

Abstract
BACKGROUND AND PURPOSE
There is much evidence supporting the role of β2-adrenoceptors (β2AR) in angiogenesis but the mechanisms underlying their effects have not been elucidated. Hence, we studied post-ischaemic angiogenesis in the hindlimb (HL) of β2AR knock-out mice (β2AR−/−) in vivo and explored possible molecular mechanisms in vitro.
EXPERIMENTAL APPROACH
Femoral artery resection (FAR) was performed in wild-type and β2AR−/− mice and adaptive responses to chronic HL ischaemia were explored; blood flow was measured by ultrasound and perfusion of dyed beads, bone rarefaction, muscle fibrosis and skin thickness were evaluated by immunoflourescence and morphometric analysis. Intrafemoral delivery of an adenovirus encoding the human β2AR (ADβ2AR) was used to reinstate β2ARs in β2AR−/− mice. Molecular mechanisms were investigated in mouse-derived aortic endothelial cells (EC) in vitro, focusing on NFκB activation and transcriptional activity.
RESULTS
Angiogenesis was severely impaired in β2AR−/− mice subjected to FAR, but was restored by gene therapy with ADβ2AR. The proangiogenic responses to a variety of stimuli were impaired in β2AR−/− EC in vitro. Moreover, removal of β2ARs impaired the activation of NFκB, a transcription factor that promotes angiogenesis; neither isoprenaline (stimulates βARs) nor TNFα induced NFκB activation in β2AR−/− EC. Interestingly, cAMP response element binding protein (CREB), a transcription factor that counter regulates NFκB, was constitutively increased in β2AR−/− ECs. ADβ2AR administration restored β2AR membrane density, reduced CREB activity and reinstated the NFκB response to isoprenaline and TNFα.
CONCLUSIONS AND IMPLICATIONS
Our results suggest that β2ARs control angiogenesis through the tight regulation of nuclear transcriptional activity.
Keywords: adrenergic signalling, gene therapy, ischaemic hindlimb, NFκB activity
Introduction
Little is known about the role of the β2-adrenoceptor (β2AR) in the vasculature. Recently, our group and others have begun to elucidate the mechanisms of β2AR control of vascular functions (Lembo et al., 1997; Ferro et al., 1999) and showed that this receptor can activate eNOS in an Akt-dependent manner and induce release of nitric oxide (NO) at the endothelium (Iaccarino et al., 2002; 2004;). We have also shown that the adenoviral-mediated endothelial overexpression of the β2AR regulates post-ischaemic angiogenesis (Iaccarino et al., 2005), to the extent that β2ARs can correct impaired angiogenesis in animal models of cardiovascular disease such as the spontaneously hypertensive rat (Iaccarino et al., 2002; 2005;). The mechanisms underlying β2AR-regulated angiogenesis have still not been elucidated. One possible explanation is that receptors such as the β2AR are able to regulate the transcriptional activity of the cell, in order to promote the release of proangiogenic cytokines such as vascular endothelial growth factor (VEGF). Indeed, β2AR overexpression results in VEGF accumulation in the culture medium of endothelial cells. VEGF has a transcriptional regulation that is controlled by the hypoxia responsive transcription factor-1 (HIF-1), and also by NFκB, which is known to be activated downstream by tumour necrosis factor receptors (TNF-R) superfamily-members and some G protein-coupled, seven transmembrane (7TM) receptors (GPCRs) (Ye, 2001).
The role of NFκB in angiogenesis is well established, and is linked to its ability to regulate inflammatory cytokine production in many cellular types; these include the endothelium, infiltrating macrophages, vascular smooth muscle cells and pericytes, and skeletal muscle cells. In this context, receptor and transcription factors are major players, allowing the finely tuned response of different cell types. Therefore, NFκB represents a particularly apt subject for our investigation on the signal transduction mechanisms underlying the proangiogenic effects of β2ARs.
The β2AR knock-out (β2AR−/−) mouse model has already proved useful to unveil specific molecular features of the β2AR (Chruscinski et al., 1999; Shenoy et al., 2006). We therefore exploited such a model to investigate the adaptive responses to chronic hindlimb (HL) ischaemia in a β2AR-negative (β2AR−/−) situation. Furthermore, using adenoviral-mediated gene transfer, we reconstituted membrane β2AR expression in the ischaemic HL. In vitro, we explored the possible mechanisms accounting for β2AR's beneficial effects on angiogenesis, focusing particularly on NFκB activation and transcriptional activity.
Methods
Mouse strain and surgical procedures
Previously described β2AR−/− and β2AR+/+ mice (age 14–18 weeks) were used in this study (Chruscinski et al., 1999). Founders were provided by courtesy of Brian Kobilka, Stanford University, CA. Mice were bred in heterozygousity and homozygous β2AR−/− or β2AR+/+ male littermates were used as the study and control population. All procedures were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee and the Federico II University Ethical Committee for Animal Research. Mice were anaesthetized with a mixture of ketamine (100 mg·kg−1) and xylazine (3 mg·kg−1) and the right common femoral artery was isolated and removed. In a group of β2AR−/− mice (n = 20), we placed a silastic catheter into the femoral artery distal to the resection, through which a solution containing 1011 tvp of either an adenovirus encoding for LacZ or the human β2AR gene was infused into the HL and allowed to remain there for 30 min while the saphenous vein was temporarily occluded (Santulli et al., 2009a). Afterwards, the virus was removed through the catheter, the common femoral artery removed and the wound closed in layers. With this manoeuvre, we ascertained that endothelial cells, vascular smooth muscle cells and skeletal myocytes of the hindlimb express the transgene carried by the viral vector, as found previously (Santulli et al., 2009a). Mice were checked daily for fur loss, skin lesions (blistering), necrosis, self-inflicted amputations of the ischaemic hindlimb. Surgical aftercare and distress surveillance were performed according to institution's guidelines.
Blood flow determination
Blood flow (BF) in the posterior tibial artery of ischaemic and non-ischaemic HL was evaluated by ultrasound (US) (using a VisualSONICS VeVo 770 imaging system with a 710 MHz scanhead) in isofluorane-anaesthetized mice (2% v·v−1) immediately after surgery and at days 3, 7, 10 and 14 thereafter. We measured maximal velocity (Vmax) and maximal diameter of the vessel. After calculation of the vessel area, BF was calculated using the formula: BF = Vmax/vessel area (Santulli et al., 2009a). Data are expressed as ischaemic to non-ischaemic ratio. Fourteen days after surgery, mice were anaesthetized as above, and a PE 10 catheter was placed into the abdominal aorta through the left common carotid, as previously described (Iaccarino et al., 2002). Maximal vasodilatation was obtained by administration of nitroglycerin (2 µg i.a.) followed by injection of 3 × 106 orange-dyed beads (15 µm diameter, Triton Technologies, San Diego, CA, USA). Animals were then killed by cervical dislocation, samples of the gastrocnemius muscle from the ischaemic and non-ischaemic HL were collected and frozen with liquid nitrogen and stored at −80°C. Next, the samples were homogenized and digested according to manufacturer protocol; the beads were collected and suspended in DMTF. The release of dye was assessed by light absorption at 450 nm (Santulli et al., 2009a). Data are expressed as ischaemic to non-ischaemic muscle ratio.
Immunofluorescence and morphology
At 14 days, we used B mode US for morphological analysis of the ischaemic and controlateral hindlimbs. In particular, we evaluated bone rarefaction, muscle fibrosis and skin thickness, all processes that are associated with HL ischaemia (Santulli et al., 2009b).
The anterior tibial muscle was isolated and harvested for immuonostaining as described previously (Zhou et al., 2003). Specimens were fixed in 4% paraformaldehyde and then embedded in paraffin. A series of cross-sections (6 µm) were obtained. Rat anti-CD31 antibody (1:50, BD Pharmingen, CA) and rabbit anti-von Willebrand (vW) factor (1:50, DAKO, Carpinteria, CA) were used as primary antibodies for double staining of endothelial cells. As a negative control, normal rat and rabbit IgG were used instead of the primary antibody. The primary antibodies were recognized by Alexa Fluor 594 goat anti-rat (Green) and Alexa Fluor 488 goat anti-rabbit (Blue) secondary antibodies (1:100), respectively (Molecular Probes, Eugene, Oregan). Nuclei were counterstained with VECTASHIELD mounting medium with DAPI (Red) (Vector, Burlingame, CA). Immunofluorescence was visualized under a fluorescence microscope (Olympus IX71, Olympus, Center Valley, CA, USA) and the number of capillaries per 20 fields was measured on each section by two independent operators (M.C., R.H.Z.), blinded to treatment. Another series of tissue sections were stained with haematoxylin/eosin (H&E) for morphological analysis.
βAR radioligand binding
Membrane fractions were obtained from quadricep muscle homogenates by centrifugation as previously described (Iaccarino et al., 1998). Total receptor density was assessed by βAR radioligand binding studies using the non-selective βAR antagonist [125I]-cyanopindolol (125I-CYP), as described previously (Iaccarino et al., 2001b). The percentage of β2ARs was calculated from the high affinity binding subpopulation using GraphPad Prism.
Adenoviral constructs
We used adenoviral vectors encoding for the human wild-type β2AR gene (Adβ2AR) and the LacZ (control virus) as previously described (Iaccarino et al., 2002; 2005; Ciccarelli et al., 2007).
Cell culture
Aortic endothelial cells (ECs) from β2AR−/− and β2AR+/+ mice were isolated as previously described (Iaccarino et al., 2002). Vessels were cut into rings, placed on matrigel, incubated in DMEM supplemented with 20% FBS and EC growth supplement (10 mg · 100 mL−1), and incubated at 37°C in 5% CO2. After 7 days, aortic rings were removed, and the ECs remaining on matrigel were expanded in DMEM containing 10% FBS.
Western blotting
Cells were deprived of serum overnight, exposed to agonists and lysed in RIPA/SDS buffer (50 mmol·L−1 Tris-HCl, pH 7.5, 150 mmol·L−1 NaCl, 1% NP-40, 0.25% deoxycholate, 9.4 mg·50 mL−1 sodium orthovanadate, 20% SDS). Protein concentration was determined using a BSA assay kit (Pierce, Thermoscientific, Rockford, IL, USA). IκBα was immunoprecipitated from total lysates with anti-IκBα antibody and protein A/G agarose. Immunocomplexes or total lysates were electrophoresed by SDS/PAGE and transferred to a nitrocellulose filter. Total IκBα and β2AR were visualized by specific antibodies (Santacruz, Santa Cruz, CA, USA), anti-rabbit horseradish peroxidase–conjugated secondary antibody (Santacruz) and standard chemiluminescence (Pierce). Autoradiographies were then digitalized and densitometry quantification performed using dedicated software (ImageQuant, GE HealthCare, Milano, Italy). Data are presented as arbitrary densitometry units (ADU) after normalization for actin. In other experiments, cells were infected with Adβ2AR at a rate of 20:1.
VEGF quantification
Aortic ECs from β2AR−/− and β2AR+/+ mice were deprived of serum overnight and then stimulated with isoprenaline (Iso) for 6 h. Culture medium was collected and VEGF was immunoprecipitated with anti-VEGF antibody (Santacruz) and protein A/G agarose (Santacruz). After being extensively washed, the immunocomplexes were electrophoresed by SDS/PAGE and transferred to nitrocellulose; VEGF was visualized by specific antibody (Santacruz), anti-rabbit HRP-conjugated secondary antibody (Santacruz) and standard chemiluminescence (Pierce). For our analysis, we examined the Western blot band corresponding to VEGF 164 isoform.
Cell transfection and luciferase assay
Transient transfection was performed using Lipofectamine 2000 (Invitrogen, Paisley, UK) according to manufacturer's instruction. Aortic ECs from β2AR−/− and β2AR+/+ mice were transfected with plasmid expression vectors coding cAMP response element binding protein (CREB) and IκB plasmids (Sorriento et al., 2009; 2010;) used for signal transduction studies, or with a κB-luciferase reporter and β-galactosidase for NFκB activity. In this latter case, 24 h after transfection, cells were deprived of serum overnight and stimulated with TNFα (20 ng·mL−1), as positive control, and Iso (10−7 mol·L−1) for 1, 3 and 6 h. Lysates were analysed using the luciferase assay system with reporter lysis buffer from Promega and measured in a β-counter. Relative luciferase activity was normalized against the co-expressed β-galactosidase activity to overcome variations in transfection efficiency between samples. In other experiments, cells were stimulated with Iso (10−7 mol·L−1) or TNFα (20 ng·mL−1) for 3 h (Santulli et al., 2009b).
Tube formation assay
When plated on matrigel (Becton Dickinson, Bedford, MA, USA), ECs organize themselves into a network-like structure, resembling sinusoids of immature vessels (Ciccarelli et al., 2008). Six-well multidishes were coated with growth factor-reduced matrigel (10 mg·mL−1) according to the manufacturer's instructions. Control and β2AR−/− ECs (2 × 105) were incubated at 37°C for 12 h in 1 mL of DMEM medium. Tube formation was defined as a structure exhibiting a length four times its width. Network formation was observed using an inverted phase-contrast microscope (ZEISS). Representative fields were taken, and the average of the total number of complete tubes formed by cells was counted in 15 random fields by two independent investigators (D.S and E.C.).
RT PCR
Total RNA was isolated from ECs deprived of serum overnight or mouse hindlimb muscle using Trizol reagent (Invitrogen) and cDNA was synthesized by means of Thermo-Script RT-PCR System (Invitrogen), following the manufacturer's instruction. After reverse transcription, real-time quantitative polymerase chain reaction (RT-PCR) was performed with the SYBR Green Real Time PCR master mix kit (Applied Biosystems, Carlsbad, CA, USA). The reaction was visualized with SYBR Green Analysis (Applied Biosystem) software on a StepOne thermocycler (Applied Biosystem). Primers for VEGF-165 and GAPDH gene analysis were as previously described (Sorriento et al., 2009).
Statistical analysis
Data are presented as mean ± SEM. Each experiment was performed from three to five times. P values were calculated by Student's t-test or two-way anova as appropriate. For distribution statistics, the chi-squared test was performed.
The nomenclature conforms to the British Journal of Pharmacology's ‘Guide to Receptors and Channels’ (Alexander et al., 2009).
Results
In vivo post-ischaemic angiogenesis
Blood perfusion evaluation
As shown in Figure 1A, compared to β2AR proficient controls, hindlimbs from β2AR−/− mice present a significantly lower βAR membrane-density. Intravascular delivery of adenovirus leads to the infections mostly of vascular cells, including endothelium and VSMC, and perivascular fibroblasts and skeletal myocytes as shown by LacZ staining (Figure S1A and B). Administration by this route of Adβ2AR in β2AR−/− mice restores βAR density in the hindlimb (Figure 1A). Gross morphology and functional analysis of mice following chronic ischaemia indicate a higher occurrence of necrosis, autoamputation and limping in β2AR−/− mice as compared to β2AR+/+ controls (Figure 1B). β2AR restoration by Adβ2AR was also able to prevent this symptom (Figure 1B). US evaluation of hindlimb perfusion (blood flow, BF) immediately after femoral artery removal shows an absence of flow in ischaemic HL in all groups of mice (data not shown). Over two weeks, BF was partially restored in β2AR+/+ while no improvement was observed in β2AR−/− mice (Figure 1C). A similar result was obtained with the dyed beads perfusion analysis (Figure 1D). As assessed by both techniques, the Adβ2AR restores blood perfusion through the ischaemic hindlimb (Figure 1C and D). In line with these results, chronic ischaemia appeared to induce a capillary rarefaction that was higher in the β2AR−/− compared to the β2AR+/+ mice, and Adβ2AR reversed this effect in the β2AR−/− (Figure 1E). β1AR mRNA levels were not affected by the β2AR removal, nor by Adβ2AR gene therapy (Figure S1C). We thus evaluated the production of reactive VEGF in the ischaemic hindlimb. Consistent with the perfusion data, VEGF gene expression was upregulated in the ischaemic hindlimbs of β2AR+/+ mice, while it was blunted in the β2AR−/− mice both before and after ischaemia (Figure 1F). In the latter, Adβ2AR gene therapy restored the VEGF level in basal conditions as well during chronic ischaemia (Figure 1F).
Figure 1.

β2AR knock-out exacerbated the symptoms of ischaemia and impaired perfusion and angiogenesis in ischaemic hindlimbs of mice. (A) Effects of adenoviral-mediated gene transfer on βAR levels (*P < 0.05 β2AR−/− vs. β2AR+/+; # P < 0.05, β2AR−/−/ADβ2AR vs. β2AR−/−, n = 3 to 5). (B) Ischaemia-induced skin lesions in β2AR−/− and β2AR+/+ hindlimbs (HLs) (*P < 0.05 β2AR−/− vs. β2AR+/+, χ2test, n = 10 per group). (C) HL blood flow over 14 days in β2AR+/+, β2AR−/−, and Adβ2AR-treated β2AR−/− ischaemic HL, as measured by US Doppler. ADβ2AR ameliorated blood perfusion in the ischaemic HL (*P < 0.05 β2AR−/− vs. β2AR+/+; # P < 0.05, ADβ2AR vs. β2AR−/−; n = 10 per group). (D) Blood perfusion in the ischaemic HL as evaluated by dyed beads dilution method (*P < 0.05 β2AR−/− vs. β2AR+/+; # P < 0.05, β2AR−/−/Adβ2AR vs. β2AR−/−; n = 10 per group). (E) HL muscle capillary density. (*P < 0.05 ischaemic vs. non-ischaemic muscle; #P < 0.05 β2AR−/−vs. β2AR+/+; ! P < 0.05, β2AR−/−/Adβ2AR vs. β2AR−/−; n = 5 per each group). (F) VEGF gene expression in ischaemic gastrocnemius muscle 3 days after femoral artery ligation and resection (*P < 0.05 vs. non-ischaemic, n = 3; ! P < 0.01 vs. non-ischaemic β2AR +/+; # P < 0.01 vs. ischaemic β2AR +/+; n = 3; $ P < 0.01 vs. non-ischaemic β2AR−/−; @ P < 0.01 vs. ischaemic β2AR−/− alone; n = 3).
Evaluation of angiogenic phenotypes in vitro
Matrigel assay and VEGF production in primary cultures of ECs
In order to study angiogenesis in vitro, we tested the ability of mouse EC primary cultures to organize into a network when plated on a matrigel substrate. β2AR gene deletion inhibited the ability of ECs to form into vascular tubes compared to wild-type cells (Figure 2A). To verify the relevance of NFκB to the pro-angiogenic phenotype of EC, we transfected β2AR+/+ cells with the NFκB inhibitor IκB, 48 h before plating cells on matrigel. As expected, IκB blocked the tubular formation of β2AR+/+ endothelial cells on matrigel (Figure 2B). We further investigated the pro-angiogenic phenotype of β2AR−/− ECs by evaluating VEGF production in basal conditions and after isoprenaline treatment. As shown in Figures 2C and 6 h of stimulation with Iso increased VEGF levels in β2AR+/+ but not in β2AR−/− EC. A similar result was obtained when VEGF mRNA levels were determined by RT-PCR in the same conditions (Figure 2D).
Figure 2.

β2AR gene deletion inhibits VEGF production and vascular tube formation in vitro. (A) Vascular tube formation of endothelial cells (ECs) was assessed on matrigel. β2AR−/− and β2AR+/+ control ECs (2 × 105) were incubated at 37°C for 12 h in 1 mL DMEM. Representative fields were taken, and the average of the total number of complete tubes was counted in 15 random fields by two independent investigators (*P < 0.05 vs. β2AR+/+ EC, 3 experiments). (B) Vascular tube formation of β2AR+/+ ECs (2 × 105) transfected with an empty Vector or a plasmid encoding for IkB was assessed on matrigel. Cells were treated as above. (*P < 0.05 vs. Vector, 3 experiments). (C) VEGF production in ECs measured by Western blot of extracellular medium. (*P < 0.05 vs. β2AR+/+; n = 3 per group). (D) VEGF mRNA in ECs measured by RT-PCR. (*P < 0.05 vs. β2AR+/+; n = 3 per group).
β2AR effects on NFκB signalling
To investigate the ability of β2ARs to modulate VEGF production and angiogenesis, we tested the possibility that β2AR may regulate the activity of the NFκB transcription factor. Indeed, in β2AR+/+ EC, βAR stimulation with Iso induced a time-dependent degradation of the endogenous NFκB inhibitor, IκBα (Figure 3A). Consistent with this result, the luciferase assays demonstrated a time-dependent increase in NFκB transcriptional activity after Iso stimulation (Figure 3B). As expected, TNFα also increased NFκB transcriptional activity in β2AR+/+ EC, and the overexpression of IκB inhibited the Iso-induced increase in NFκB activity (Figure 3B).
Figure 3.

Effects of β2AR gene deletion on NFκB signalling. (A) IκBα levels visualized by Western blot in β2AR+/+ ECs. Isoprenaline (Iso, 10−7M) was kept in the tissue culture medium for the time indicated. Densitometry units (ADU) (results normalized to actin response) are depicted in graphs. (*P < 0.05 vs. control, n = 3). (B) Effects of TNFα (20 ng·mL−1) for 6 h or Iso (10−7M) for 1, 3 and 6 h on NFκB transcription activity in β2AR+/+ EC and in β2AR+/+ EC transfected with the IκB plasmid (*P < 0.05 vs. control, # P < 0.05 vs. control; ! P < 0.05 Iso + IκB vs. IκB; n = 3 to 5). (C) IκBα levels assessed by Western blot in β2AR−/− and β2AR+/+ ECs stimulated with Iso (10−7M, 1 h) or TNFα (20 ng·mL−1, 1 h) (*P < 0.05 vs. control; # P < 0.05 vs. β2AR+/+; n = 3 to 5). (D) NFκB activity in EC induced by Iso (10−7M) and TNFα (20 ng·mL−1) (*P < 0.05 vs. control; # P < 0.05 vs. β2AR +/+; n = 3 to 5). (E) ECs were stimulated with Iso (10−7M) and TNFα (20 ng·mL−1). CREB phosphorylation was visualized by Western blot, digitalized and corrected for endogenous CREB. (*P < 0.05 vs. control; # P < 0.05 vs. β2AR +/+, n = 3). (F) EC were stimulated as in (E), and NFκB phosphorylation was visualized by WB, digitalized and corrected for total NFκB. (*P < 0.05 vs. control; # P < 0.05 vs. β2AR +/+, n = 3).
In ECs, β2AR gene deletion inhibits Iso-induced IκBα downregulation (Figure 3C), confirming that β2AR may regulate NFκB activation in response to Iso. Surprisingly, β2AR gene deletion also inhibited TNFα-induced IκBα downregulation (Figure 3C). Accordingly, restoration of β2ARs by means of Adβ2AR infection corrected both Iso and TNFα–mediated IκBα degradation to levels comparable to the ones observed in β2AR+/+ EC (Figure 3C), confirming the importance of β2ARs in NFκB endothelial signalling. In accord with this, in β2AR−/− EC, both Iso and TNFα-induced NFκB-activity were blocked, and Adβ2AR restored the responses to both agonists (Figure 3D). These data suggest that β2AR knock-out may have a general impact on cytokine transcription.
Recently, CREB, another β2AR-controlled transcription factor, has been shown to down-regulate the activity of NFκB (Ye, 2001). Indeed, CREB binding protein (CBP) and the related cofactor p300 are co-activators able to regulate the activity of transcription factors, and CREB and NFκB have been shown to compete for limiting amounts of CBP/p300 (Ye, 2001). In fact, the recruitment of these co-activators by CREB reduces their availability for NFκB. Thus, we evaluated CREB and NFκB activation by Western blot in β2AR−/− ECs stimulated with Iso or TNFα. β2AR gene deletion increased CREB phosphorylation both in basal conditions and after stimulation with Iso or TNFα and the restoration of β2ARs by adenovirus-mediated gene transfer decreased p-CREB levels (Figure 3E). Reciprocal results were obtained with NFκB. Indeed, compared to β2AR proficient controls, in β2AR−/− ECs, Iso and TNFα failed to induce NFκB phosphorylation while Adβ2AR infection restored the responses to both agonists (Figure 3F).
Identification of signal transduction components
To determine whether CREB upregulation is a common mechanism of inhibition for β2AR and TNFR stimulation of NFκB, we assessed pNFκB after stimulation with Iso or TNFα in β2AR+/+ EC trasfected with CREB or IkB. Indeed, both substances resulted in the inhibition of NFκB activation to both agonists (Figure 4A). Given the ability of the β2AR to couple to both Gs and Gi, we tested the possibility that Gi is indeed involved in maintaining low levels of CREB within the cell. To test this hypothesis, we treated cells with the Gi inhibitor Pertussis toxin for 18 h and then stimulated β2AR+/+ ECs with Iso. Gi inhibition was accompanied by the inhibition of Iso-induced IκB downregulation (Figure 4B) and NFκB phosphorylation (Figure 4C). Furthermore, PTX increased the basal level of pCREB and prevented Iso-induced dephosphorylation of CREB (Figure 4D).
Figure 4.
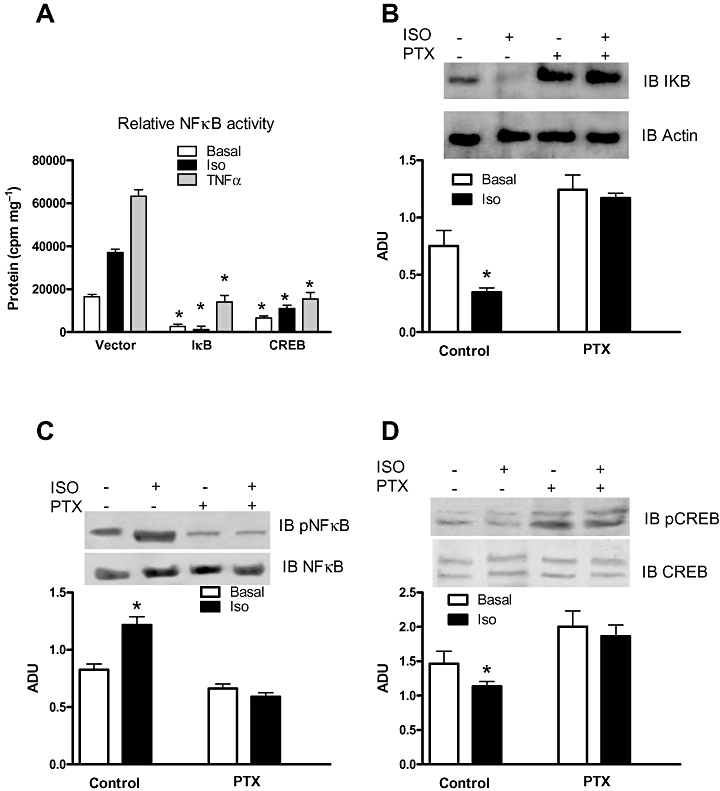
Molecular mechanisms involved in β2AR activation of NFκB. (A) Effects of transgenic expression of IκB and CREB in β2AR +/+ EC on NFκB activation in response to Iso (10−7M, 3 h) and TNFα (20 ng·mL−1). *P < 0.05 vs. Vector, n = 3. (B) Evaluation by Western blot of β2AR +/+ EC of the effects of pertussis toxin (PTX, 10−4M) on Iso-induced IκB downregulation. *P < 0.05 vs. basal, n = 4; ADU: arbitrary densitometric units. (C) Evaluation by Western blot of β2AR+/+ EC of the effects of pertussis toxin (PTX, 10−4M) on Iso-induced NFκB phosphorylation. *P < 0.05 vs. basal, n = 4. (D) Evaluation by Western blot of β2AR+/+ EC of the effects of pertussis toxin (PTX, 10−4M) on Iso-induced CREB dephosphorylation. *P < 0.05 vs. basal, n = 4.
Finally, we have presented these new findings in a cartoon as shown in Figure 5.
Figure 5.

In the wild-type cell (left), the presence of the β2AR causes a tonic stimulation that fosters the activation of NFκB, which, by translocating to the nucleus upon activation, competes for CBP with CREB. Furthermore, the β2AR exerts a tonic inhibition on cAMP production, by coupling to Gi. Once the receptor is removed (right), the inhibitory effect on cAMP is released, unbalancing the nuclear transcription towards CREB, which binds more CBP, thus making NFκB binding to the same cofactor unlikely to occur. pP65/p50: NFκB; TNFR: TNFα receptor.
Discussion
The results of our study provide compelling evidence that the removal of the β2AR impairs angiogenesis both in vitro and in vivo. This finding extends previous knowledge regarding the ability of β2AR overexpression to enhance angiogenesis in vivo, and it further supports the knowledge of the ability of β2AR to induce VEGF and cytokine production. Indeed, our work adds novel findings concerning the signalling that connects β2ARs and the nuclear transcriptional activity of NFκB.
Evidence has been obtained suggesting that the adrenergic system is activated within ischaemic regions (Newton et al., 1997) but the physiological relevance of this response is unclear. A teleological explanation would suggest that this activation is needed to better adapt to the ischaemic stress. Indeed, the regenerative properties of catecholamines have been implicated by the observation that catecholamine-deficient mice undergo inappropriate embryo development (Zhou et al., 1995). During postnatal life, catecholamines participate in cardiac and vascular remodelling and in tissue regeneration in response to various stresses (Iaccarino et al., 2001a; Ciccarelli et al., 2008). Adrenoceptors are the effectors of catecholamines, but their role appears to be redundant, since correct embryo development occurs in all of the AR knock-out models, and in adult life, mice with deletion of AR genes do not present basal phenotypic alterations. Under stress conditions, though, or in the presence of catecholamine challenge, a perturbation in the phenotype of KO mice has been noted (Rohrer et al., 1999). In particular, previous studies in βAR−/− mice have shown that β2ARs are mostly involved in vascular and metabolic mechanisms (Chruscinski et al., 1999; Rohrer et al., 1999). Our study extends this notion and is the first to illustrate the requirement for endogenous β2ARs in neoangiogenesis following chronic ischaemia, by providing the following evidence: (i) β2AR−/− mice present impaired tolerance to chronic ischaemia; (ii) this phenotype is rescued by the local restoration of β2AR membrane-density, induced by adenoviral-mediated gene transfer; (iii) a functional relationship appears to exist between the β2AR and NFκB, the transcription factor involved in ischaemia-induced cytokine production.
These results accord with our recent observation that overexpression of the β2AR enhances the adaptive pro-angiogenic response to ischaemia (Iaccarino et al., 2005). Importantly, the present results add to the hypothesis that other endogenously expressed βAR subtypes cannot undertake this role of β2ARs in ischaemic neo-angiogenesis. Intimate differences in the signalling capabilities of this receptor make it unique, and probably this is the reason for our findings. Removal of this receptor alters the intracellular signal transduction pathways to such an extent that the trascriptional status of the cell is modified. Our in vitro study revealed that angiogenesis is impaired in β2AR−/− EC, probably due to the impaired production of cytokines such as VEGF. The main transcription factors modulating VEGF expression are hypoxia-inducible factor-1α (HIF-1α) (Marti et al., 2000) and NFκB (Kiriakidis et al., 2003). HIF-1α activates the transcription of target genes in response to hypoxia and is inactive in basal conditions due to ubiquitination and degradation. NFκB, in contrast, can be activated following stimulation of different receptors. The ability of GPCRs to activate this transcription factor has been established in different cell types, mainly in the immune system. With regard to the βARs, the evidence showing that βAR agonists are able to induce NFκB activation is controversial. In monocytes, Farmer and Pugin (2000) showed that a number of βAR agonists had inhibitory effects on LPS-induced TNFα and IL-8 production, and proposed that cAMP through PKA is the mediator of such inhibition (Farmer and Pugin, 2000). In contrast, Chandrasekar et al. (2004) showed that in cardiac-derived ECs, Iso induces NFκB promoter activity in a β2AR-dependent manner. These authors concluded that β2ARs induce NFκB activation in a cAMP-independent manner, through the activation of Gi, Pi3K, Akt and IKK with IκBα degradation. Our results accord with the latter findings, since in β2AR+/+ EC, Iso induced IκBα degradation and enhanced NFκB transcriptional activity in a time-dependent manner. Furthermore, our data indicate that the physical presence of β2ARs is needed to activate NFκB; indeed, β2AR gene deletion inhibited NFκB activity in response to both GPCRs and TNF-Rs. Gαs and Gαi signalling pathways have opposite effects on NFκB: Gs-dependent signalling, induces PKA-dependent CREB activation and consequently inhibition of NFκB activity (Ye, 2001). In contrast, Gi activates NFκB by inhibiting/removing IκB. Our hypothesis is that the lack of β2ARs that have the ability to couple to Gi tilts the balance towards Gs-dependent signalling, which would lead to PKA-induced CREB activation and CBP/p300 recruitment, making the latter unavailable for NFκB activation (Figure 5). Indeed, validation of such a hypothesis derives from our studies with the pertussis toxin, showing the relevance of Gi for β2AR-induced activation of NFκB. Irrespective of this result, the observation that regardless of its mechanism of action β2AR influenced not only isoprenaline, but also TNFα-dependent activation of NFκB is strongly suggestive of a pivotal role played by β2ARs in the instruction of signals required for the fine-tuning of NFκB transcriptional activity. We have performed our experiments in the endothelial cells, but it is most likely that the β2AR can regulate NFκB activity also in other cell types. In particular it would be interesting to evaluate the skeletal muscle, which expresses a large number of β2ARs and is also an important source of VEGF.
In conclusion, our data indicate that the β2AR is important in mediating production of key pro-angiogenic cytokines, such as VEGF. The impairment of the signal transduction of this receptor results in imparment of angiogenesis in response to chronic ischaemia. Our data add a novel piece to the puzzling paradigm of those pathophysiological conditions that are characterized by increased adrenergic neural drive, impaired β adrenoceptor signalling and impaired organ function such as myocardial ischaemia. Under these conditions, the reduction of β2ARs signalling is detrimental not only for the cardiac function but also for the development of an adequate compensatory neoangiogenesis, which would worsen the blood supply to the ischaemic heart and accelerate the progression of myocardial dysfunction. The use of therapeutic strategies aimed at improving βAR signalling, may therefore achieve double efficacy, by ameliorating myocardial function and by hastening compensatory angiogenesis.
Acknowledgments
Fundings to GI from Italian Ministry of Research, PRIN 20074MSWYW.
Glossary
Abbreviations
- ADβ2AR
gene therapy with adenovirus encoding the human β2AR
- ADU
arbitrary densitometry units
- β2AR
β2 adrenoceptor
- β2AR−/−
β2AR knock-out mice
- BF
blood flow
- CREB
cAMP response element binding protein
- EC
endothelial cell
- US
ultrasound
- VEGF
vascular endothelial growth factor
Conflict of interest
The authors declare that they have no competing financial interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Teaching Materials; Figs 1–5 as PowerPoint slide.
Figure S1 Efficient gene delivery via femoral artery catheterization (A-B). Recombinant LacZ Adenovirus was delivered to the ischemic hindlimb via catheterization as described in the Methods section and results in gene transduction of capillaries (A) as well as perivascular muscle fibers (B). Muscle β1AR mRNA levels measured by RT-PCR (C). Hindlimb muscles processed as described in methods showing unmodified β1AR mRNA levels among β2AR +/+, β2AR−/− and Adβ2AR treated mice.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th Edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar B, Marelli-Berg FM, Tone M, Bysani S, Prabhu SD, Murray DR. Beta-adrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3K, Akt, IKK, and NF-kappaB. Biochem Biophys Res Commun. 2004;319:304–311. doi: 10.1016/j.bbrc.2004.04.185. [DOI] [PubMed] [Google Scholar]
- Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the beta2 adrenergic receptor gene. J Biol Chem. 1999;274:16694–16700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- Ciccarelli M, Cipolletta E, Santulli G, Campanile A, Pumiglia K, Cervero P, et al. Endothelial beta2 adrenergic signaling to AKT: role of Gi and SRC. Cell Signal. 2007;19:1949–1955. doi: 10.1016/j.cellsig.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Ciccarelli M, Santulli G, Campanile A, Galasso G, Cervero P, Altobelli GG, et al. Endothelial alpha1-adrenoceptors regulate neo-angiogenesis. Br J Pharmacol. 2008;153:936–946. doi: 10.1038/sj.bjp.0707637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer P, Pugin J. Beta-adrenergic agonists exert their ‘anti-inflammatory’ effects in monocytic cells through the IkappaB/NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol. 2000;279:L675–L682. doi: 10.1152/ajplung.2000.279.4.L675. [DOI] [PubMed] [Google Scholar]
- Ferro A, Queen LR, Priest RM, Xu B, Ritter JM, Poston L, et al. Activation of nitric oxide synthase by beta 2-adrenoceptors in human umbilical vein endothelium in vitro. Br J Pharmacol. 1999;126:1872–1880. doi: 10.1038/sj.bjp.0702512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Tomhave ED, Lefkowitz RJ, Koch WJ. Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by beta-adrenergic receptor stimulation and blockade. Circulation. 1998;98:1783–1789. doi: 10.1161/01.cir.98.17.1783. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Barbato E, Cipoletta E, Fiorillo A, Trimarco B. Role of the sympathetic nervous system in cardiac remodeling in hypertension. Clin Exp Hypertens. 2001a;23:35–43. doi: 10.1081/ceh-100001195. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Barbato E, Cipolleta E, Esposito A, Fiorillo A, Koch WJ, et al. Cardiac betaARK1 upregulation induced by chronic salt deprivation in rats. Hypertension. 2001b;38:255–260. doi: 10.1161/01.hyp.38.2.255. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Cipolletta E, Fiorillo A, Annecchiarico M, Ciccarelli M, Cimini V, et al. Beta(2)-adrenergic receptor gene delivery to the endothelium corrects impaired adrenergic vasorelaxation in hypertension. Circulation. 2002;106:349–355. doi: 10.1161/01.cir.0000022690.55143.56. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Ciccarelli M, Sorriento D, Cipolletta E, Cerullo V, Iovino GL, et al. AKT participates in endothelial dysfunction in hypertension. Circulation. 2004;109:2587–2593. doi: 10.1161/01.CIR.0000129768.35536.FA. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, et al. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res. 2005;97:1182–1189. doi: 10.1161/01.RES.0000191541.06788.bb. [DOI] [PubMed] [Google Scholar]
- Kiriakidis S, Andreakos E, Monaco C, Foxwell B, Feldmann M, Paleolog E. VEGF expression in human macrophages is NF-kappaB-dependent: studies using adenoviruses expressing the endogenous NF-kappaB inhibitor IkappaBalpha and a kinase-defective form of the IkappaB kinase 2. J Cell Sci. 2003;116:665–674. doi: 10.1242/jcs.00286. [DOI] [PubMed] [Google Scholar]
- Lembo G, Iaccarino G, Vecchione C, Barbato E, Izzo R, Fontana D, et al. Insulin modulation of an endothelial nitric oxide component present in the alpha2- and beta-adrenergic responses in human forearm. J Clin Invest. 1997;100:2007–2014. doi: 10.1172/JCI119732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, et al. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. doi: 10.1016/S0002-9440(10)64964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton GE, Adelman AG, Lima VC, Seidelin PH, Schampaert E, Parker JD. Cardiac sympathetic activity in response to acute myocardial ischemia. Am J Physiol. 1997;272:H2079–2084. doi: 10.1152/ajpheart.1997.272.5.H2079. [DOI] [PubMed] [Google Scholar]
- Rohrer DK, Chruscinski A, Schauble EH, Bernstein D, Kobilka BK. Cardiovascular and metabolic alterations in mice lacking both beta1- and beta2-adrenergic receptors. J Biol Chem. 1999;274:16701–16708. doi: 10.1074/jbc.274.24.16701. [DOI] [PubMed] [Google Scholar]
- Santulli G, Ciccarelli M, Palumbo G, Campanile A, Galasso G, Ziaco B, et al. In vivo properties of the proangiogenic peptide QK. J Transl Med. 2009a;7:41. doi: 10.1186/1479-5876-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Cipolletta E, Campanile A, Maione S, Trimarco V, Marino M, et al. Deletion of the CaMK4 Gene in Mice Determines a Hypertensive Phenotype. Circulation. 2009b;120:S613–S613. [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Sorriento D, Campanile A, Santulli G, Leggiero E, Pastore L, Trimarco B, et al. A new synthetic protein, TAT-RH, inhibits tumor growth through the regulation of NFkappaB activity. Mol Cancer. 2009;8:97. doi: 10.1186/1476-4598-8-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B, Iaccarino G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-{kappa}B-Dependent hypertrophic gene expression. Hypertension. 2010;56:696–704. doi: 10.1161/HYPERTENSIONAHA.110.155960. [DOI] [PubMed] [Google Scholar]
- Ye RD. Regulation of nuclear factor kappaB activation by G-protein-coupled receptors. J Leukoc Biol. 2001;70:839–848. [PubMed] [Google Scholar]
- Zhou QY, Quaife CJ, Palmiter RD. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature. 1995;374:640–643. doi: 10.1038/374640a0. [DOI] [PubMed] [Google Scholar]
- Zhou RH, Lee TS, Tsou TC, Rannou F, Li YS, Chien S, et al. Stent implantation activates Akt in the vessel wall: role of mechanical stretch in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:2015–2020. doi: 10.1161/01.ATV.0000095161.06906.ED. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.