

Abstract
BACKGROUND AND PURPOSE
Aortic valve stenosis (AVS) is associated with significant cardiovascular morbidity and mortality. To date, no therapeutic modality has been shown to be effective in retarding AVS progression. We evaluated the effect of angiotensin-converting enzyme inhibition with ramipril on disease progression in a recently developed rabbit model of AVS.
EXPERIMENTAL APPROACH
The effects of 8 weeks of treatment with either vitamin D2 at 25 000 IU for 4 days a week alone or in combination with ramipril (0.5 mg·kg−1) on aortic valve structure and function were examined in New Zealand white rabbits. Echocardiographic aortic valve backscatter (AVBS) and aortic valve : outflow tract flow velocity ratio were utilized to quantify changes in valve structure and function.
KEY RESULTS
Treatment with ramipril significantly reduced AVBS and improved aortic valve : outflow tract flow velocity ratio. The intravalvular content of the pro-oxidant thioredoxin-interacting protein was decreased significantly with ramipril treatment. Endothelial function, as measured by asymmetric dimethylarginine concentrations and vascular responses to ACh, was improved significantly with ramipril treatment.
CONCLUSIONS AND IMPLICATIONS
Ramipril retards the development of AVS, reduces valvular thioredoxin-interacting protein accumulation and limits endothelial dysfunction in this animal model. These findings provide important insights into the mechanisms of AVS development and an impetus for future human studies of AVS retardation using an angiotensin-converting enzyme inhibitor.
Keywords: aortic valve stenosis, endothelial function, redox stress, ACE inhibitor, thioredoxin-interacting protein, asymmetric dimethylarginine, echocardiography
Introduction
Aortic valve stenosis (AVS) is a major cause of cardiac morbidity and mortality (Aronow et al., 1999; Otto et al., 1999), and is associated with significant health-care costs in the Western community. Understanding the pathogenesis of AVS in humans is inherently restricted by the inability to examine aortic valves serially. However, models of the development of AVS have been developed, utilizing cell culture of valve fibroblasts (Jian et al., 2003; Kennedy et al., 2009) and a variety of animal models (Drolet et al., 2003; Rajamannan et al., 2005; Cuniberti et al., 2006; Weiss et al., 2006; Ngo et al., 2008). In view of the previously prevailing hypothesis that AVS might result primarily from an ‘atherogenesis-like’ process (Rajamannan, 2009), many of these models have utilized (partially or entirely) hypercholesterolaemia as a basis for the induction of aortic valve calcification (Drolet et al., 2003; Rajamannan et al., 2005; Weiss et al., 2006; Hekimian et al., 2009). However, hypercholesterolaemia in such models has never been proven to be sufficient to induce AVS (Drolet et al., 2003; Rajamannan et al., 2005; Weiss et al., 2006). Indeed, recent clinical studies have failed to show either any influence of cholesterol-lowering therapy on AVS progression (Cowell et al., 2005; Rossebo et al., 2008; Chan et al., 2010), or any association between hypercholesterolaemia and aortic valve disease in an aging population (Ngo et al., 2009).
In a rabbit model of AVS, we have recently shown that vitamin D2 alone for 8 weeks induced the development of AVS with calcification and inflammatory infiltration of the aortic valve, which was coupled to changes in valve echogenicity as well as transvalvular haemodynamic changes (Ngo et al., 2008), hallmark features of human AVS. Furthermore, this model exhibits marked elevation of intravalvular thioredoxin-interacting protein (TXNIP) (an endogenous inhibitor of the antioxidant thioredoxin, which plays a central role in regulating redox stress) (World et al., 2006).
There is evidence to suggest that the activation of angiotensin II (Ang II)-producing systems may be involved in the pathogenesis of AVS. Angiotensin-converting enzyme (ACE) (O'Brien et al., 2002) has been detected on stenotic valve lesions but not on normal valves. Furthermore, there is also evidence of local production of Ang II by chymase tryptase, and cathepsin G from activated mast cells identified on stenotic valve lesions (Helske et al., 2004; 2006;). Furthermore, interventions targeting the renin-angiotensin-aldosterone system may exert salutary effects on endothelial function, which is likely to be relevant to the pathophysiology of AVS, given that AVS is associated with valvular endothelial dysfunction (Chirkov et al., 2006) and systemic endothelial dysfunction (as determined from biochemical evidence; Ngo et al., 2007). Gkizas et al. (2010) have recently demonstrated that eplerenone limits the degenerative changes within the valve matrix of a rabbit model of aortic valve disease. Similarly, Osman et al. (2007) have demonstrated that β2-adrenoceptor stimulation, which induces endothelial release of nitric oxide (NO) (Figueroa et al., 2009), decreased calcification of human aortic valve interstitial cells.
However, to date, no randomized prospective study either in animal models or in a clinical setting has shown retardation of development of haemodynamically defined AVS by either ACE inhibitors, angiotensin-receptor blockers or aldosterone antagonists. The principal objective of the current study was therefore to determine whether the ACE inhibitor, ramipril, could modify the development of AVS in an animal model of AVS (Ngo et al., 2008). We also planned to examine the effects of ramipril on vascular endothelial function, and on intravalvular accumulation of TXNIP.
Methods
Experimental protocol
Male New Zealand White rabbits (2–2.5 kg) were treated with vitamin D2 alone (n = 14) or together with ramipril (n = 16) for 8 weeks. Vitamin D2 25 000 IU was administered on 4 days per week (n = 14), as previously described (Ngo et al., 2008). Ramipril was administered in drinking water at approximately 0.5 mg·kg−1 daily. Additionally, untreated rabbits were used as normal controls, for comparison (n = 6).
At baseline and after 8 weeks of treatment, echocardiography was performed under sedation with ketamine (3.5 mg·kg−1) and xylazine (5 mg·kg−1). Blood samples were taken at baseline, 4 and 8 weeks for the measurement of serum creatinine, calcium, phosphate and total cholesterol. Plasma asymmetric dimethylarginine (ADMA), a biochemical marker of endothelial dysfunction, was assayed at baseline and after 8 weeks' treatment.
After 8 weeks, the animals were killed under anaesthesia with ketamine/xylazine; hearts and thoracic aortae were rapidly removed and placed in ice-cold Krebs solution and aortic valve leaflets were excised within 30 min.
The experimental protocol was approved by the institutional animal ethics committee and conforms to the National Health and Medical Research Council of Australia guidelines for animal usage for experimentation.
Preparation of solutions
Vitamin D2 (Ergocalciferol 40 000 IU·g−1, Sigma Pharmaceuticals, South Croydon, Victoria, Australia) powder stored under nitrogen was weighed and dissolved in ethanol (100 µL), prior to dilution with tap water to a final volume of 55 mL for each rabbit. Vitamin D2 solution was prepared fresh daily. For the untreated control group, an equal amount of ethanol was added to drinking water.
Ramipril (crushed Tritace® 5 mg, Aventis Pharmaceuticals, Bridgewater, NJ, USA) was suspended in ethanol (100 µL). Ramipril (±vitamin D2) solution was diluted to a final volume of 55 mL. All rabbits received water and normal chow ad libitum.
Echocardiographic measurements
Ultrasound images were obtained with a 5 MHz sector phased-array probe, operated at 6.7 MHz, connected to a Vivid 5, GE Vingmed (Horten, Norway). Aortic valve backscatter (AVBS) was utilized as the primary measure of increased valve echogenicity as previously described (Ngo et al., 2004). Peak left ventricular outflow tract flow (LVOTv) and transaortic valve flow (AVv) were measured utilizing continuous-wave Doppler recordings from the highest velocity available; these were used as measures of AVS. The ratio of AVv/LVOTv was used to compare changes in haemodynamics between the two groups. Reproducibility between different observers of AVv/LVOTv was 3.3% (n = 6). Left ventricular wall thicknesses as measured by interventricular septal dimension (indexed for body weight) were measured from 2D-guided M-mode echocardiographic tracings. Echocardiographic imaging and analyses were performed by observers blinded to the treatments.
Evaluation of aortic valve echogenicity
Valve echogenicity was quantified by the determination of AVBS in rabbits, performed as previously described (Ngo et al., 2004; 2008;). Two-dimensional ultrasonic backscatter images of the aortic valves were obtained from standard parasternal-like long-axis view over three cardiac cycles with a zoom of 2 cm. Three consecutive scans were performed per animal. A total of 10 square-shaped regions of interest (five on the anterior leaflet and five on the posterior leaflets) were obtained by placing 7 × 7 pixel sample volumes on the valves. Backscatter values from the blood pool adjacent to the left ventricular outflow tract and the aortic blood pool were used as reference values, as previously described (Ngo et al., 2008).
Aortic valve characterization
TXNIP protein was assessed immunohistochemically using goat anti-TXNIP (D-15) antibody (SC 33098, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) as previously described (Ngo et al., 2008). Sections were incubated with the primary antibody diluted 1:200 with phosphate-buffered saline (PBS) overnight at 4°C; rabbit anti-goat biotinylated (DAKO, Carpinteria, CA, USA), diluted 1:200 with PBS was used as secondary antibody and then with an avidin-biotin peroxidase complex (Vector, Burlingame, CA, USA), diluted 1:200 with PBS. Localization of the peroxidase conjugates was achieved by using diaminobenizidine tetrahydrochloride as a chromagen for 1 min. Sections incubated with 1:10 normal goat serum, instead of the primary antiserum, served as negative controls. Immunostaining for TXNIP was quantified using computer-assisted image analysis, as previously described (Ngo et al., 2008).
Alazarin red S was utilized to quantify aortic valve calcification and RAM-11 to quantify macrophage infiltration as previously described (Ngo et al., 2008).
Real-time RT-PCR for TXNIP quantification
Rabbit aortic valves were snap-frozen and RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Real-time PCR was used to assess transcript levels with negative controls included in each run. Specific primers TXNIP and 28S for the use of SYBR Green are TXNIP forward AGGATTCTGTGAAGGTGATG; TXNIP reverse GCCTCTGACTGATGACAACT; 28S forward TGGTAAGGAAACCAGCTCTA; 28S reverse ATGCAGTATCAGCCAGTCTT. Primers specificity in real-time PCR reactions was confirmed using RT-PCR. Briefly, total RNA (1 µg) was treated with DNase I (Invitrogen) and then RNA was used in 25 uL of real-time PCR reaction including Brilliant SYBR Green One-step QRT-PCR Master Mix according to manufacturer's instructions (Stratagene, La Jolla, CA, USA). Real-time quantitations were performed on 7000 Sequence Detection System thermal Cycler (Applied Biosystems, Mulgrave, Victoria, Australia). The calculation of relative change in mRNA was performed using the delta-delta method as previously described (Qi et al., 2006; 2007;), with normalization for the housekeeping gene 28S.
Endothelial function
Plasma ADMA concentrations
Plasma concentrations of ADMA, an endogenous inhibitor of NO synthase, were measured by high-performance liquid chromatography with fluorescence detection using AccQ-Fluor as the derivatizing reagent as previously described (Heresztyn et al., 2004; Ngo et al., 2008).
Isolated aortic preparations
The thoracic aortae were cut into 2–3 mm rings and mounted under 2 g tension in organ baths as previously described (Ngo et al., 2008). Contractile responses were expressed as a percentage of the potassium physiological saline solution response. In aortic rings precontracted with phenylephrine to 70% of maximal, relaxation concentration-response curves were obtained for ACh and sodium nitroprusside, as measures of endothelium-dependent and direct NO-mediated relaxation respectively. Concentration-response curves were analysed by GraphPad Prism version 4 (GraphPad Software, Inc., La Jolla, CA, USA).
Study analysis/statistics
All statistical data involved comparisons between the vitamin D2 and vitamin D2/ramipril groups. The primary (null) hypothesis tested in this study was that ramipril would not affect AVS induced by vitamin D2 alone, as measured by AVv/LVOTv ratio and AVBS after 8 weeks; these analyses were performed using two-way analysis of variance (anova) and Student's unpaired t-test respectively. For a study with 15 animals in each group, on the basis of our previously published results (Ngo et al., 2008), there was approximately 80% power to detect 1.50 SD difference between group means (approximately 50% reduction of AVBS), corresponding to 6 dB of AVBS. Two-way anova was used to compare other echocardiographic parameters and biochemical parameters as well as comparing plasma ADMA concentrations over the 8 weeks of treatment. All other differences between groups were expressed as changes between baseline and after 8 weeks of treatment, and examined using unpaired t-test. Comparisons between non-parametric data were performed using Mann–Whitney U-test. The untreated rabbit (control) group was subjected to no formal statistical analysis as examinations of this group were subsidiary objectives of the study. Correlations between parameters of endothelial function and AVBS were performed utilizing linear regression. Real-time RT-PCR results are expressed as a fold change compared with the control value. Results are expressed as mean ± SEM, unless otherwise stated. A level of probability of P < 0.05 was accepted as statistically significant. All statistical analyses were performed using Graphpad Prism version 4 (GraphPad Software, Inc.).
Results
Effects of ramipril on echocardiographic parameters
In untreated rabbits (control group), changes in AVv/LVOTv and AVBS over 8 weeks were similar to those previously documented by us (Ngo et al., 2008): AVv/LVOTv increased marginally, while mean AVBS at 8 weeks was 4.7 ± 0.7 dB. Rabbits treated with vitamin D2 developed similar degrees of AVS to those previously described over an 8 week period (Ngo et al., 2008), as shown in Figure 1. Treatment with ramipril significantly retarded AVS development (F = 5.6, P = 0.01), as assessed by comparison of changes in AVv/LVOTv (Figure 2A). After 8 weeks, there was also significant diminution of AVBS values (P < 0.05) in the vitamin D2/ramipril-treated rabbits versus the vitamin D2 alone-treated group (Figure 2B). Ramipril also limited the development of left ventricular hypertrophy compared with vitamin D2 alone group (F = 10.8, P < 0.01) over 8 weeks (Figure 2C).
Figure 1.
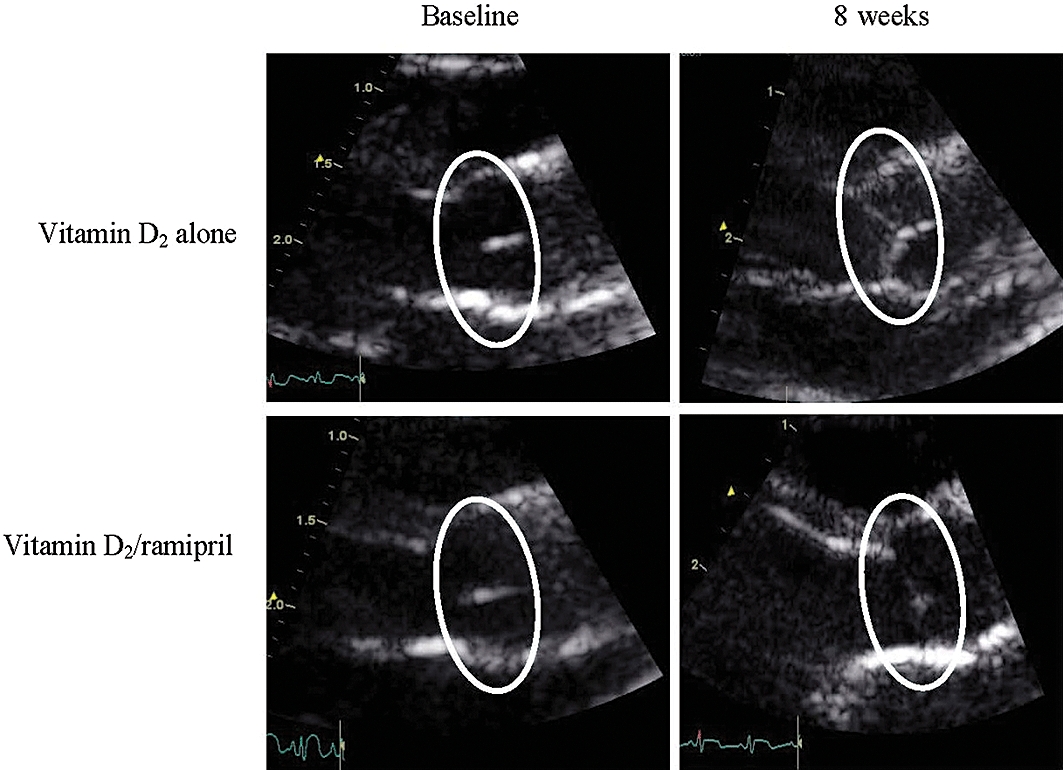
Echocardiographic views of rabbit aortic valves at baseline and after 8 weeks of treatment. Zoom pictures (2 cm) of parasternal-like long axis view of AV leaflets. Typical examples for vitamin-D2 group and vitamin D2/ramipril groups are shown at baseline and after 8 weeks. Regions of interest show position of valve. Note increased echogenicity in the vitamin D2 group (in comparison with vitamin D2/ramipril valves) at 8 weeks.
Figure 2.
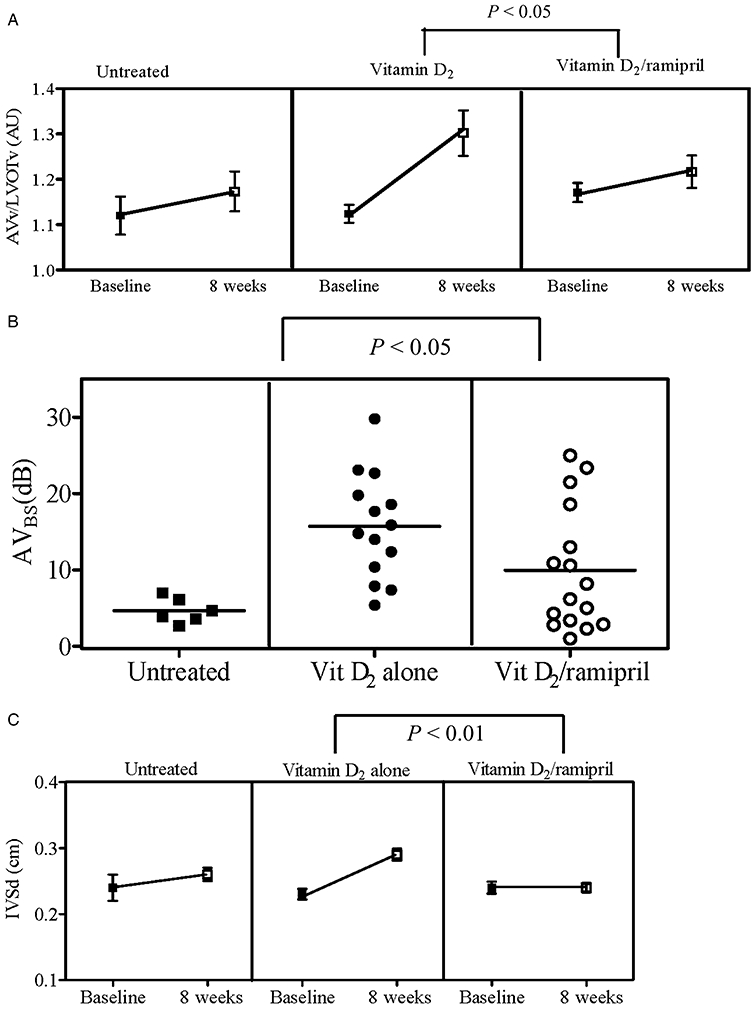
(A) Comparison of transvalvular flow between groups. In untreated rabbits, AVv/LVOTv was 1.12 ± 0.04 (baseline) versus 1.17 ± 0.04 (8 weeks); and AVBS 4.7 ± 0.7 dB after 8 weeks. There was a statistically significant difference in the time-treatment interaction between the vitamin D2 (n = 14) and vitamin D2/ramipril (n = 16) groups [F = 5.6; P = 0.02; two-way analysis of variance (anova)]. (B) AVBS comparison between groups. Mean AVBS in the vitamin D2 alone group was higher compared with the vitamin D2/ramipril group (15.7 ± 1.8 dB vs. 9.9 ± 2.0 dB, respectively, P < 0.05; unpaired t-test). Mean AVBS for the untreated group was 4.7 ± 0.7 dB. (C) Interventricular septal dimension (IVSd) comparison between groups. Significant retardation of left ventricular hypertrophy as evident by a decrease in IVSd in the vitamin D2/ramipril treated group versus vitamin D2 treated alone over the 8 weeks of treatment was observed (F = 10.8, P < 0.01; two-way anova). AVBS, aortic valve backscatter score; AVv, aortic valve velocity; LVOTv, left ventricular outflow tract flow velocity.
Valve characterization
Alazarin red S scores were significantly (Spearman's rank test: P < 0.001) correlated with AVBS, while there was a borderline correlation (Spearman's rank test: P = 0.07) between AVBS and RAM-11 scores (Figure 3A).
Figure 3.
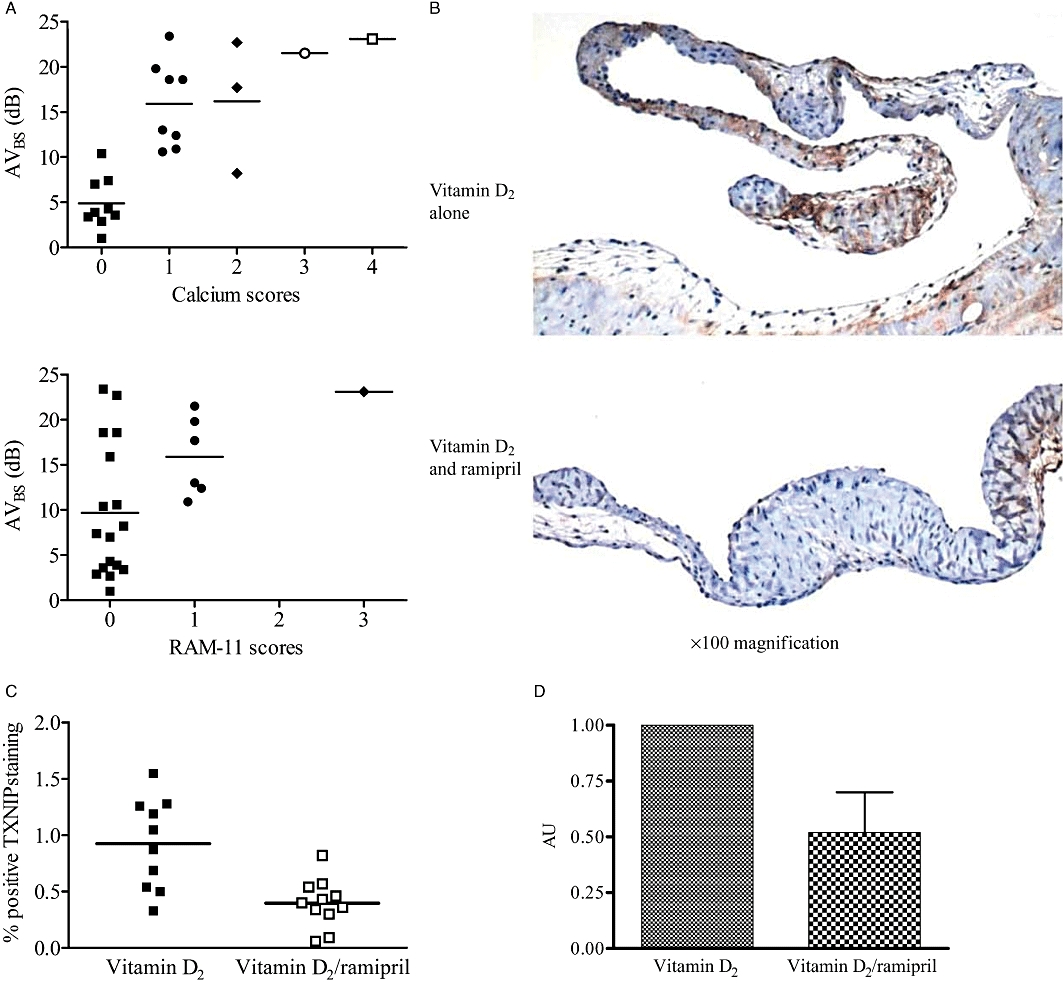
(A) Histological staining with Alizarin red S, and immunohistochemistry with RAM-11 for macrophages. Valves with lower AVBS scores had a significantly reduced calcium content (P < 0.001) and tended to have a reduced inflammatory milieu (P = 0.07). (B) Ramipril co-treatment reduced the TXNIP immuno-positive staining in valve tissue. (C) Ramipril co-treatment (n = 11) significantly reduced valvular TXNIP compared with vitamin D2 treatment alone (n = 10) (0.4 ± 0.06 vs. 0.9 ± 0.1, respectively, P = 0.001; unpaired t-test). (D) Real-time RT-PCR results showed that the vitamin D2/ramipril (n = 4) group had a significantly reduced TXNIP mRNA compared with the vitamin D2 alone group (n = 3) (P = 0.02). AVBS, aortic valve backscatter score; TXNIP, thioredoxin-interacting protein.
TXNIP immunostaining in vitamin D2-treated rabbits was similar to that previously observed in this model (Ngo et al., 2008). Concomitant treatment with ramipril markedly attenuated TXNIP accumulation, both as assessed via quantitative immunostaining and via real-time RT-PCR (Figures 3B–D).
Biochemistry
Treatment with vitamin D2 in combination with ramipril was associated with a non-significant trend towards a fall in serum creatinine (0.15 ± 0.014 mmol·L−1 versus 0.11 ± 0.02 mmol·L−1; F = 1.9, P = 0.19) and a significant decrease in calcium/phosphate product concentrations (9.9 ± 0.6 vs. 7.8 ± 0.6; F = 10.6, P = 0.004).
Vascular endothelial function
Plasma ADMA concentrations
As previously observed (Boger et al., 1997; Ngo et al., 2008), ADMA concentrations decreased with age in both groups of animals. Treatment with ramipril reduced ADMA concentrations compared with vitamin D2 treatment alone (F = 7.9, P < 0.01; Figure 4A); after 8 weeks, the ADMA concentrations were similar to those observed previously in untreated rabbits (Ngo et al., 2008).
Figure 4.
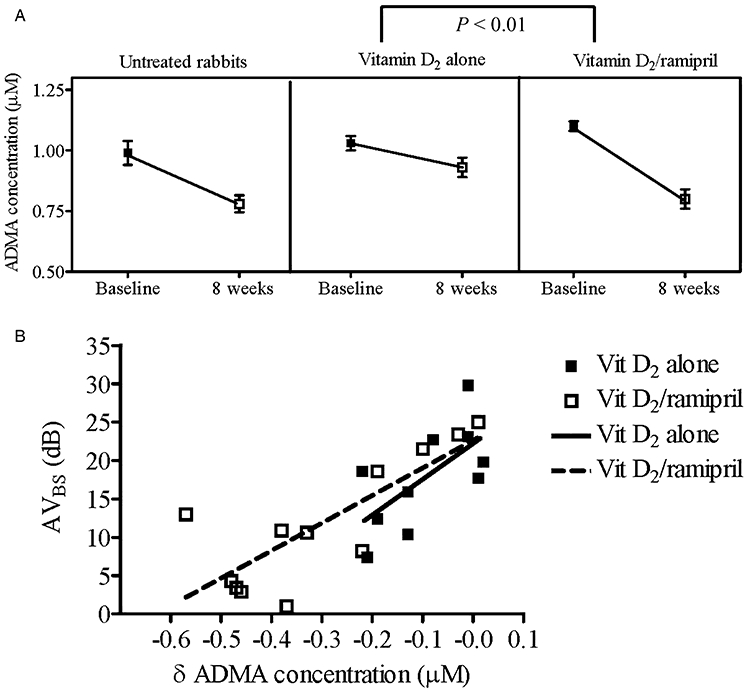
(A) Comparison of ADMA concentrations between the vitamin D2 (n = 10) and vitamin D2/ramipril (n = 12) groups over the 8 weeks of treatment. As previously demonstrated (Ngo et al., 2008), in untreated rabbits ADMA concentrations decrease substantially with aging (0.99 ± 0.05 to 0.78 ± 0.04 µM). Two-way analysis of variance showed there was statistical difference in ADMA concentrations between the two groups over time (F = 9.5; P = 0.006). (B) For both vitamin D2 alone and vitamin D2/ramipril, there were negative correlations between AVBS and changes in ADMA concentration (r2 = 0.4 and r2 = 0.6, respectively, both P < 0.05). ADMA, asymmetric dimethylarginine; AVBS, aortic valve backscatter score.
NO-mediated vasodilatation
As shown in Figure 5A, treatment with vitamin D2/ramipril was associated with significant increases in maximal vasodilator responses to ACh in isolated aortic ring preparations (P < 0.05). Responses to SNP were also significantly improved with ramipril treatment (P < 0.05).
Figure 5.
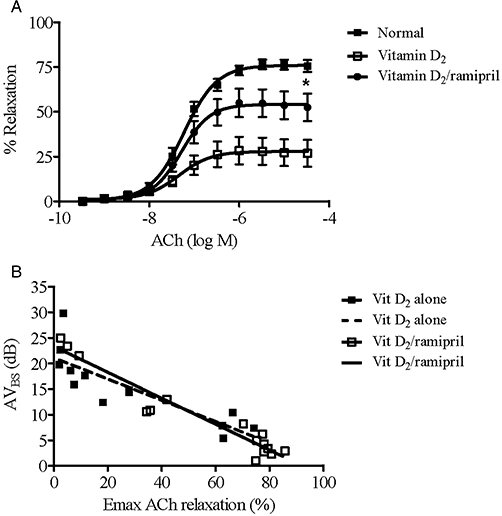
(A) Maximal ACh-induced relaxation responses of aortic rings from untreated rabbits (n = 6); vitamin D2 group (n = 13); and vitamin D2/ramipril group (n = 15). Maximal relaxation responses to ACh in untreated rabbits were 76.0 ± 1.5%. Relaxation was significantly greater in the vitamin D2/ramipril group versus the vitamin D2 alone group (P = 0.02). (B) Negative correlations between AVBS and maximum ACh-induced vasorelaxation (r2 = 0.7 and r2 = 0.9, respectively, P < 0.001 for both). AVBS, aortic valve backscatter score.
Correlations between endothelial function parameters and AVBS
Given that NO exerts an inhibitory effect both on TXNIP expression (Schulze et al., 2006) and on valve matrix calcification (Kennedy et al., 2009), we sought correlations between the extent of endothelial dysfunction in the presence and absence of ramipril treatment and AVBS scores. There were significant negative correlations between AVBS and both change in ADMA concentrations (δ ADMA) for both treatment groups (vitamin D2 alone, r2 = 0.6 and vitamin D2/ramipril, r2 = 0.8, respectively, both P < 0.001) (Figure 4B); and maximum ACh-mediated vasorelaxation (vitamin D2 alone, r2 = 0.8 and vitamin D2/ramipril, r2 = 0.9, respectively, both P < 0.001) (Figure 5B).
Discussion
The current study demonstrates that concomitant ramipril therapy retarded the development of vitamin D2-induced AVS in the rabbit model, as measured by changes in transvalvular flow profile and AVBS. This retardation of AVS development was associated with a reduction in TXNIP accumulation within the valve matrix, and the preservation of vascular endothelial function, as assessed both physiologically (ACh responses) and biochemically (ADMA concentrations). These findings therefore represent the first definitive demonstration that ACE inhibitors may retard the development of AVS.
A number of recent findings support the concept that TXNIP represents a fundamentally important modulator of intracellular oxidative stress, which is under the negative control of NO (Schulze et al., 2006; Kanagasingam et al., 2009; Shaked et al., 2009). Increased expression of TXNIP is associated with the stimulation of profibrotic mechanisms (Kobayashi et al., 2003; Advani et al., 2009). Furthermore, it has recently become clear that TXNIP functions to link oxidative stress with inflammatory activation (Schroder et al., 2010; Zhou et al., 2010), which is a prominent feature of early AVS (Miller et al., 2008). Indeed, increased TXNIP tissue concentrations are both reactive oxygen species (ROS)-stimulated and promote further ROS release by inhibiting thioredoxin activity (Yamawaki et al., 2003; World et al., 2006; Berk, 2007; Schroder et al., 2010). In view of the role of ADMA as an inhibitor of NO synthesis (Leiper and Vallance, 1999), it might be expected that increased ADMA concentrations would also be associated with increased TXNIP expression; however, this has not been reported previously. Although this currently demonstrated endothelial dysfunction with elevations of ADMA/TXNIP associations and AVS development does not prove causation, it provides a potential physiological basis for the observed effects of ramipril. Furthermore, in cell culture models of aortic valve calcification, we have demonstrated that both NO (Kennedy et al., 2009) and TXNIP modulate calcification (Kanagasingam et al., 2009).
Evidence for a potential therapeutic role for ACE inhibitors in AVS comes from three categories of findings: increased generation of Ang II within diseased valve matrix, pathophysiological actions of Ang II which might contribute to the development of AVS and limited in vivo studies. Apart from the specific generation of Ang II in stenotic valve lesions (Helske et al., 2004; 2006;), Ang II exerts multiple effects, which may be conducive to the development of fibrosis and calcification. Specifically, Ang II exerts pro-inflammatory effects, mainly by increasing the expression and activation of NAD(P)H oxidase (Cai et al., 2003). The associated production of ROS leads to an increased redox stress (Das et al., 2004), ‘scavenging’ of NO (Cai and Harrison, 2000), activation of apoptosis (Dimmeler and Zeiher, 2000) and the associated monocyte/macrophage infiltration (Brasier et al., 2002; Cathcart, 2004), which subsequently promotes accumulation of transforming growth factor-β1 (Saito et al., 2005) and may contribute to fibrosis and calcification. These data are consistent with recent findings related to the association of redox stress with AVS. As regards in vivo evidence, Arishiro et al. (2007), using a hypercholesterolaemic rabbit model, showed that olmesartan significantly reduced atherosclerotic changes in aortic valves. Simolin et al. (2009) also recently reported that enalapril reduced AV thickening in mice with renal insufficiency. In retrospective, non-randomized human studies, inconsistent benefits of ACE inhibitors have been found: O'Brien et al. (2005) found that the use of ACE inhibitors was associated with lower aortic valve calcium scores, while Rosenhek et al. (2004) demonstrated a non-significant trend for ACE inhibitors to be associated with the reduction of peak velocity and pressure gradient. Despite this considerable body of evidence, no other previous studies have been performed to test the hypothesis that ACE inhibitors retard AVS progression.
The current data also shed further light on the relationship between the development of AVS and potential impairment of NO effect, as measured in this study by ADMA concentrations and impaired NO-mediated vasodilatation. As AVS is characterized by the progressive loss of valve endothelium (Harasaki et al., 1978; Riddle et al., 1980; Otto et al., 1994), valvular endothelial function cannot be assessed. The extent of change in ADMA concentrations with time and the impairment of ACh-induced relaxation were correlated to the extent of AVBS in individual animals. The preservation of NO effect in early AVS might also be relevant to the limitation of the associated cardiovascular morbidity risk (Otto et al., 1999). Furthermore, these data are consistent with our previous findings, obtained in a cell culture model (Kennedy et al., 2009), in this model (Ngo et al., 2008) and in studies of advanced human AVS (Chirkov and Horowitz, 2007; Ngo et al., 2007). The effects of ramipril observed in the present study are also consistent with results from previous investigations with other ACE inhibitors with regard to ADMA kinetics (Ito et al., 2001; Chen et al., 2002) and endothelial function (Pitt et al., 2001).
The observed changes in vascular endothelial function and valvular TXNIP content might arise either from decreased Ang II generation and/or decreased bradykinin degradation. For example, the inhibition of NAD(P)H oxidase expression would limit NO ‘scavenging’ while bradykinin would stimulate NOS activation (Skidgel et al., 2006). In turn, NO has been shown to suppress TXNIP expression (Schulze et al., 2006). Thus, the suppression of TXNIP effect, and therefore of intracellular redox stress, could potentially modulate valvular calcium deposition in the setting of early AVS. No previous studies have examined the effects of ACE inhibitors on TXNIP physiology. However, the current data do not demonstrate a primary role for TXNIP in the development of AVS.
The results of the current study should also be considered in light of the recent publication of three negative clinical investigations of statin therapy in AVS (Cowell et al., 2005; Rossebo et al., 2008; Chan et al., 2010). As previously observed, hypercholesterolaemia, both in animal models (Drolet et al., 2003; Rajamannan et al., 2005; Weiss et al., 2006) and in humans (Ngo et al., 2009), is minimally associated with the development of AVS. The current experiments provide a basis for a ‘cholesterol-independent’ schema for the pathogenesis of the condition and for its limitation.
The results of the current study are subjected to several caveats. First, the development of AVS was induced by the exposure to supraphysiological doses of vitamin D2 and therefore does not precisely recapitulate human AVS, despite the marked similarities, as previously described (Ngo et al., 2008). Secondly, this model, despite the increase of transvalvular flow, is essentially one of ‘mild’ AVS; therefore, no conclusions can be drawn on the effects of ramipril in more advanced cases of the disease. On the other hand, the rabbit model of AVS reflects the major histological features of early clinical AVS, including inflammatory infiltration, lipid deposition and calcification (as well as valve thickening and narrowing). The extent of retardation of AVS, and the salvage of endothelial function, varied markedly in ramipril-treated rabbits: we cannot exclude the possibility that intake of ramipril was variable. Finally, it is possible, although unlikely, on the basis of epidemiological data (Ngo et al., 2009), that the hypotensive and afterload-reducing effects of ramipril might have contributed directly to the retardation of AVS development, and also to the retardation of the development of left ventricular hypertrophy. However, the effects of ramipril on intravalvular TXNIP accumulation, valve AVBS and valve AVv/LVOTv all suggest direct, rather than indirect effects on the valve itself.
The current data provide a strong rationale for the inception of a randomized trial of ACE inhibition as a strategy for the limitation of AVS in humans. While the evidence showing the benefits of ACE inhibitors in the prevention of cardiovascular death and myocardial infarction is unquestionable, the results of the current study add AVS as a potential therapeutic area where ACE inhibition may be of pivotal importance.
Acknowledgments
This work was supported in part by research grants from the National Health and Medical Research Council and National Heart Foundation of Australia.
Glossary
Abbreviations
- ACE
angiotensin converting enzyme
- ACEi
angiotensin converting enzyme inhibitor
- ADMA
asymmetric dimethylarginine
- Ang II
angiotensin II
- ARB
angiotensin-receptor blocker
- AVBS
aortic valve backscatter
- AVS
aortic valve stenosis
- AVv
aortic valve velocity
- IVSd
interventricular septal dimension
- LVOTv
left ventricular outflow tract flow velocity
- NAD(P)H
nicotinamide adenine dinucleotide phosphate
- PCR
polymerase chain reaction
- ROS
reactive oxygen species
- SeCr
serum creatinine
- SNP
sodium nitroprusside
- TGF-β1
transforming growth factor-β1
- TXNIP
thioredoxin-interacting protein
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Advani A, Gilbert RE, Thai K, Gow RM, Langham RG, Cox AJ, et al. Expression, localization, and function of the thioredoxin system in diabetic nephropathy. J Am Soc Nephrol. 2009;20:730–741. doi: 10.1681/ASN.2008020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arishiro K, Hoshiga M, Negoro N, Jin D, Takai S, Miyazaki M, et al. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits. J Am Coll Cardiol. 2007;49:1482–1489. doi: 10.1016/j.jacc.2006.11.043. [DOI] [PubMed] [Google Scholar]
- Aronow WS, Ahn C, Shirani J, Kronzon I. Comparison of frequency of new coronary events in older subjects with and without valvular aortic sclerosis. Am J Cardiol. 1999;83:599–600. doi: 10.1016/s0002-9149(98)00922-9. A598. [DOI] [PubMed] [Google Scholar]
- Berk BC. Novel approaches to treat oxidative stress and cardiovascular diseases. Trans Am Clin Climatol Assoc. 2007;118:209–214. [PMC free article] [PubMed] [Google Scholar]
- Boger RH, Bode-Boger SM, Brandes RP, Phivthong-ngam L, Bohme M, Nafe R, et al. Dietary L-arginine reduces the progression of atherosclerosis in cholesterol-fed rabbits: comparison with lovastatin. Circulation. 1997;96:1282–1290. doi: 10.1161/01.cir.96.4.1282. [DOI] [PubMed] [Google Scholar]
- Brasier AR, Recinos A, 3rd, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257–1266. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- Cathcart MK. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:23–28. doi: 10.1161/01.ATV.0000097769.47306.12. [DOI] [PubMed] [Google Scholar]
- Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121:306–314. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- Chen JW, Hsu NW, Wu TC, Lin SJ, Chang MS. Long-term angiotensin-converting enzyme inhibition reduces plasma asymmetric dimethylarginine and improves endothelial nitric oxide bioavailability and coronary microvascular function in patients with syndrome X. Am J Cardiol. 2002;90:974–982. doi: 10.1016/s0002-9149(02)02664-4. [DOI] [PubMed] [Google Scholar]
- Chirkov YY, Horowitz JD. Impaired tissue responsiveness to organic nitrates and nitric oxide: a new therapeutic frontier? Pharmacol Ther. 2007;116:287–305. doi: 10.1016/j.pharmthera.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Chirkov YY, Mishra K, Chandy S, Holmes AS, Kanna R, Horowitz JD. Loss of anti-aggregatory effects of aortic valve tissue in patients with aortic stenosis. J Heart Valve Dis. 2006;15:28–33. [PubMed] [Google Scholar]
- Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- Cuniberti LA, Stutzbach PG, Guevara E, Yannarelli GG, Laguens RP, Favaloro RR. Development of mild aortic valve stenosis in a rabbit model of hypertension. J Am Coll Cardiol. 2006;47:2303–2309. doi: 10.1016/j.jacc.2005.12.070. [DOI] [PubMed] [Google Scholar]
- Das DK, Maulik N, Engelman RM. Redox regulation of angiotensin II signaling in the heart. J Cell Mol Med. 2004;8:144–152. doi: 10.1111/j.1582-4934.2004.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Zeiher AM. Reactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factors. Regul Pept. 2000;90:19–25. doi: 10.1016/s0167-0115(00)00105-1. [DOI] [PubMed] [Google Scholar]
- Drolet MC, Arsenault M, Couet J. Experimental aortic valve stenosis in rabbits. J Am Coll Cardiol. 2003;41:1211–1217. doi: 10.1016/s0735-1097(03)00090-1. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Poblete I, Fernandez R, Pedemonte C, Cortes V, Huidobro-Toro JP. NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009;297:H134–H143. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- Gkizas S, Koumoundourou D, Sirinian X, Rokidi S, Mavrilas D, Koutsoukos P, et al. Aldosterone receptor blockade inhibits degenerative processes in the early stage of calcific aortic stenosis. Eur J Pharmacol. 2010;642:107–112. doi: 10.1016/j.ejphar.2010.05.048. [DOI] [PubMed] [Google Scholar]
- Harasaki H, Hanano H, Tanaka J, Tokunaga K, Torisu M. Surface structure of the human cardiac valve. A comparative study of normal and diseased valves. J Cardiovasc Surg (Torino) 1978;19:281–290. [PubMed] [Google Scholar]
- Hekimian G, Passefort S, Louedec L, Houard X, Jacob MP, Vahanian A, et al. High-cholesterol + vitamin D2 regimen: a questionable in-vivo experimental model of aortic valve stenosis. J Heart Valve Dis. 2009;18:152–158. [PubMed] [Google Scholar]
- Helske S, Lindstedt KA, Laine M, Mayranpaa M, Werkkala K, Lommi J, et al. Induction of local angiotensin II-producing systems in stenotic aortic valves. J Am Coll Cardiol. 2004;44:1859–1866. doi: 10.1016/j.jacc.2004.07.054. [DOI] [PubMed] [Google Scholar]
- Helske S, Syvaranta S, Kupari M, Lappalainen J, Laine M, Lommi J, et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006;27:1495–1504. doi: 10.1093/eurheartj/ehi706. [DOI] [PubMed] [Google Scholar]
- Heresztyn T, Worthley MI, Horowitz JD. Determination of l-arginine and NG, NG – and NG, NG' -dimethyl-L-arginine in plasma by liquid chromatography as AccQ-Fluor fluorescent derivatives. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;805:325–329. doi: 10.1016/j.jchromb.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Ito A, Egashira K, Narishige T, Muramatsu K, Takeshita A. Renin-angiotensin system is involved in the mechanism of increased serum asymmetric dimethylarginine in essential hypertension. Jpn Circ J. 2001;65:775–778. doi: 10.1253/jcj.65.775. [DOI] [PubMed] [Google Scholar]
- Jian B, Narula N, Li QY, Mohler ER, 3rd, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465. doi: 10.1016/s0003-4975(02)04312-6. discussion 465–466. [DOI] [PubMed] [Google Scholar]
- Kanagasingam NS, Horowitz JD, Kennedy JA. Transforming growth factor-β1 increases thioredoxin-interacting protein in calcifying aortic valve cells: attenuation by nitric oxide. Heart Lung Circ. 2009;18:S193. [Google Scholar]
- Kennedy JA, Hua X, Mishra K, Murphy GA, Rosenkranz AC, Horowitz JD. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol. 2009;602:28–35. doi: 10.1016/j.ejphar.2008.11.029. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Uehara S, Ikeda T, Itadani H, Kotani H. Vitamin D3 up-regulated protein-1 regulates collagen expression in mesangial cells. Kidney Int. 2003;64:1632–1642. doi: 10.1046/j.1523-1755.2003.00263.x. [DOI] [PubMed] [Google Scholar]
- Leiper J, Vallance P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc Res. 1999;43:542–548. doi: 10.1016/s0008-6363(99)00162-5. [DOI] [PubMed] [Google Scholar]
- Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo DT, Wuttke RD, Turner S, Marwick TH, Horowitz JD. Quantitative assessment of aortic sclerosis using ultrasonic backscatter. J Am Soc Echocardiogr. 2004;17:1123–1130. doi: 10.1016/j.echo.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Ngo DT, Heresztyn T, Mishra K, Marwick TH, Horowitz JD. Aortic stenosis is associated with elevated plasma levels of asymmetric dimethylarginine (ADMA) Nitric Oxide. 2007;16:197–201. doi: 10.1016/j.niox.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Ngo DT, Stafford I, Kelly DJ, Sverdlov AL, Wuttke RD, Weedon H, et al. Vitamin D(2) supplementation induces the development of aortic stenosis in rabbits: interactions with endothelial function and thioredoxin-interacting protein. Eur J Pharmacol. 2008;590:290–296. doi: 10.1016/j.ejphar.2008.05.051. [DOI] [PubMed] [Google Scholar]
- Ngo DT, Sverdlov AL, Willoughby SR, Nightingale AK, Chirkov YY, McNeil JJ, et al. Determinants of occurrence of aortic sclerosis in an aging population. JACC Cardiovasc Imaging. 2009;2:919–927. doi: 10.1016/j.jcmg.2009.03.016. [DOI] [PubMed] [Google Scholar]
- O'Brien KD, Shavelle DM, Caulfield MT, McDonald TO, Olin-Lewis K, Otto CM, et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106:2224–2230. doi: 10.1161/01.cir.0000035655.45453.d2. [DOI] [PubMed] [Google Scholar]
- O'Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM, et al. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005;165:858–862. doi: 10.1001/archinte.165.8.858. [DOI] [PubMed] [Google Scholar]
- Osman L, Chester AH, Sarathchandra P, Latif N, Meng W, Taylor PM, et al. A novel role of the sympatho-adrenergic system in regulating valve calcification. Circulation. 2007;116(Suppl 11):I282–I287. doi: 10.1161/CIRCULATIONAHA.106.681072. [DOI] [PubMed] [Google Scholar]
- Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341:142–147. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- Pitt B, O'Neill B, Feldman R, Ferrari R, Schwartz L, Mudra H, et al. The QUinapril Ischemic Event Trial (QUIET): evaluation of chronic ACE inhibitor therapy in patients with ischemic heart disease and preserved left ventricular function. Am J Cardiol. 2001;87:1058–1063. doi: 10.1016/s0002-9149(01)01461-8. [DOI] [PubMed] [Google Scholar]
- Qi W, Chen X, Twigg S, Polhill TS, Gilbert RE, Pollock CA. Tranilast attenuates connective tissue growth factor-induced extracellular matrix accumulation in renal cells. Kidney Int. 2006;69:989–995. doi: 10.1038/sj.ki.5000189. [DOI] [PubMed] [Google Scholar]
- Qi W, Chen X, Gilbert RE, Zhang Y, Waltham M, Schache M, et al. High glucose-induced thioredoxin-interacting protein in renal proximal tubule cells is independent of transforming growth factor-beta1. Am J Pathol. 2007;171:744–754. doi: 10.2353/ajpath.2007.060813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamannan NM. Calcific aortic stenosis: lessons learned from experimental and clinical studies. Arterioscler Thromb Vasc Biol. 2009;29:162–168. doi: 10.1161/ATVBAHA.107.156752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, et al. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91:806–810. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle JM, Magilligan DJ, Jr, Stein PD. Surface topography of stenotic aortic valves by scanning electron microscopy. Circulation. 1980;61:496–502. doi: 10.1161/01.cir.61.3.496. [DOI] [PubMed] [Google Scholar]
- Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, et al. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–1295. doi: 10.1161/01.CIR.0000140723.15274.53. [DOI] [PubMed] [Google Scholar]
- Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- Saito K, Ishizaka N, Aizawa T, Sata M, Iso-o N, Noiri E, et al. Iron chelation and a free radical scavenger suppress angiotensin II-induced upregulation of TGF-beta1 in the heart. Am J Physiol Heart Circ Physiol. 2005;288:H1836–H1843. doi: 10.1152/ajpheart.00679.2004. [DOI] [PubMed] [Google Scholar]
- Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- Schulze PC, Liu H, Choe E, Yoshioka J, Shalev A, Bloch KD, et al. Nitric oxide-dependent suppression of thioredoxin-interacting protein expression enhances thioredoxin activity. Arterioscler Thromb Vasc Biol. 2006;26:2666–2672. doi: 10.1161/01.ATV.0000248914.21018.f1. [DOI] [PubMed] [Google Scholar]
- Shaked M, Ketzinel-Gilad M, Ariav Y, Cerasi E, Kaiser N, Leibowitz G. Insulin counteracts glucotoxic effects by suppressing thioredoxin-interacting protein production in INS-1E beta cells and in Psammomys obesus pancreatic islets. Diabetologia. 2009;52:636–644. doi: 10.1007/s00125-009-1274-2. [DOI] [PubMed] [Google Scholar]
- Simolin MA, Pedersen TX, Bro S, Mayranpaa MI, Helske S, Nielsen LB, et al. ACE inhibition attenuates uremia-induced aortic valve thickening in a novel mouse model. BMC Cardiovasc Disord. 2009;9:10. doi: 10.1186/1471-2261-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skidgel RA, Stanisavljevic S, Erdos EG. Kinin- and angiotensin-converting enzyme (ACE) inhibitor-mediated nitric oxide production in endothelial cells. Biol Chem. 2006;387:159–165. doi: 10.1515/BC.2006.021. [DOI] [PubMed] [Google Scholar]
- Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- World CJ, Yamawaki H, Berk BC. Thioredoxin in the cardiovascular system. J Mol Med. 2006;84:997–1003. doi: 10.1007/s00109-006-0109-6. [DOI] [PubMed] [Google Scholar]
- Yamawaki H, Haendeler J, Berk BC. Thioredoxin: a key regulator of cardiovascular homeostasis. Circ Res. 2003;93:1029–1033. doi: 10.1161/01.RES.0000102869.39150.23. [DOI] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.