [Google Scholar]
- Hartwig W, Werner J, Jimenez RE, Z'Graggen K, Weimann J, Lewandrowski KB, et al. Trypsin and activation of circulating trypsinogen contribute to pancreatitis-associated lung injury. Am J Physiol. 1999;277:G1008–G1016. doi: 10.1152/ajpgi.1999.277.5.G1008. [DOI] [PubMed] [Google Scholar]
- Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med. 1999;5:221–225. doi: 10.1038/5587. [DOI] [PubMed] [Google Scholar]
- Jones SA, Dewald B, Clark-Lewis I, Baggiolini M. Chemokine antagonists that discriminate between interleukin-8 receptors. Selective blockers of CXCR2. J Biol Chem. 1997;272:16166–16169. doi: 10.1074/jbc.272.26.16166. [DOI] [PubMed] [Google Scholar]
- Kawabata S, Miura T, Morita T, Kato H, Fujikawa K, Iwanaga S, et al. Hghly sensitive peptide-4-methylcoumaryl-7-amide substrates for blood-clotting proteases and trypsin. Eur J Biochem. 1988;172:17–25. doi: 10.1111/j.1432-1033.1988.tb13849.x. [DOI] [PubMed] [Google Scholar]
- Kelly M, Hwang JM, Kubes P. Modulating leukocyte recruitment in inflammation. J Allergy Clin Immunol. 2007;120:3–10. doi: 10.1016/j.jaci.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Tada S, Nakamoto N, Toda K, Horikawa H, Kurita S, et al. Rho/Rho kinase is a key enzyme system involved in the angiotensin II signaling pathway of liver fibrosis and steatosis. J Gastroenterol Hepatol. 2007;22:2022–2033. doi: 10.1111/j.1440-1746.2006.04735.x. [DOI] [PubMed] [Google Scholar]
- Klein M, Paul R, Angele B, Popp B, Pfister HW, Koedel U. Protein expression pattern in experimental pneumococcal meningitis. Microbes Infect. 2006;8:974–983. doi: 10.1016/j.micinf.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Kusama K, Nozu F, Awai T, Tanaka S, Honma I, Tsunoda Y, et al. Deactivation of ROCK-II by Y-27632 enhances basolateral pancreatic enzyme secretion and acute pancreatitis induced by CCK analogues. Biochem Biophys Res Commun. 2003;305:339–344. doi: 10.1016/s0006-291x(03)00758-7. [DOI] [PubMed] [Google Scholar]
- Kyriakides C, Jasleen J, Wang Y, Moore FD, Ashley SW, Jr, Hechtman HB. Neutrophils, not complement, mediate the mortality of experimental hemorrhagic pancreatitis. Pancreas. 2001;22:40–46. doi: 10.1097/00006676-200101000-00007. [DOI] [PubMed] [Google Scholar]
- Laschke MW, Menger MD, Wang Y, Lindell G, Jeppsson B, Thorlacius H. Sepsis-associated cholestasis is critically dependent on P-selectin-dependent leukocyte recruitment in mice. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1396–G1402. doi: 10.1152/ajpgi.00539.2006. [DOI] [PubMed] [Google Scholar]
- Laschke MW, Dold S, Jeppsson B, Schilling MK, Menger MD, Thorlacius H. Rho-Kinase Inhibitor Attenuates Cholestasis-Induced CXC Chemokine Formation, Leukocyte Recruitment, and Hepatocellular Damage in the Liver. J Surg Res. 2008;159:666–673. doi: 10.1016/j.jss.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Laschke MW, Dold S, Menger MD, Jeppsson B, Thorlacius H. The Rho-kinase inhibitor Y-27632 inhibits cholestasis-induced platelet interactions in the hepatic microcirculation. Microvasc Res. 2009;78:95–99. doi: 10.1016/j.mvr.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Lee S, Bowrin K, Hamad AR, Chakravarti S. Extracellular matrix lumican deposited on the surface of neutrophils promotes migration by binding to beta2 integrin. J Biol Chem. 2009;284:23662–23669. doi: 10.1074/jbc.M109.026229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Klintman D, Liu Q, Sato T, Jeppsson B, Thorlacius H. Critical role of CXC chemokines in endotoxemic liver injury in mice. J Leukoc Biol. 2004;75:443–452. doi: 10.1189/jlb.0603297. [DOI] [PubMed] [Google Scholar]
- Lindkvist B, Wierup N, Sundler F, Borgstrom A. Long-term nicotine exposure causes increased concentrations of trypsinogens and amylase in pancreatic extracts in the rat. Pancreas. 2008;37:288–294. doi: 10.1097/MPA.0b013e31816a7744. [DOI] [PubMed] [Google Scholar]
- Mangalmurti NS, Xiong Z, Hulver M, Ranganathan M, Liu XH, Oriss T, et al. Loss of red cell chemokine scavenging promotes transfusion-related lung inflammation. Blood. 2009;113:1158–1166. doi: 10.1182/blood-2008-07-166264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithöfer K, Fernández-del Castillo C, Rattner D, Warshaw AL. Subcellular kinetics of early trypsinogen activation in acute rodent pancreatitis. Am J Physiol. 1998;274:G71–G79. doi: 10.1152/ajpgi.1998.274.1.G71. [DOI] [PubMed] [Google Scholar]
- Monson KM, Dowlatshahi S, Crockett ET. CXC-chemokine regulation and neutrophil trafficking in hepatic ischemia-reperfusion injury in P-selectin/ICAM-1 deficient mice. J Inflamm (Lond) 2007;4:11. doi: 10.1186/1476-9255-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol. 2008;155:444–454. doi: 10.1038/bjp.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor CM, Rubbia-Brandt L, Hadengue A, Jordan M, Morel P, Frossard JL. Role of macrophage inflammatory peptide-2 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. Lab Invest. 2003;83:471–478. doi: 10.1097/01.lab.0000063928.91314.9f. [DOI] [PubMed] [Google Scholar]
- Quaid GA, Cave C, Robinson C, Williams MA, Solomkin JS. Preferential loss of CXCR-2 receptor expression and function in patients who have undergone trauma. Arch Surg. 1999;134:1367–1371. doi: 10.1001/archsurg.134.12.1367. [DOI] [PubMed] [Google Scholar]
- Rahman M, Zhang S, Chew M, Ersson A, Jeppsson B, Thorlacius H. Platelet-derived CD40L (CD154) mediates neutrophil upregulation of Mac-1 and recruitment in septic lung injury. Ann Surg. 2009;250:783–790. doi: 10.1097/SLA.0b013e3181bd95b7. [DOI] [PubMed] [Google Scholar]
- Regner S, Manjer J, Appelros S, Hjalmarsson C, Sadic J, Borgstrom A. Protease activation, pancreatic leakage, and inflammation in acute pancreatitis: differences between mild and severe cases and changes over the first three days. Pancreatology. 2008;8:600–607. doi: 10.1159/000161011. [DOI] [PubMed] [Google Scholar]
- Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- Rios-Santos F, Alves-Filho JC, Souto FO, Spiller F, Freitas A, Lotufo CM, et al. Down-regulation of CXCR2 on neutrophils in severe sepsis is mediated by inducible nitric oxide synthase-derived nitric oxide. Am J Respir Crit Care Med. 2007;175:490–497. doi: 10.1164/rccm.200601-103OC. [DOI] [PubMed] [Google Scholar]
- Ryschich E, Kerkadze V, Deduchovas O, Salnikova O, Parseliunas A, Marten A, et al. Intracapillary leucocyte accumulation as a novel antihaemorrhagic mechanism in acute pancreatitis in mice. Gut. 2009;58:1508–1516. doi: 10.1136/gut.2008.170001. [DOI] [PubMed] [Google Scholar]
- Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol. 1999;276:G835–G842. doi: 10.1152/ajpgi.1999.276.4.G835. [DOI] [PubMed] [Google Scholar]
- Santen S, Wang Y, Laschke MW, Menger MD, Jeppsson B, Thorlacius H. Rho-kinase signalling regulates CXC chemokine formation and leukocyte recruitment in colonic ischemia-reperfusion. Int J Colorectal Dis. 2010;25:1063–1070. doi: 10.1007/s00384-010-0997-3. [DOI] [PubMed] [Google Scholar]
- Satoh S, Utsunomiya T, Tsurui K, Kobayashi T, Ikegaki I, Sasaki Y, et al. Pharmacological profile of hydroxy fasudil as a selective Rho kinase inhibitor on ischemic brain damage. Life Sci. 2001;69:1441–1453. doi: 10.1016/s0024-3205(01)01229-2. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Rattner DW, Lewandrowski K, Compton CC, Mandavilli U, Knoefel WT, et al. A better model of acute pancreatitis for evaluating therapy. Ann Surg. 1992;215:44–56. doi: 10.1097/00000658-199201000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif R, Dawra R, Wasiluk K, Phillips P, Dudeja V, Kurt-Jones E, et al. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut. 2009;58:813–819. doi: 10.1136/gut.2008.170423. [DOI] [PubMed] [Google Scholar]
- Shin HK, Salomone S, Potts EM, Lee SW, Millican E, Noma K, et al. Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab. 2007;27:998–1009. doi: 10.1038/sj.jcbfm.9600406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotani S, Shimada M, Suehiro T, Soejima Y, Yosizumi T, Shimokawa H, et al. Involvement of Rho-kinase in cold ischemia-reperfusion injury after liver transplantation in rats. Transplantation. 2004;78:375–382. doi: 10.1097/01.tp.0000128618.41619.e7. [DOI] [PubMed] [Google Scholar]
- Slotta JE, Braun OO, Menger MD, Thorlacius H. Fasudil, a Rho-kinase inhibitor, inhibits leucocyte adhesion in inflamed large blood vessels in vivo. Inflamm Res. 2006;55:364–367. doi: 10.1007/s00011-006-6013-2. [DOI] [PubMed] [Google Scholar]
- Slotta JE, Laschke MW, Menger MD, Thorlacius H. Rho-kinase signalling mediates endotoxin hypersensitivity after partial hepatectomy. Br J Surg. 2008;95:976–984. doi: 10.1002/bjs.6082. [DOI] [PubMed] [Google Scholar]
- Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, et al. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol. 2005;32:504–510. doi: 10.1165/rcmb.2004-0009OC. [DOI] [PubMed] [Google Scholar]
- Tekamp-Olson P, Gallegos C, Bauer D, McClain J, Sherry B, Fabre M, et al. Cloning and characterization of cDNAs for murine macrophage inflammatory protein 2 and its human homologues. J Exp Med. 1990;172:911–919. doi: 10.1084/jem.172.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorlacius K, Slotta JE, Laschke MW, Wang Y, Menger MD, Jeppsson B, et al. Protective effect of fasudil, a Rho-kinase inhibitor, on chemokine expression, leukocyte recruitment, and hepatocellular apoptosis in septic liver injury. J Leukoc Biol. 2006;79:923–931. doi: 10.1189/jlb.0705406. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Gao CF, Wei D, Wang C, Ding SQ. Acute pancreatitis: etiology and common pathogenesis. World J Gastroenterol. 2009;15:1427–1430. doi: 10.3748/wjg.15.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XP, Li ZJ, Zhang J. Inflammatory mediators and microcirculatory disturbance in acute pancreatitis. Hepatobiliary Pancreat Dis Int. 2009;8:351–357. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.