

Abstract
BACKGROUND AND PURPOSE
GPR35 is a poorly characterized G protein-coupled receptor at which kynurenic acid has been suggested to be the endogenous ligand. We wished to test this and develop assays appropriate for the study of this receptor.
EXPERIMENTAL APPROACH
Human and rat orthologues of GPR35 were engineered and expressed and assays developed to assess interaction with β-arrestin-2, activation of Gα13 and agonist-induced internalization.
KEY RESULTS
GPR35-β-arrestin-2 interaction assays confirmed that both the endogenous tryptophan metabolite kynurenic acid and the synthetic ligand zaprinast had agonist action at each orthologue. Zaprinast was substantially more potent than kynurenic acid at each and both agonists displayed substantially greater potency at rat GPR35. Two novel thiazolidinediones also displayed agonism and displayed similar potency at each GPR35 orthologue. The three ligand classes acted orthosterically with respect to each other, suggesting overlapping binding sites and, consistent with this, mutation to alanine of the conserved arginine at position 3.36 or tyrosine 3.32 in transmembrane domain III abolished β-arrestin-2 recruitment in response to each ligand at each orthologue.
CONCLUSIONS AND IMPLICATIONS
These studies indicate that β-arrestin-2 interaction assays are highly appropriate to explore the pharmacology of GPR35 and that Gα13 activation is an alternative avenue of signal generation from GPR35. Arginine and tyrosine residues in transmembrane domain III are integral to agonist recognition and function of this receptor. The potency of kynurenic acid at human GPR35 is sufficiently low, however, to question whether it is likely to be the true endogenous ligand for this receptor.
Keywords: GPR35, G protein-coupled receptor, kynurenic acid, zaprinast, species orthologues
Introduction
Although G protein-coupled receptors (GPCRs) are the largest and most studied group of transmembrane polypeptides and have been the most tractable group of proteins for the development of small molecule therapeutic medicines (Grigoriadis et al., 2009; Lundstrom, 2009), a substantial number remain poorly characterized. More than 80 members of the rhodopsin-like or class A family of GPCRs remain orphans (Xiao et al., 2008; Harmar et al., 2009), in that endogenously generated ligands that alter their function remain unknown. Moreover, for a number of others, reported pairings with endogenous ligand(s) that activate them remain uncertain, unconfirmed or of questionable physiological significance (Harmar et al., 2009; Yin et al., 2009). In certain cases, surrogate chemical ligands have been identified via high throughput screens and such compounds provide an initial means to explore the function of such GPCRs. However, it is possible that such compounds may act in a manner and at sites distinct from endogenous ligand(s), as has been shown in the case of allosteric agonists (Milligan et al., 2009).
GPR35 was first identified from human genomic DNA as an open reading frame corresponding to a 309 amino acid polypeptide, located on chromosome 2 (O'Dowd et al., 1998). It is most closely related to GPR23, which has been suggested (Noguchi et al., 2003), but not confirmed (Yin et al., 2009), to be a receptor for lysophosphatidic acid, and to the so-called third cannabinoid receptor, GPR55 (Brown, 2007). There appears to be an alternate form of GPR35 with an extended N-terminal sequence and the expression of GPR35 in NIH3T3 cells has been reported to have transforming activity, consistent with indications that GPR35 may play a role in gastric cancers (Okumura et al., 2004). Expression of GPR35 is reported to be predominantly in the gastro-intestinal tract and immune tissues (O'Dowd et al., 1998; Wang et al., 2006; Barth et al., 2009), suggesting further potential roles in immuno-modulation, and it has also been reported to be expressed in pancreatic islets (Leonard et al., 2005).
Although better established as a non-selective antagonist at ionotropic NMDA and AMPA/kainate glutamate receptors where it blocks kainic acid-induced neurotoxicity (Stone and Darlington, 2002), the tryptophan metabolite kynurenic acid has also been shown to have agonist action at GPR35 (Wang et al., 2006), and measures of levels of kynurenic acid indicate it to be present in micromolar concentration in rat small intestine (Kuc et al., 2008). Such data are potentially consistent with kynurenic acid being an endogenous agonist for GPR35 (Wang et al., 2006; Barth et al., 2009). Furthermore, it has been shown that levels of kynurenic acid can be elevated markedly in the bile in conditions such as cholecystolithiasis and obstructive jaundice (Paluszkiewicz et al., 2009). Zaprinast, commonly employed as an inhibitor of cGMP phosphodiesterases (Corbin and Francis, 2002), has also been reported to have agonist action at GPR35 (Taniguchi et al., 2006). Despite these observations, there is no information on the mechanism of binding of either of these ligands to GPR35 or even whether they might bind to similar sites.
Previous studies on the pharmacology of GPR35 have tended to employ either promiscuous and/or chimeric G proteins (Kostenis et al., 2005) to generate easy to measure ligand-induced signals (Leonard et al., 2005). Although a common approach within the pharmaceutical industry (Milligan and Rees, 1999; Kostenis et al., 2005), such studies may provide limited insight into the natural G protein coupling profile of GPCRs. As noted earlier, GPR35 is related to GPR55, an unusual GPCR in that it has been shown to have marked selectivity to activate Gα13 (Ryberg et al., 2007). The Gα12/Gα13 group of heterotrimeric G proteins (Worzfeld et al., 2008) are significantly less studied than the other subgroups, not least because most approaches to measure their activation are either indirect or complex. A further approach to assess GPCR function and pharmacology that has become widely used in recent times is to monitor agonist-induced interactions between a GPCR and a β-arrestin (Pfleger et al., 2007; van der Lee et al., 2009). By employing both GPR35-β-arrestin-2 interaction assays and additionally demonstrating activation of Gα13, we now confirm that both zaprinast and the endogenous tryptophan metabolite kynurenic acid have agonist action at GPR35, able to activate both human and rat orthologues of the receptor. Zaprinast is substantially more potent than kynurenic acid at both orthologues and, indeed, the potency of kynurenic acid at human GPR35 is sufficiently low to question whether it is likely to be the true endogenous ligand for this receptor. Differences in the potency of these two ligands, and the higher potency of both ligands at the rat compared with the human orthologue in the β-arrestin-2 interaction assays, were mirrored by the relative potency of the ligands in their capacity to cause internalization of the two orthologues. We also show that two novel thiazolidinediones have agonist action and similar potency at the two orthologues of GPR35. Finally, we demonstrate key roles for arginine and tyrosine residues on the predicted inward facing side of transmembrane domain III in the binding/function of each of the three agonist classes.
Methods
Drugs, chemical reagents and other materials
Materials for cell culture were from Sigma-Aldrich (Gillingham, Dorset, UK) or Invitrogen (Paisley, Strathclyde, UK). Polyethylenimine (PEI), linear MW-25000 was from Polysciences Inc. (Warrington, PA, USA). Zaprinast [2-(2-propyloxyphenyl)-8-azapurin-6-one], kynurenic acid and dipyridamole were from Tocris Bioscience (Bristol, UK). Ethyl 4-hydroxy-2-quinolinecarboxylate, anti-FLAG M2 monoclonal antibody, anti-FLAG polyclonal antiserum (from rabbit) and anti-goat IgG peroxidase (from rabbit) were from Sigma-Aldrich. Compound 10 ((Z)-[4-(2,4-dioxo-thiazolidin-5-ylidenemethyl)-phenoxy] acetic acid) was from ChemBridge Corporation (San Diego, CA USA) and Compound 3 ((Z)-5-(3-chloro-benzylidene)-thiazolidine-2,4-dione) from Ambinter SARL (Paris, France). β-Arrestin PathHunter™ assay technology was licensed from DiscoveRx (Birmingham, UK). Anti-mouse and anti-rabbit IgG horseradish peroxidase linked antisera (from sheep) were from GE Healthcare (Amersham, Buckinghamshire, UK). The antiserum directed against green fluorescent protein (GFP) and cross-reactive with enhanced yellow fluorescent protein (eYFP) was produced in-house. The active-state dependent Gα13 antibody was from NewEast Biosciences (Malvern, PA, USA). cDNA encoding Gln226Leu Gα13 was purchased from the Missouri S&T cDNA resource centre (Missoula, MI, USA). cDNA encoding eYFP was supplied by Clontech (CA, USA).
Test systems used
DNA constructs
Epitope tagging of GPR35
cDNAs corresponding to the short, 927 base pair version of human GPR35 (originally from dorsal root ganglion), and the 921 base pair sequence (from colon) from Rattus norvegicus corresponding, respectively, to open reading frames of 309 and 307 amino acids were obtained from Pfizer (Taniguchi et al., 2006) and used as PCR templates. A FLAG epitope (amino acid sequence DYKDDDDK) was introduced in the N-terminal end of both human and rat GPR35 cDNA by PCR using the following primers: human sense, 5′GCGAAGCTTGCCACCATGGATTACAAGGATGACGACGATAAGAATGGCACCTACAACACC 3′, rat sense, 5′CGCGGATCCGCCACCATGGATTACAAGGAT GACGACGATAAGAACAATACAAATTGTAGCATC 3′; and human antisense: 5′GGCCGCGGCCGCCTCTAGAATTAGGCGAGGG 3′, rat antisense: 5′GGCCGCGGCCGCCGGTGAGGCTCAGGCTCTG 3′. The HindIII, BamHI and NotI restriction sites used for cloning are underlined. The resulting cDNA was subsequently cloned in-frame into the HindIII/BamHI and NotI sites of an eYFP-pcDNA5/FRT/TO plasmid yielding the final N-terminal epitope- and C-terminal fluorescent protein-tagged constructs FLAG-humanGPR35-eYFP-pcDNA5/FRT/TO, and FLAG-ratGPR35-eYFP-pcDNA5/FRT/TO. The integrity of each fusion was confirmed by DNA sequencing.
Site-directed mutagenesis of human and rat GPR35
Using the Stratagene QuikChange method, primers were used to introduce mutations into FLAG-tagged human GPR35-eYFP or FLAG-tagged rat GPR35-eYFP in either pcDNA3 or pcDNA5/FRT/TO. Template was digested with DpnI to leave mutated plasmid and sequencing was carried out to confirm the introduction of the mutations.
Construction of β-arrestin2-Renilla luciferase
A full-length cDNA encoding Renilla luciferase 6 (Rluc; 312 amino acids) (Lorenz et al., 1991), generated by PCR amplification of a Renilla luciferase-containing vector plasmid (pRLCMV), was digested with XbaI and NhoI, and the resulting 1-kilobase XbaI-NhoI fragment was subcloned into a bovine β-arrestin2-pcDNA3 vector, resulting in β-arrestin2-Rluc-pcDNA3.
Construction of G protein chimeras
Chimeric Gαq/Gα12 and Gαq/Gα13 G proteins were generated by replacing the C-terminal 5 amino acids of Gαq with the equivalent sequence from Gα12 or Gα13. Gαq/Gα12 sense primer; GCTATCGACGGTACCGCCACCATGACTCTGGAGTCCATCATGGCGTGC, and antisense primer; GTCGATAGCGCGGCCGCTCACTGCAGCATGATGTCCTTCAGGTTCTCCTGCAGGATGGTGTCCTTGACGGCTGCAAAGAC. The Gαq/Gα13 sense primer was the same as for Gαq/Gα12, and antisense primer; GTCGATAGCGCGGCCGCTCACTGTAGCATAAGCTGCTTGAGGTTGTCATGCAGGATGGTGTCCTTGACGGCTGCAAAGAC. The KpnI and NotI restriction sites used for cloning are underlined. The resulting cDNA was cloned into pcDNA3 and confirmed by DNA sequencing.
Cell culture and transfection
Flp-In™ T-REx™ 293 cells (Canals et al., 2006; Canals and Milligan, 2008; Stoddart et al., 2008a) (Invitrogen) were maintained in Dulbecco's modification of Eagle's medium without sodium pyruvate (Invitrogen), supplemented with 10% (v/v) foetal calf serum, 1% penicillin/streptomycin mixture, and 10 µg·mL−1 blasticidin at 37°C in a 5% CO2 humidified atmosphere. PathHunter™ HEK 293 β-arrestin2 Eco-FECT cells (DiscoveRx) were maintained in Dulbecco's modification of Eagle's medium supplemented with 0.292 g·L−1 L-glutamine, 10% (v/v) foetal calf serum, 1% penicillin/streptomycin mixture, 200 µg·mL−1 hygromycin B, and 250 µg·mL−1 zeocin at 37°C in a 5% CO2 humidified atmosphere. HEK293T cells were maintained in Dulbecco's modification of Eagle's medium supplemented with 0.292 g·L−1 L-glutamine and 10% (v/v) newborn calf serum at 37°C in a 5% CO2 humidified atmosphere. Transfection was performed using 1 mg·mL−1 PEI, linear MW-25000. Cells were plated, grown until 60–80% confluent then transfected with 5 µg of required plasmid DNA and PEI (ratio 1:6 DNA : PEI), diluted in 150 mM NaCl, pH 7.4. After incubation at room temperature for 10 min, the mixture was added to HEK 293T cells. Cells were incubated for 24 h then transferred to 96-well plates coated with poly-D-lysine. In all experiments, the total amount of DNA transfected was equalized between constructs by the addition of the empty expression vector pcDNA3.
Generation of Flp-In™ T-REx™ 293 cells stably expressing forms of GPR35
To generate Flp-In™ T-REx™ 293 cells able to inducibly express FLAG-tagged human GPR35-eYFP, FLAG-tagged rat GPR35-eYFP or mutants of these receptors, the cells were transfected with a mixture containing the desired cDNA in pcDNA5/FRT/TO vector and the pOG44 vector (1:9) using Effectene (Qiagen), according to the manufacturer's instructions. After 48 h, the medium was changed to medium supplemented with 200 µg·mL−1 hygromycin B to initiate selection of stably transfected cells. Following isolation of resistant cells, expression of the appropriate construct from the Flp-In™ locus was induced by treatment with up to 100 ng·mL−1 doxycycline for 24 h.
Experimental design
Endoglycosidase treatment
Endoglycosidase treatment (Canals et al., 2006) was carried out overnight at 37°C using peptide N-glycosidase F (NGaseF) (Roche Diagnostics), at a final concentration of 1 unit·µL−1.
Gα13 activation assay
Gα13 activation assays were performed using a Gα13 activation assay kit (NewEast Biosciences) according to the manufacturer's instructions. Flp-In™ T-REx™ 293 cells able to express FLAG-tagged human GPR35-eYFP or FLAG-tagged rat GPR35-eYFP in response to doxycycline were induced and incubated for varying times with either vehicle or agonist. Cells were immediately washed with ice-cold phosphate-buffered saline and prepared in ice-cold lysis buffer. Lysates were incubated with the anti-active-state Gα13 mouse monoclonal antibody. Gα13-GTP was then recovered using protein A/G agarose and the precipitated Gα13-GTP was detected following SDS-PAGE by immunoblot analysis using a conformation insensitive anti-Gα13 rabbit polyclonal antiserum. Parallel immunoblots of cell lysates with the polyclonal anti-Gα13 antiserum assessed total cell levels of this G protein.
Measurements made
Single cell Ca2+ imaging
HEK 293T cells were plated onto poly-D-lysine coated coverslips and transfected with either FLAG-tagged human GPR35-eYFP or FLAG-tagged rat GPR35-eYFP along with chimeric Gαq/Gα12, Gαq/Gα13 or wild type Gαq using PEI. Single cell calcium assays were then performed as described previously (Stoddart et al., 2008a).
Cell lysates and western blotting
Cell lysates were obtained by harvesting the cells with ice-cold radioimmuno-precipitation assay (RIPA) buffer (50 mM HEPES, 150 mM NaCl, 1% Triton X-100, and 0.5% sodium deoxycholate supplemented with 10 mM NaF, 5 mM EDTA, 10 mM NaH2PO4, 5% ethylene glycol, and Complete protease inhibitor mixture (Roche Diagnostics, West Sussex, UK), pH 7.4). Cell extracts were then centrifuged for 30 min at 14 000×g, and the supernatant was recovered. Samples were subjected to SDS-PAGE analysis using 4–12% BisTris gels (NuPAGE, Invitrogen) and MOPS buffer. After electrophoresis, proteins were transferred onto nitrocellulose membranes that were incubated in a solution of 5% non-fat milk and 0.2% Tween-20 in Tris-buffered saline at 4°C on a rotating shaker overnight to block non-specific binding sites. The membrane was then incubated for 3 h at room temperature with anti-FLAG monoclonal antibody or anti-GFP antiserum and detected using horseradish peroxidase-linked anti-rabbit IgG secondary antiserum (GE Healthcare, Buckinghamshire, UK), or horseradish peroxidase-linked anti-goat IgG secondary antiserum (Sigma). Immunoblots were developed by application of enhanced chemiluminescence solution (Pierce).
Cell-surface biotinylation studies
For cell-surface biotinylation, cells were grown in six-well plates coated with poly-D-lysine and induced to express the appropriate GPR35 construct as described above. Ligands were added to cells for 30 min then the cells were washed with ice-cold borate buffer (10 mM boric acid, 154 mM NaCl, 7.2 mM KCl, and 1.8 mM CaCl2, pH 9.0) and incubated on ice with 1 mL of 0.8 mM EZ-Link sulphosuccinimidyl 2-(biotinamido)ethyl-1,3-dithiopropionate (Pierce) in borate buffer for 15 min. The cells were then rinsed with a solution of 0.192 M glycine and 25 mM Tris, pH 8.3, to quench the excess biotin, and lysed with RIPA buffer. Lysates were centrifuged for 30 min at 14 000×g, and the supernatant was recovered. An aliquot of the lysate was saved for Western blotting. Cell-surface biotinylated proteins were isolated using 50 µL of ImmunoPure immobilized streptavidin (Pierce). After 1 h of incubation at 4°C with constant rotation, samples were centrifuged, and the streptavidin beads were washed three times with RIPA buffer. Finally, the biotinylated proteins were eluted with 50 µL of SDS sample buffer for 1 h at 37°C, and SDS-PAGE and Western blotting were performed.
Cell surface elisa
Receptor expression in HEK293T cells was measured 48 h after transfection using PEI (as described above). From a 10 cm dish, cells were seeded at 50 000 cells per well into poly-D-lysine coated 96 well plates. After 24 h, the medium was replaced with 20 mM HEPES /DMEM (pH 7.4) containing anti-FLAG antiserum (1:1000) and cells were incubated for 30 min. The cells were washed twice with 20 mM HEPES/Dulbeccos' modified Eagles' medium and then incubated with anti-mouse horseradish peroxidase-conjugated IgG at a 1:5000 dilution as secondary antibody and 1 µM Hoechst 33 342 nuclear stain (Boehringer GmbH, Mannheim, Germany) to determine cell number. Cells were washed twice in PBS, incubated with Sureblue 3′,3′,5,5′-tetramethylbenzidine liquid substrate reagent (Insight Biotechnology) for 5 min at room temperature and the absorbance at 620 nm measured using a Victor2 Multilabel Counter (Packard Bioscience). The absorbance was normalized to the cell number in the well.
Live cell epifluorescence microscopy
Cells expressing the appropriate receptors tagged with eYFP were grown on poly-D-lysine coated coverslips. The coverslips were placed into a microscope chamber containing physiological saline solution (130 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, and 10 mM d-glucose, pH 7.4). Fluorescent images were acquired using a Nikon TE2000-E inverted microscope (Nikon Instruments, Melville, NY) equipped with an oil immersion Plan Fluor lens and a cooled digital CoolSNAPHQ charge-coupled device camera (Photometrics, Tucson AZ, USA).
DiscoveRx PathHunter™β-Arrestin assay
FLAG-tagged human and rat GPR35 were cloned into the Prolink vector (DiscoveRx) for receptor-Prolink fusion protein production (Yin et al., 2009). Parental HEK293 cells that stably express β-arrestin2-β-gal-EA (HEK 293-BAEA) (Yin et al., 2009) were transiently transfected with required receptor using PEI. Cells were incubated 24 h then 30 000 cells were seeded onto poly-D-lysine coated 96 well plates and incubated overnight. Medium was exchanged prior to compound addition then cells were incubated 60 min at 37°C. Detection reagents (DiscoveRx) were then added to each well and incubated for a further 60 min at room temperature. Plates were read on a Victor2 Multilabel Counter (Packard Bioscience) using a standard luminescence protocol.
BRET
HEK293T cells were transfected with required receptor tagged with eYFP and β-arrestin2 tagged with Renilla luciferase 6 (Lorenz et al., 1991) (ratio 4:1), using PEI. An additional transfection was performed with only the Renilla luciferase construct and empty expression vector pcDNA3. From 10 cm dishes, cells were seeded at 50 000 cells per well into poly-D-lysine coated 96 well plates. After 24 h, cells were washed twice with Hank's Balanced Salt Solution (pH 7.4), and coelentrazine-h (Promega, Southampton, UK) was added to a final concentration of 5 µM. Cells were incubated in darkness for 10 min at 37°C before addition of ligands. Cells were incubated for a further 5 min at 37°C before reading on a Mithras LB940 plate reader (Berthold Technology, Bad Wildbad, Germany), that allows sequential reading of emission signals detected at 485 nm and 530 nm. The BRET ratio was then calculated as emission at 530 nm·emission−1 at 485 nm. Net BRET was defined as the 530 nm/485 nm ratio of cells co-expressing Rluc and eYFP minus the BRET ratio of cells expressing only the Renilla luciferase construct in the same experiment. This value was multiplied by 1000 to obtain mBRET units.
Data analysis and statistical procedures
All experiments were performed at least three times. Where appropriate, statistical significance was established and accepted if P < 0.05.
Results
Both rat (r) and human (h) orthologues of the poorly characterized GPCR GPR35 were modified to incorporate an N-terminal FLAG epitope tag. These were further modified by C-terminal, in-frame fusion of enhanced yellow fluorescent protein (eYFP) to generate FLAG-rGPR35-eYFP and FLAG-hGPR35-eYFP. Each of these constructs was cloned into the Flp-In™ locus of Flp-In™ T-REx™ 293 cells and pools of antibiotic-resistant cells isolated. In the absence of the inducer doxycycline, neither construct could be detected via fluorescence microscopy (Figure 1A and B), while after treatment with doxycycline essentially all cells expressed the appropriate receptor as monitored by fluorescence corresponding to eYFP (Figure 1A and B). Imaging studies indicated that each construct was located extensively at the plasma membrane, while the greater brightness of FLAG-rGPR35-eYFP expressing cells following addition of a maximally effective concentration of doxycycline (100 ng·mL−1) suggested that the rat orthologue might be expressed at higher levels than the human form (Figure 1A and B). The inducible nature of constructs located at the Flp-In™ locus of these cells meant, however, that expression levels of each orthologue of GPR35 could be titrated by varying the concentration of doxycycline in the culture medium (Figure 1A and B). Furthermore, immunoblotting of membranes prepared from cells treated with varying concentrations of doxycycline confirmed this as well as that higher amounts of the rat orthologue were indeed expressed in response to equal concentrations of the inducer (Figure 1C).
Figure 1.
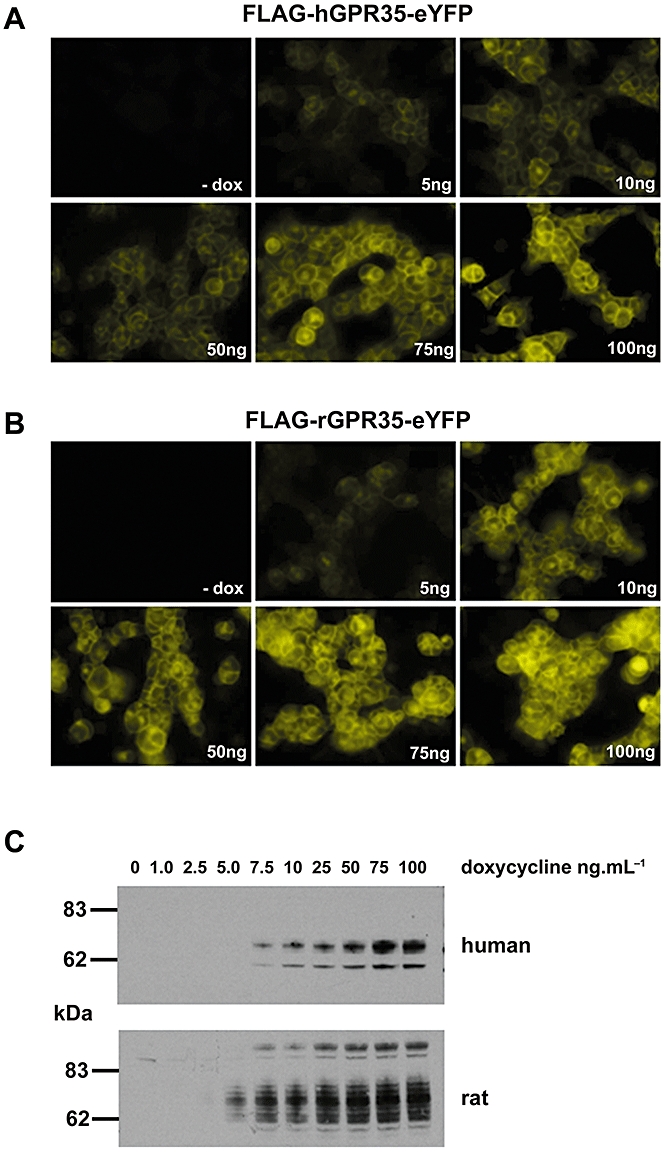
Expression of FLAG-hGPR35-eYFP and FLAG-rGPR35-eYFP. (A, B) Flp-In™ T-REx™ 293 cells harbouring either FLAG-hGPR35-eYFP (A) or FLAG-rGPR35-eYFP (B) at the Flp-In™ locus were maintained in the absence (- dox) or presence of varying concentrations (ng·mL−1) of doxycycline for 24 h and then imaged. (C) Membranes prepared from cells akin to those of (A, B) were resolved by SDS-PAGE and immunoblotted with anti-FLAG (5 µg of protein was loaded per lane). Upper panel human GPR35, lower panel rat GPR35.
Immunoblotting of membranes isolated from uninduced and doxycycline-induced cells with either anti-FLAG (Figure S1) or anti-green fluorescent protein (GFP) (Figure S1) antisera demonstrated the same pattern of detected proteins, confirming the full length expression of both the rGPR35 and hGPR35 constructs (Figure S1). FLAG-hGPR35-eYFP was detected predominantly as a doublet of apparent Mr 62 and 74 kDa while FLAG-rGPR35-eYFP was present as a diffuse series of polypeptides with apparent Mr between 62 and 74 kDa as well as a discrete polypeptide with apparent Mr of some 100 kDa (Figure S1). Both constructs were heterologously N-glycosylated because treatment of membranes expressing FLAG-hGPR35-eYFP with NGaseF resulted in the generation of a single predominant band of 55 kDa (Figure S1), while equivalent treatment of membranes expressing FLAG-rGPR35-eYFP resulted in a pair of immunodetected polypeptides with apparent masses of 47 kDa and 53 kDa and maintenance of the species with Mr close to 100 kDa (Figure S1).
Although hGPR35 has been reported to respond to the tryptophan metabolite kynurenic acid and cause a substantial increase in binding of [35S]-GTPγS to pertussis toxin-sensitive G proteins (Wang et al., 2006), in preliminary studies, we were able to observe only very modest increases in binding of [35S]-GTPγS following either transient expression of FLAG-hGPR35 in HEK293T cells or following production of membranes from the Flp-In™ T-REx™ 293 cells described above (not shown). This was also the case when employing zaprinast, a synthetic ligand that, although generally employed as a selective cGMP phosphodiesterase inhibitor (Corbin and Francis, 2002), has also been shown to have agonist activity at GPR35 (Taniguchi et al., 2006). In phylogenetic trees of GPCR sequences expressed in man and rodents (Fredriksson et al., 2003; Fredriksson and Schioth, 2005), GPR35 resides close to GPR55, a GPCR with unusual G protein selectivity in that it appears to activate Gα13 specifically (Ryberg et al., 2007). To assess if GPR35 might also interact with Gα13, we adopted two distinct approaches. In the first, we transiently co-transfected into HEK293T cells a chimeric Gαq/Gα13 G protein in which the C-terminal five amino acids of Gαq were replaced with the equivalent sequence of Gα13 along with either FLAG-rGPR35-eYFP or FLAG-hGPR35-eYFP and then performed single cell Ca2+ imaging studies. In both cases, addition of zaprinast resulted in a rapid and substantial increase in [Ca2+]i (Figure 2A). By contrast, introduction into the cells of full length Gαq along with the receptor constructs resulted in no increase in [Ca2+]i in response to zaprinast, although subsequent addition of ATP, to activate endogenously expressed P2Y purinoceptors, confirmed that these cells were capable of generating a Ca2+ response to an appropriately coupled stimulus (Emkey and Rankl, 2009). These results indicated that the effect of zaprinast and GPR35 required the receptor recognition element of Gα13 (Figure 2A). Similar studies that co-expressed an equivalent Gαq/Gα12 chimera along with the orthologues of GPR35 also failed to generate an increase in [Ca2+]i in response to zaprinast (Figure 2B) although, again, the inherent responsiveness of the cells was demonstrated by subsequent addition of ATP (Figure 2B). In a second approach, we transfected Flp-In™ T-REx™ 293 cells induced to express either FLAG-hGPR35-eYFP or FLAG-rGPR35-eYFP with either wild-type Gα13 or, as a positive control, Gln226Leu Gα13 which is a constitutively active form of this G protein. Subsequently, samples were immunoprecipitated with a conformation-selective Gα13 antibody able to identify only the GTP-bound, active state, and then immunoblotted with a Gα13 antibody able to identify all forms of this G protein. Gln226Leu Gα13 was recovered effectively by this approach and zaprinast produced an increase in GTP-bound wild type Gα13 (Figure 3) that was more pronounced in the cells expressing human GPR35 (Figure 3A and B) than the rat orthologue (Figure 3C and D). Control immunoblots with the non-activation state-dependent anti-Gα13 antibody confirmed equivalent expression of wild-type Gα13 in the +/− zaprinast experiments as well as expression of Gln226Leu Gα13 (Figure 3). Time-course studies indicated that, in cells expressing FLAG-hGPR35-eYFP, levels of GTP-bound Gα13 continued to increase in the presence of zaprinast over a 30 min period (Figure 3B); while in cells expressing FLAG-rGPR35-eYFP, zaprinast-induced increases in GTP-bound Gα13 appeared to reach maximal levels within 10–15 min (Figure 3D). The extent of expression of Gα13 compared to non-transfected control cells is shown in Figure 3E.
Figure 2.
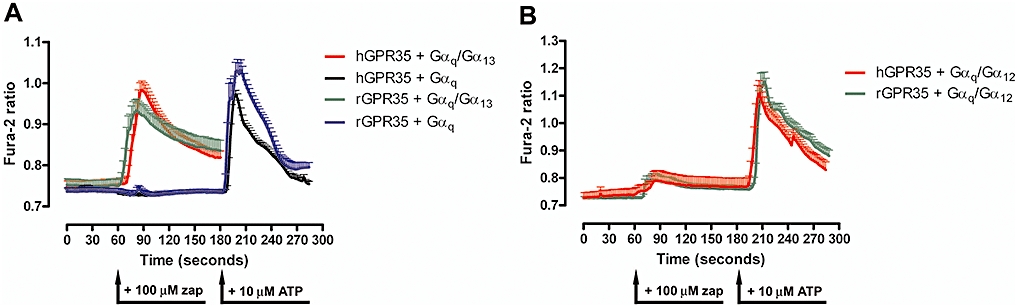
Orthologues of GPR35 elevate [Ca2+]i via a Gαq/Gα13 chimera but not a Gαq/Gα12 chimera. (A) HEK293T cells were transfected with either human GPR35 or rat GPR35 and either full length Gαq or a Gαq/Gα13 chimera. Single cell Ca2+ imaging studies were performed and effects of zaprinast (zap) were recorded in each case. In cells that did not respond to zaprinast (black, blue), ATP was added subsequently to activate endogenously expressed P2Y purinoceptors. (B) Similar studies were performed on cells transfected to express human GPR35 or rat GPR35 and a Gαq/Gα12 chimera. As in (A), cells transfected to express the Gαq/Gα12 chimera, although producing little response to zaprinast, were subsequently challenged with ATP to confirm that [Ca2+]i could be elevated in these cells. At least 20 cells were examined for each condition and data represent means ± SEM.
Figure 3.
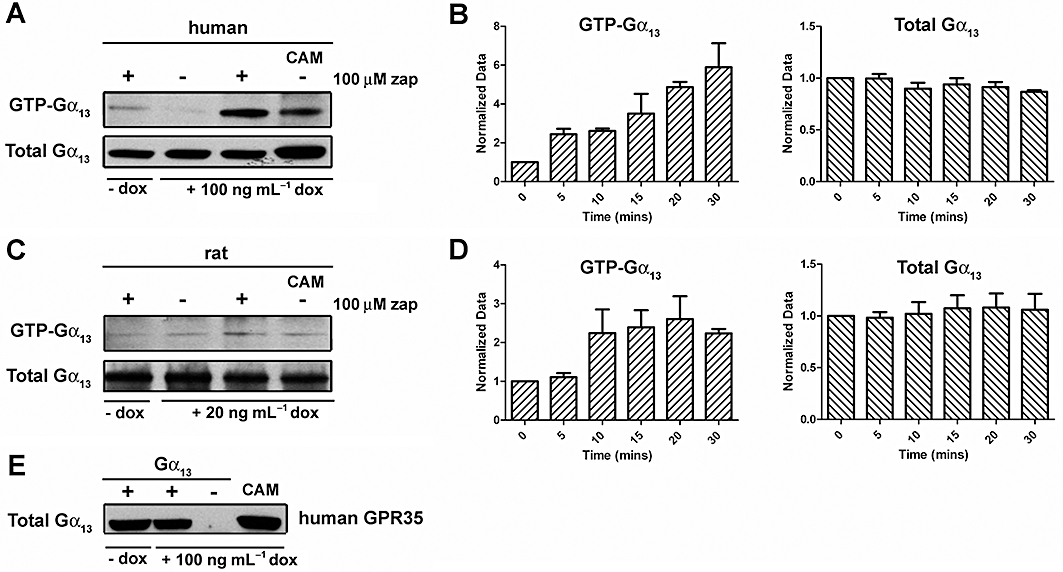
Orthologues of GPR35 promote activation of Gα13. Studies with a Gα13 antibody that interacts selectively with GTP-Gα13. Cells harbouring either FLAG-hGPR35-eYFP (A, B) or FLAG-rGPR35-eYFP (C, D) were either untreated (- dox) or treated (+ dox) with doxycycline (concentrations as noted). Cells were then transfected with either wild type Gα13 or a constitutively active Gln226Leu Gα13 variant (CAM). Cells were treated with or without 100 µM zaprinast as noted for 30 min. Samples were immunoprecipitated with an active-state Gα13 antibody and these samples (GTP-Gα13) and cell lysates (Total Gα13) were resolved by SDS-PAGE and subsequently immunoblotted with a polyclonal Gα13 antiserum. In (B and D) the relative amount of GTP-Gα13 and total levels of Gα13 were assessed by analysis of such immunoblots following different periods of exposure to 100 µM zaprinast (means ± SEM, n = 3). (E) Transfection with either wild-type or CAM Gα13 results in a substantial enhancement of expression. Cells induced to express FLAG-hGPR35-eYFP were transfected transiently with either wild-type Gα13 or the CAM variant. An empty vector transfection (-Gα13) was also performed. Lysates from cells were resolved by SDS-PAGE and immunoblotted with the polyclonal Gα13 antiserum.
Because of the relatively poor capacity to generate and quantify large amounts of data using such Gα13 activation studies, we also developed and employed a pair of GPR35-β-arrestin-2 interaction assays. Initially, we employed the PathHunter™β-galactosidase complementation-based (van der Lee et al., 2009; Yin et al., 2009) GPR35-β-arrestin-2 interaction assay. Rat and human GPR35 modified at the C-terminus to incorporate the Prolink™ fragment of β-galactosidase were each introduced into HEK293-BAEA (β-arrestin enzyme acceptor) cells stably expressing β-arrestin-2 C-terminally tagged with the remaining sequence of β-galactosidase. Although kynurenic acid produced limited complementation of β-galactosidase activity (not shown), in both cases a substantial, concentration-dependent increase in β-galactosidase activity was recorded in response to zaprinast (Figure 4A). Interestingly, the potency of zaprinast in the cells transfected to express the rGPR35 construct (pEC50 = 8.79 ± 0.12, mean ± SEM, n = 4) was more than 1000-fold greater (P < 0.0001) than in cells transfected to express the hGPR35 construct (pEC50 = 5.42 ± 0.14, n = 4) (Figure 4A). Parallel studies took advantage of the eYFP tag added to the C-terminus of the species orthologues of GPR35 to establish a bioluminescence resonance energy transfer (BRET)-based β-arrestin-2 interaction assay (Hamdan et al., 2005; Kocan and Pfleger, 2009) by co-transfecting HEK293T cells with FLAG-hGPR35-eYFP and β-arrestin-2 C-terminally tagged with Renilla luciferase. In this assay, kynurenic acid also displayed very limited effects and, indeed, these were not detectable at concentrations of the ligand below 10−4 M (Figure 4B). By contrast, zaprinast produced a robust, concentration-dependent increase in BRET, reflecting hGPR35-β-arrestin-2 interactions, with pEC50 = 5.42 ± 0.07, n = 8) (Figure 4B). Furthermore, when using FLAG-rGPR35-eYFP as the energy acceptor zaprinast was again both an agonist and substantially more potent (P < 0.0001) in producing interactions with β-arrestin-2-Renilla luciferase (pEC50 = 7.13 ± 0.16, n = 8) (Figure 4B). This was also the case for kynurenic acid as an agonist at FLAG-rGPR35-eYFP in this assay (P < 0.004) with measured pEC50 = 4.18 ± 0.06, n = 8 (Figure 4B). The importance of the acidic nature of kynurenic acid for function was evident because ethyl 4-hydroxy-2-quinolinecarboxylate (also known as kynurenic acid ethyl ester) was inactive at both FLAG-rGPR35-eYFP and FLAG-hGPR35-eYFP (Figure 4B). Although interactions between both FLAG-hGPR35-eYFP and FLAG-rGPR35-eYFP and β-arrestin-1-Renilla luciferase could also be measured in response to zaprinast the signal to background ratio was lower than for β-arrestin-2-Renilla luciferase (data not shown) and, therefore, in subsequent studies, we employed β-arrestin-2-Renilla luciferase. As well as GPR55, other receptors that lie in the same general arm of the GPCR phylogenetic tree as GPR35 include the free fatty acid receptors FFA1-3 (also called GPR40, GPR43 and GPR41, respectively) (Stoddart et al., 2008b; Milligan et al., 2009). As well as medium- and longer-chain free fatty acids, FFA1 is activated by a series of thiazolidine-2-4,dione ligands, including the clinically employed drug rosiglitazone (Smith et al., 2009). Although rosiglitazone produced a FLAG-rGPR35-eYFP/β-arrestin-2-Renilla luciferase BRET response, it displayed only low potency in this assay (data not shown), and as it could not be used at concentrations above 10−4 M, we were unable to extrapolate data to obtain meaningful and robust measures of EC50. However, other thiazolidine-2-4,diones, including (Z)-[4-(2,4-dioxo-thiazolidin-5-ylidenemethyl)-phenoxy] acetic acid (compound 10) and (Z)-5-(3-chloro-benzylidene)-thiazolidine-2,4-dione (compound 3) displayed significantly higher potency (compound 10, pEC50 = 5.13 ± 0.14, n = 3, compound 3, pEC50 = 5.08 ± 0.28,n = 3) at FLAG-rGPR35-eYFP (Figure 4C). Moreover, unlike zaprinast and kynurenic acid, both these thiazolidine-2-4,diones were at least as potent at human GPR35 in this assay (compound 10, pEC50 = 5.86 ± 0.08, n = 3; compound 3, pEC50 = 5.08 ± 0.25, n = 3) (Figure 4C).
Figure 4.
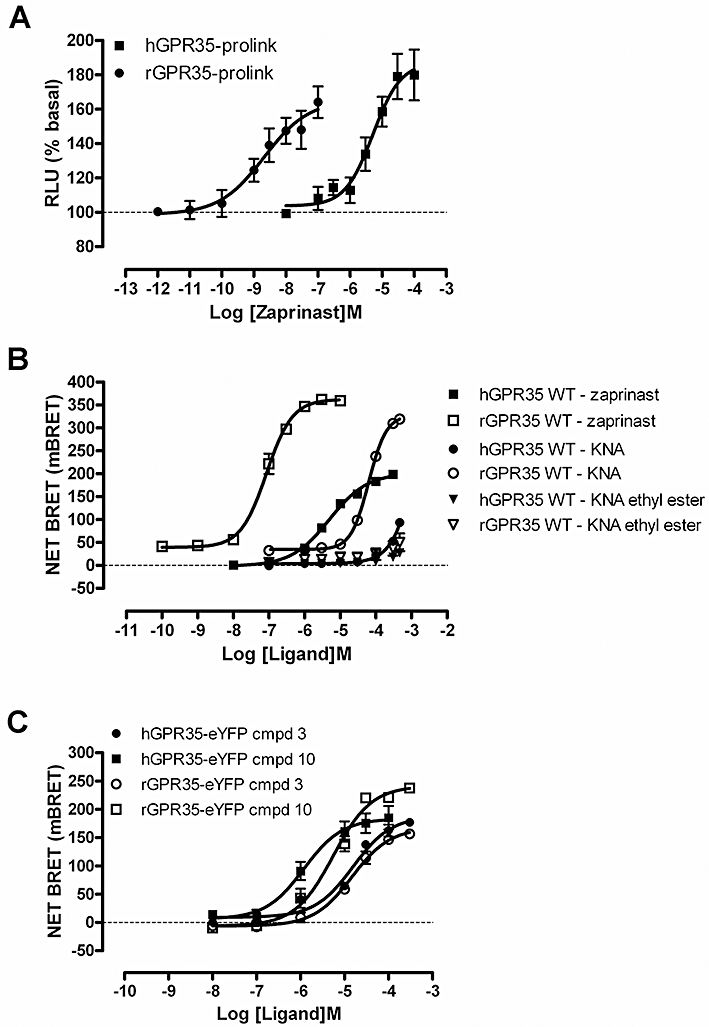
Analysis of GPR35-β-arrestin-2 interactions. (A) Human and rat forms of GPR35 C-terminally tagged with the Prolink™ fragment of β-galactosidase were transiently introduced into HEK 293-BAEA cells stably expressing β-arrestin-2 linked to the remaining element of β-galactosidase. Cells were treated with varying concentrations of zaprinast for 60 min, as suggested by the manufacturer, and β-galactosidase activity reflecting complementation of the enzyme produced via GPR35-β-arrestin-2 interaction was measured. (B) FLAG-hGPR35-eYFP or FLAG-rGPR35-eYFP was co-transfected with β-arrestin-2-Renilla luciferase into HEK293T cells. BRET signals were monitored after treatment of the cells for 5 min with varying concentrations of zaprinast, kynurenic acid or kynurenic acid ethyl ester. (C) Studies akin to those of (B) were conducted using varying concentrations of compound 3 and compound 10 using FLAG-hGPR35-eYFP or FLAG-rGPR35-eYFP.
Unlike kynurenic acid, zaprinast does not contain a formal negative charge (Figure 5A), although it does contain an acid bioisostere (a substituent or group with similar physical or chemical properties that imparts similar biological properties to a chemical compound). Furthermore, we have previously shown that the interaction of thiazolidine-2-4,diones, such as rosiglitazone, with FFA1 requires the same key positively charged arginine residues that are also known to interact with the carboxylate moiety of free fatty acids ligands (Smith et al., 2009). We therefore tested if, given their different chemical structures, these three ligand classes potentially bound to overlapping sites in GPR35. To assess this for zaprinast and kynurenic acid, FLAG-rGPR35-eYFP was co-expressed with β-arrestin-2-Renilla luciferase and concentration-response curves to zaprinast performed in the presence of a range of constant concentrations of kynurenic acid. As a direct agonist, kynurenic acid generated BRET signals in the absence of zaprinast; but at sub-maximal concentrations, it did not alter the EC50 for zaprinast (Figure 5B). Furthermore, the maximal BRET signal achieved was the same for both ligands and there was no additivity of the effects of co-addition of maximally effective concentrations of the two ligands (Figure 5B). Reversal of the protocol, in which any effect of differing but constant concentrations of zaprinast on concentration-response curves to kynurenic acid was examined, resulted in similar observations and conclusions (Figure 5C). These results are consistent with the two ligands acting orthosterically in relation to one another. The low potency of kynurenic acid at FLAG-hGPR35-eYFP meant, however, that it was not possible to assess this question for the human orthologue. Similar conclusions were reached in equivalent studies in which concentration-response curves to zaprinast were performed in the presence of varying but fixed concentrations of compound 10 (Figure 5D). Furthermore, the relatively high potency of compound 10 at human GPR35 allowed assessment of this question also at the human orthologue (Figure 5E). There was some indication of a shift in the potency of zaprinast to higher concentrations at both rat and human GPR35 in the presence of the highest concentrations of compound 10 that could be tested (Figure 5D and E). This is at least potentially consistent with compound 10 displaying a degree of allostery as well as direct agonism (Milligan et al., 2009).
Figure 5.
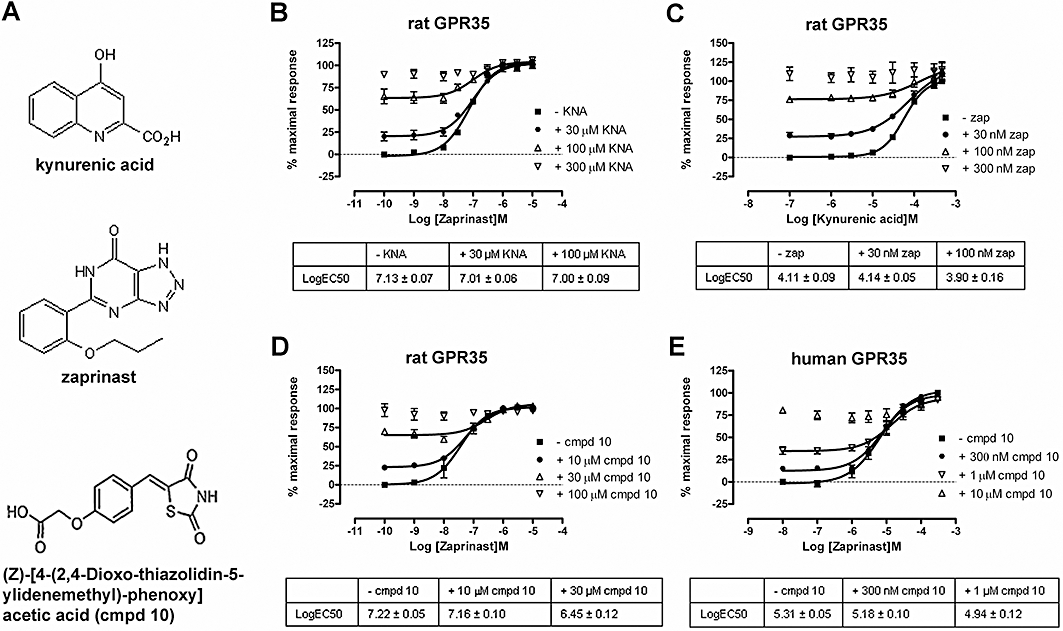
Zaprinast, kynurenic acid and compound 10 share an overlapping binding site in GPR35. (A) Kynurenic acid, zaprinast and (Z)-[4-(2,4-dioxo-thiazolidin-5-ylidenemethyl)-phenoxy] acetic acid (compound 10) are agonists at GPR35. Receptor-β-arrestin-2 interaction BRET assays were performed as in Figure 4 following co-transfection of either FLAG-rGPR35-eYFP (B, C, D) or FLAG-hGPR35-eYFP (E) and β-arrestin-2-Renilla luciferase. Concentration-response curves to zaprinast were performed in the presence of varying fixed concentrations of kynurenic acid (B) or concentration-response curves to kynurenic acid were performed in the presence of varying fixed concentrations of zaprinast (C). In (D and E), concentration-response curves to zaprinast were performed in the presence of varying fixed concentrations of compound 10. Tables provide the estimated pEC50 values for each condition. Data represent means ± SEM.
Recent studies have suggested that for a number of GPCRs able to respond to acid ligands an arginine residue in transmembrane domain III (TMD III) may play a key role (Liu et al., 2009). To assess if this was the case for GPR35, Arg97 in rGPR35 [position 3.36 in the numbering system of Ballesteros and Weinstein (1995)] was converted to Ala in the context of FLAG-rGPR35-eYFP. Following co-expression of FLAG-Arg97Ala-rGPR35-eYFP and β-arrestin-2-Renilla luciferase, no increase in BRET signal could be recorded to kynurenic acid (Figure 6A), zaprinast (Figure 6B) or compound 10 (Figure 6C). Mutation of the equivalent residue (Arg100) in hGPR35 also abolished response to zaprinast and compound 10 as well as the small effect of kynurenic acid (Figure 6A and C). Although the Arg 3.36 Ala mutations abolished the response to the ligands, this was not due to reduced global expression of the modified receptors. Direct measurement of eYFP fluorescence showed the mutants to be expressed as well as wild-type (Figure 6D). Intact cell surface elisa studies, suggested a trend towards reduction in cell surface delivery of the mutants compared to the wild-type receptor (Figure 6E) but, only in the case of rat Arg 3.36 Ala, was this statistically significant. One turn of the predicted helix of TMD III above Arg 3.36, at position 3.32, is a tyrosine residue in both the rat and human orthologues (Figure 7). Mutation of this residue to Ala in FLAG-rGPR35-eYFP also resulted in a complete loss of function to each of the three ligands (Figure 7A, B and D). This was also the case for both zaprinast and compound 10 at the FLAG-hGPR35-eYFP mutant (Figure 7C and D). To gain more insight into the role of Tyr 3.32, this residue was also mutated to Leu in both orthologues. Interestingly, for the Tyr 3.32 Leu substitution in rat GPR35, the potency (pEC50 = 5.35 ± 0.06,n = 4) of zaprinast was reduced (P < 0.0001) some 60-fold but close to maximal efficacy was maintained (Figure 7A); while for both kynurenic acid (Figure 7B) and compound 10 (Figure 7D), very limited activity could be recorded at ligand concentrations that were practical to employ. At concentrations of kynurenic acid above 10−4 M, there was some indication that the Tyr 3.32 Leu mutant of FLAG-rGPR35-eYFP also generated a weak response (Figure 7B), but it was not possible to fit such data adequately to obtain accurate potency values. The equivalent mutations in FLAG-hGPR35-eYFP both resulted in complete loss of function to zaprinast (Figure 7C) but, with the lower potency of this ligand at FLAG-hGPR35-eYFP compared with FLAG-rGPR35-eYFP, this may simply reflect an inability to use sufficiently high concentrations of zaprinast to discriminate between the Ala and Leu mutations rather than inherently indicating a distinct mechanism of binding/function. Equally, compound 10 lacked measurable activity at Tyr 3.32 Leu hGPR35 (Figure 7D). In all cases, total expression levels of the constructs were unaffected by the introduced mutations (Figure 7E) and, although cell surface delivery of FLAG-hGPR35-eYFP mutants tended to be lower than for the wild-type constructs (Figure 7F), this did not reach statistical significance.
Figure 6.
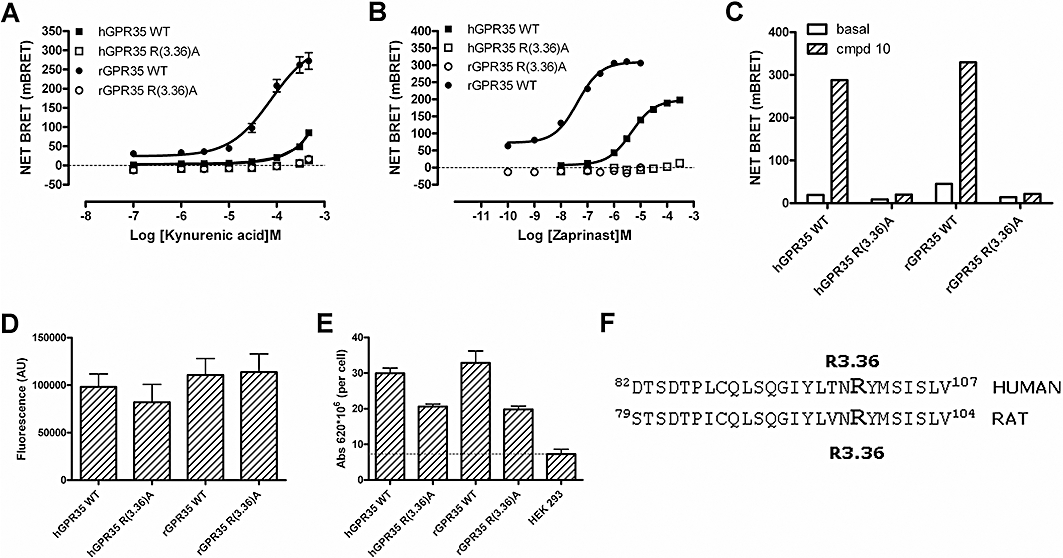
Mutation of arginine 3.36 eliminates agonist function of zaprinast, kynurenic acid and compound 10. (A, B) Receptor-β-arrestin-2 interaction BRET assays in response to varying concentrations of kynurenic acid (A) or zaprinast (B) were performed as in Figure 4 using either wild-type or Arg3.36Ala mutants of either FLAG-hGPR35-eYFP or FLAG-rGPR35-eYFP. The effects of the Arg3.36Ala mutations on response to a single concentration (10−4 M) of compound 10 that was maximally effective at the wild-type GPR35 orthologues was also assessed (C). Total levels of GPR35 expressed, as monitored by eYFP fluorescence above non-transfected cells, were unaltered by the Arg3.36Ala mutation (D), while cell surface delivery was monitored by anti-FLAG elisa (E) of the forms of GPR35. * Less than wild-type P < 0.05. (F) The amino acid sequence of a section of transmembrane domain III from both human and rat GPR35 is shown with Arg3.36 highlighted.
Figure 7.
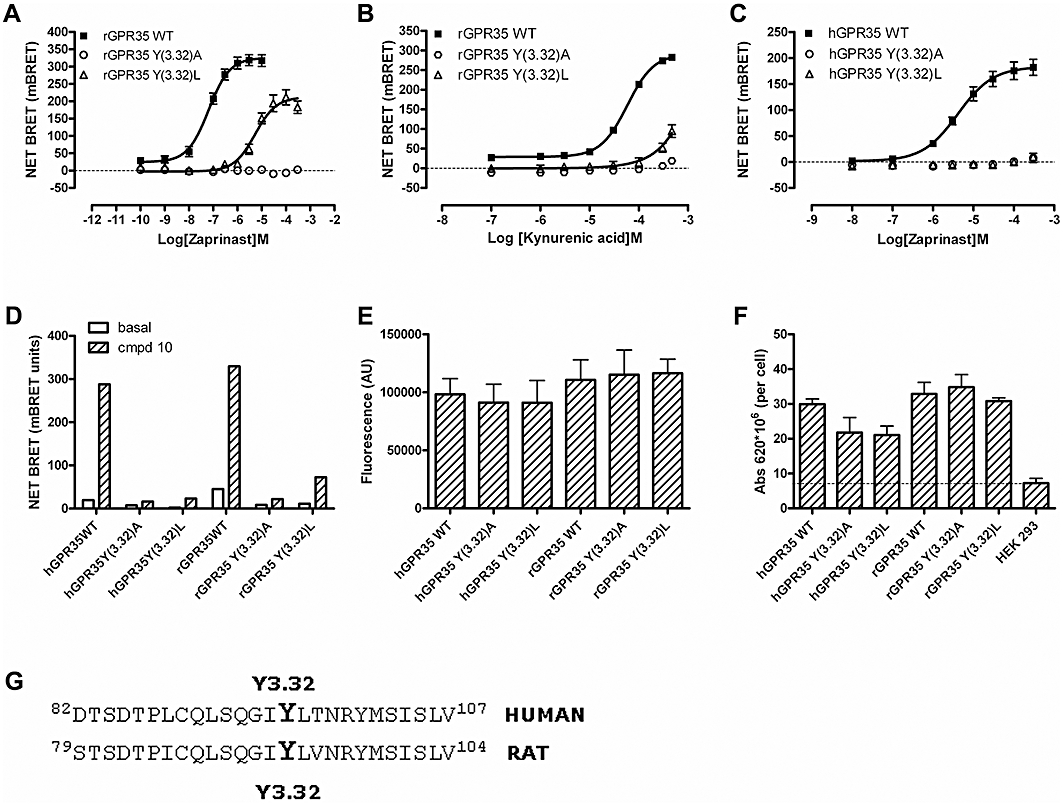
The role of tyrosine 3.32 of GPR35 in ligand function. Receptor-β-arrestin-2 interaction BRET assays were performed in response to varying concentrations of zaprinast (A, C) or kynurenic acid (B) following co-transfection of FLAG-rGPR35-eYFP, FLAG-Tyr3.32Ala rGPR35-eYFP or FLAG-Tyr3.32Leu rGPR35-eYFP (A, B) of the equivalent forms of human GPR35 (C). Effects of mutation of Tyr 3.32 on the response to 10−4 M compound 10 was also studied (D). Total expressed levels, as monitored by eYFP fluorescence (E), of the various forms of GPR35 were unaltered by these mutations, as was cell surface delivery monitored by anti-FLAG elisa (F). (G) The amino acid sequence of a section of transmembrane domain III from both human and rat GPR35 is shown with Tyr3.32 highlighted.
Interactions between GPCRs and β-arrestins are often a harbinger of agonist-induced internalization of the receptor (Moore et al., 2007; Marchese et al., 2008). Cells harbouring FLAG-hGPR35-eYFP at the Flp-In locus were induced to express the receptor in response to doxycycline. Imaging studies over time showed that a high concentration of zaprinast was able to cause internalization of FLAG-hGPR35-eYFP (data not shown). Because the data from such studies were difficult to quantify adequately, we employed biotinylation of cell surface FLAG-hGPR35-eYFP to detect ligand-induced cellular redistribution. Biotinylated, and therefore cell surface, FLAG-hGPR35-eYFP was highly induced by doxycycline treatment (Figure 8A) and levels of cell surface receptor were reduced by treatment with zaprinast for 30 min in a concentration-dependent manner (pEC50 = 4.85 ± 0.38, n = 3) (Figure 8B). Both compound 3 and compound 10 also produced strong internalization of GPR35 (Figure 8A). By contrast, even at 100 µM, kynurenic acid was unable to cause significant (cell surface levels 84 ± 7.9%, n = 3, not significantly different from control, one-way anova) internalization of FLAG-hGPR35-eYFP (Figure 8A). At least over this time period, treatment with zaprinast or the two novel thiazolidine-2-4,dione agonists did not cause down-regulation of FLAG-hGPR35-eYFP as total levels of the construct in cell extracts were unaltered (Figure 8A). To compare the sensitivity of internalization of FLAG-rGPR35-eYFP and FLAG-hGPR35-eYFP in response to zaprinast, cell surface anti-FLAG elisa assays were performed on cells treated with varying concentrations of zaprinast. Although at maximally effective concentrations zaprinast produced a similar extent of receptor internalization, as anticipated from the β-arrestin-2 recruitment assays, zaprinast was substantially more potent (P < 0.001) at the rat orthologue (pEC50 = 7.33 ± 0.35, n = 3) than at human GPR35 (pEC50 = 4.53 ± 0.29, n = 3) (Figure 8C).
Figure 8.
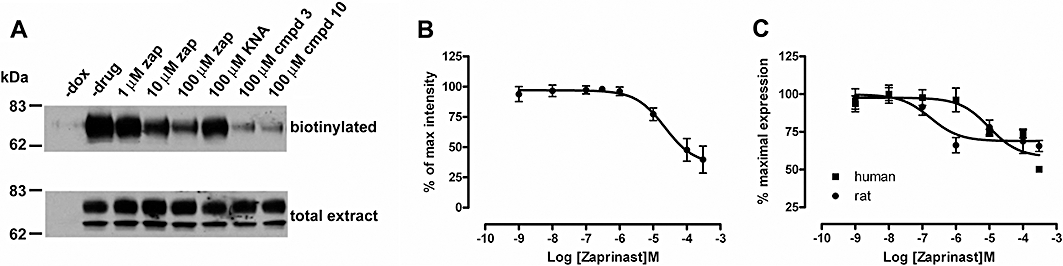
Agonist-induced internalization of GPR35 is correlated with agonist potency in receptor-β-arrestin-2 interaction BRET assays. (A) Biotinylation studies demonstrated expression at the cell surface of FLAG-hGPR35-eYFP following treatment with doxycycline (compare – dox with – drug). Following treatment with increasing concentrations of zaprinast (zap), and single concentrations of either compound 3 or compound 10 for 30 min, but not with kynurenic acid (KNA), cell surface (biotinylated) but not total levels (total extract) of GPR35 were reduced (8 µg of protein from total extract was loaded per lane). Results from a representative experiment are shown. in (B). Cell surface levels in such experiments were quantified. Data represent means ± SEM, n = 3. (C) Cells induced to express similar levels of FLAG-hGPR35-eYFP and FLAG-rGPR35-eYFP were exposed to varying concentrations of zaprinast for 30 min. Cell surface anti-FLAG elisa studies were quantified to measure internalization of the GPR35 orthologues. A representative experiment of 3 performed in triplicate is shown.
Discussion and conclusions
GPR35 is a poorly characterized GPCR that has been suggested to contribute to immune-modulation (Wang et al., 2006; Barth et al., 2009), gastric function (Okumura et al., 2004) and the regulation of insulin secretion (Leonard et al., 2005). The tryptophan metabolite kynurenic acid (Stone and Darlington, 2002) has previously been shown to have agonist action at GPR35 (Wang et al., 2006; Barth et al., 2009) and, hence, is a potential endogenous regulator of this receptor. The EC50 for kynurenic acid to elevate [Ca2+]i in CHO cells expressing either mouse or rat GPR35, and also transfected to express a mixture of chimeric G proteins, has been reported to be in the region of 10 µM, while equivalent studies with the human orthologue resulted in EC50 = 40 µM (Wang et al., 2006). These concentrations are somewhat greater than estimated levels of kynurenic acid in the rat small intestine (Kuc et al., 2008). However, the potency values reported in the screening studies are difficult to interpret in terms of ligand affinity because a lack of information on the absolute or relative expression levels of the orthologues means that nothing was known about possible receptor reserve or the propensity of the orthologues of GPR35 to interact with the G proteins provided, although receptor reserve is frequently observed for GPCR downstream signalling assays compared with more receptor proximal assays (Burt et al., 1996). Moreover, no high affinity ligands suitable for use in receptor-ligand binding studies are available for GPR35.
Interestingly, in terms of the potential for kynurenic acid to be a true endogenous regulator of GPR35, Barth et al. (2009) have recently reported potent effects of this ligand to induce firm arrest of human monocytes on ICAM-1 expressing human umbilical vein endothelial cells via a β2-integrin-mediated process, with action reported at concentrations as low as 300 nM. Furthermore, gene silencing studies were at least consistent with this reflecting a GPR35-mediated effect (Barth et al., 2009). By contrast, these authors reported much less potent effects of kynurenic acid in promoting activation of neutrophils or the firm arrest of monocytes to fibronectin via a β1-integrin-mediated process and did not assess if these effects were also inhibited by short hairpin RNA-mediated GPR35 knock-down (Barth et al., 2009).
For poorly characterized receptors, such as GPR35, that lack antagonist ligands or agonists that are of sufficient affinity to allow the generation of ligand binding data, it is a challenge, as noted earlier, to obtain affinity/potency data that are not potentially compromised by receptor expression levels and the possibility of receptor reserve. Furthermore, in the current studies it was obvious in the stable cell lines that maximally effective induction of the rat GPR35 construct resulted in higher steady-state levels than equivalent induction of the human orthologue. This was despite the two constructs being equivalent and each being cloned into the single, defined Flp-In™ locus of Flp-In™ T-REx™ 293 cells. One potential means to overcome such issues is to employ assays based on the interaction between a GPCR and a β-arrestin. Models of the roles of β-arrestins in the desensitization of agonists at GPCRs generally invoke mechanisms in which agonist-occupancy of the receptor is required to allow receptor phosphorylation by G protein-coupled receptor kinases and, subsequently, enhanced interactions with β-arrestins (Moore et al., 2007; Marchese et al., 2008). As such, the potency of agonists in GPCR-β-arrestin interaction assays is anticipated to mirror agonist occupancy of the receptor. We used the potency of kynurenic acid in GPR35-β-arrestin 2 interaction assays using both rat and human orthologues of GPR35 therefore as a surrogate measure of ligand affinity. Kynurenic acid was significantly more potent at rat GPR35 compared to human GPR35. Moreover, as the estimated potency of kynurenic acid at human GPR35 in such assays was less than millimolar, for effects of kynurenic acid in human cell systems observed at 300 nM (Barth et al., 2009) to be GPR35-mediated must either reflect the requirement for extremely low receptor occupancy to generate the relevant signal, or the presence of other proteins in certain cell types, such as monocytes, that alter the affinity of this ligand for GPR35. Indeed, the estimate of the affinity of kynurenic acid for human GPR35 produced in the current studies is more consistent with the concentrations noted by Barth et al. (2009) to be required for firm attachment of monocytes to fibronectin or activation of neutrophils (1–10 mM). Such variation between functional potency and surrogate affinity measures also questions the relevance of kynurenic acid, at least in man, as a true endogenous regulator of GPR35, not-withstanding the gene silencing data of Barth et al. (2009). It would be of considerable interest to explore if zaprinast and/or the novel thiazolidine-2-4,diones identified herein is also able to induce firm arrest of human monocytes in the model employed by Barth et al. (2009). If so, however, it would be vital to exclude that an effect of zaprinast actually reflected regulation of cGMP phosphodiesterases via parallel studies that used an alternative, chemically unrelated, cGMP phosphodiesterase inhibitor such as dipyridamole (2,6-bis(diethanolamino)-4,8-dipiperidinopyrimido[5,4-d] pyrimidine) that is not an agonist at GPR35 (data not shown). Equally, although (Z)-[4-(2,4-dioxo-thiazolidin-5-ylidenemethyl)-phenoxy] acetic acid and (Z)-5-(3-chloro-benzylidene)-thiazolidine-2,4-dione are clearly agonists at GPR35, related chemicals have a range of targets, including other GPCRs and members of the nuclear hormone receptor family, and the potential selectivity of these ligands for GPR35 remains to be investigated.
While β-arrestin interaction assays were both robust and offered the best available means to infer true ligand potency data, as a GPCR the agonist-occupancy of GPR35 was anticipated to result in activation of one or more G proteins. However, despite a number of reports on GPR35 employing either [35S]-GTPγS binding assays (Milligan, 2003; Wang et al., 2006) or demonstrating effects to be transduced via pertussis toxin-sensitive mechanisms (Ohshiro et al., 2008; Yang et al., 2010) preliminary studies indicated this to be an endpoint with very low signal to background. We considered, therefore, alternative G protein sub-types that might be activated by GPR35. One of the most closely related GPCRs to GPR35 is the lysophosphatidylinositol/atypical cannabinoid receptor GPR55 (Nevalainen and Irving, 2010), which has been suggested to couple selectively to Gα13 (Ryberg et al., 2007). With the hypothesis that this might also be the case for GPR35, we have shown both direct activation of Gα13 by GPR35 and, by the use of G protein chimeras, that the receptor is able to specify interaction with Gα13 over its other sub-family member Gα12. The availability of antibodies able to identify only the active state of the α subunit of different heterotrimeric G proteins (Lane et al., 2008) provides a novel means to explore the selectivity of GPCR function.
Recent studies that identified GPR81 as a GPCR able to be activated by lactate (Liu et al., 2009) identified a role of a TMD III arginine (position 3.36) as the charge partner for the carboxylate function of the ligand. These authors also noted the conservation of this residue in a number of other GPCRs that respond to small, acidic ligands (Liu et al., 2009) and, within the list, suggested Arg 3.36 in GPR35 as a likely partner for the acid function of kynurenic acid (Liu et al., 2009). The lack of activity, noted herein, of kynurenic acid ethyl ester at GPR35 provided further support for such a model. Furthermore, although zaprinast does not possess a formal negative charge, it does contain an acid bioisostere while thiazolidine-2,4-diones such as rosiglitazone require the same arginine residues for binding to, and activation of, the GPCR FFA1 as do free fatty acids (Smith et al., 2009). Mutation of this residue to Ala abolished the response to kynurenic acid for both rat and human GPR35. Equally, the response to both zaprinast and the novel thiazolidine-2,4-dione agonists was also attenuated. Liu et al. (2009) also noted a key role for Arg 240 (position 6.55) in GPR81, in that the response to lactate was also eliminated by mutation of this residue to Ala, while an equivalent mutation in the nicotinic acid receptor GPR109a also eliminates the response to endogenous agonists (Tunaru et al., 2005). This indicates a role for both these arginine residues in coordinating the carboxylate moiety of the respective ligands. However, GPR35 does not have an arginine residue at this position. Indeed, in both rat and human GPR35 it is a leucine. We also therefore explored contributions of other residues in TMDIII. The significantly higher potency of zaprinast compared with kynurenic acid at each orthologue studied, as well as the higher potency of zaprinast at the rat receptor compared with human GPR35, allowed more detailed analysis of the role of the tyrosine residue at position 3.32, located one turn of the helix higher than Arg 3.36, by focusing on the rat receptor. Here, although mutation to alanine resulted in ablation of response to zaprinast, the rather less extensive mutation to leucine also resulted in some 60-fold shift to the right in potency. Function in response to zaprinast could not be detected at the equivalent Tyr 3.32 Leu mutation of human GPR35, possibly because the lower potency of this ligand at the wild-type human orthologue meant that it was not practical to test the ligand at concentrations substantially more than 100 times EC50 at the wild-type. Consistent with the effects of mutation of residues 3.32 and 3.36 on the binding/function of each of zaprinast (Z)-[4-(2,4-dioxo-thiazolidin-5-ylidenemethyl)-phenoxy] acetic acid and kynurenic acid, studies that explored potential additivity of, or allosterism between, the three ligand classes in a series of co-addition experiments failed to identify any such effects, apart from a potential negative allosteric contribution of compound 10 on the action of zaprinast, and were consistent with these ligands being orthosteric agonists that share a common or overlapping binding site.
A key role of the interaction of a β-arrestin with many GPCRs is to promote clathrin-mediated internalization of the receptor as part of the cycles of receptor desensitization and resensitization and, with prolonged exposure to agonist, potential down-regulation of receptor amount (Moore et al., 2007; Marchese et al., 2008). We therefore paralleled studies on the potency of the ligands in the GPR35-β-arrestin 2 interaction studies by also monitoring the cellular distribution of GPR35 following treatment of cells with the ligands. The C-terminal fusion of eYFP was used to demonstrate both effective cell surface delivery of both orthologues of the receptor and to assess ligand-induced redistribution away from the cell surface, while analysis of cell surface GPR35 via both biotinylation and cell surface elisa provided biochemical correlates of the imaging studies. In all cases the relative potencies of zaprinast at both orthologues was consistent with those from the GPR35-β-arrestin 2 interaction experiments. This is to be anticipated as both assays are expected to be correlated with receptor occupancy.
A central issue in these and other studies on GPR35 is the nature of the assays employed. In initial studies demonstrating the agonist action of both kynurenic acid (Wang et al., 2006) and zaprinast (Taniguchi et al., 2006), key studies employed [Ca2+]i elevation assays in cells transfected to express combinations of promiscuous and chimeric G proteins. The rationale for such an approach is that little was known about GPR35, including nothing about G protein-coupling profile and selectivity, and such approaches are used widely in high throughput screening campaigns (Milligan and Rees, 1999; Kostenis et al., 2005). Although they have been an enormous boon to GPCR de-orphanization campaigns, there has been growing interest in the concept that details of ligand pharmacology can be determined by the identity of the assay employed (Kenakin, 2007; Williams and Hill, 2009). Direct measures of activation of the G12/G13 subfamily of G proteins lag well behind the other G proteins, however, reagents such as the active-state antibodies used herein offer a practical approach. As noted above, agonist-induced interaction between GPCRs and β-arrestins is almost universal and this, as well as the expectation, as also discussed earlier, that potency measures in such assays should not be hostage to issues of receptor reserve, was a determining factor in adopting such assays and these are now in widespread use in ligand identification campaigns. We employed both a BRET-based and the β-galactosidase complementation-based assay that is marketed as the PathHunter™ system (Birmingham, UK). These were both robust in allowing confirmation of previously described GPR35 agonists. However, as we also used the eYFP-tagged forms of GPR35 to assess cell surface delivery and to rapidly and non-destructively assess relative expression level of mutants of GPR35, we employed the BRET-based assays more routinely because the same eYFP tagged forms of GPR35 functioned as energy acceptor in these assays. Interestingly, although the potency of zaprinast at human GPR35 was identical in the two β-arrestin 2 interaction assays, the potency of this ligand was significantly higher at rat GPR35 in the PathHunter™ system. We have no obvious explanation for these findings but simply note the observation. A further distinction between the two assays was that kynurenic acid produced a substantially more robust signal in the BRET-based assay. Again we have no obvious explanation for this but note that Yin et al. (2009) have also reported an inability to record agonist effects when using the PathHunter™ assay for a small number of GPCRs at which ligand activity has been clearly demonstrated in other assays.
Clearly, the variation in potency at rodent and human orthologues of GPR35, particularly of zaprinast, presents a current challenge in employing pharmacological approaches to determine physiological functions of GPR35. Furthermore, the wide variation in the potency of kynurenic acid in assays of human white cell function reported by Barth et al. (2009) seems difficult to reconcile with each effect being mediated exclusively by GPR35. The identification of further GPR35 active and, potentially, selective ligands will aid in this endeavour.
Acknowledgments
SdM was funded by the Dutch Cancer Society. We thank Jose Brea and Maria I. Loza, Departamento de Farmacologia, Universidad de Santiago de Compostela, Spain for identifying sources of compounds 3 and 10.
Glossary
Abbreviations
- BRET
bioluminescence resonance energy transfer
- eYFP
enhanced yellow fluorescent protein
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- NGaseF
N-glycosidase F
- PEI
polyethylenimine
- TMD
transmembrane domain
Conflicts of interests
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Teaching Materials; Figs 1–8 as PowerPoint slide.
Figure S1 FLAG-hGPR35-eYFP and FLAG-rGPR35-eYFP are differentially N-glycosylated. Cells harbouring either FLAG-hGPR35- eYFP (h) or FLAG-rGPR35-eYFP (r) were either untreated (−) or treated (+) with doxycyline (100 ng·mL−1, as in Figure 1). Membranes isolated from these cells were either untreated, treated with NGase F (+ NGase F) or incubated in the same conditions as for NGase F but in the absence of the enzyme (− NGase F). Samples were resolved by SDS-PAGE and immunoblotted with a monoclonal anti-FLAG antibody (A) or with a goat polyclonal anti-GFP antiserum (B). 5 μg of protein was loaded per lane.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Barth MC, Ahluwalia N, Anderson TJ, Hardy GJ, Sinha S, Alvarez-Cardona JA, et al. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J Biol Chem. 2009;284:19189–19195. doi: 10.1074/jbc.M109.024042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ. Novel cannabinoid receptors. Br J Pharmacol. 2007;152:567–575. doi: 10.1038/sj.bjp.0707481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt AR, Carr IC, Mullaney I, Anderson NG, Milligan G. Agonist activation of p42 and p44 mitogen-activated protein kinases following expression of the mouse delta opioid receptor in Rat-1 fibroblasts: effects of receptor expression levels and comparisons with G-protein activation. Biochem J. 1996;320:227–235. doi: 10.1042/bj3200227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Milligan G. Constitutive activity of the cannabinoid CB1 receptor regulates the function of co-expressed Mu opioid receptors. J Biol Chem. 2008;283:11424–11434. doi: 10.1074/jbc.M710300200. [DOI] [PubMed] [Google Scholar]
- Canals M, Jenkins L, Kellett E, Milligan G. Up-regulation of the angiotensin II type 1 receptor by the MAS proto-oncogene is due to constitutive activation of Gq/G11 by MAS. J Biol Chem. 2006;281:16757–16767. doi: 10.1074/jbc.M601121200. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Francis SH. Pharmacology of phosphodiesterase-5 inhibitors. Int J Clin Pract. 2002;56:453–459. [PubMed] [Google Scholar]
- Emkey R, Rankl NB. Screening G protein-coupled receptors: measurement of intracellular calcium using the fluorometric imaging plate reader. Methods Mol Biol. 2009;565:145–158. doi: 10.1007/978-1-60327-258-2_7. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Schioth HB. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol Pharmacol. 2005;67:1414–1425. doi: 10.1124/mol.104.009001. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Grigoriadis DE, Hoare SR, Lechner SM, Slee DH, Williams JA. Drugability of extracellular targets: discovery of small molecule drugs targeting allosteric, functional, and subunit-selective sites on GPCRs and ion channels. Neuropsychopharmacology. 2009;34:106–125. doi: 10.1038/npp.2008.149. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Audet M, Garneau P, Pelletier J, Bouvier M. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen. 2005;10:463–475. doi: 10.1177/1087057105275344. [DOI] [PubMed] [Google Scholar]
- Harmar AJ, Hills RA, Rosser EM, Jones M, Buneman OP, Dunbar DR, et al. IUPHAR-DB: the IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Res. 2009;37:D680–D685. doi: 10.1093/nar/gkn728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- Kocan M, Pfleger KD. Detection of GPCR/beta-arrestin interactions in live cells using bioluminescence resonance energy transfer technology. Methods Mol Biol. 2009;552:305–117. doi: 10.1007/978-1-60327-317-6_22. [DOI] [PubMed] [Google Scholar]
- Kostenis E, Waelbroeck M, Milligan G. Techniques: promiscuous Galpha proteins in basic research and drug discovery. Trends Pharmacol Sci. 2005;26:595–602. doi: 10.1016/j.tips.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kuc D, Zgrajka W, Parada-Turska J, Urbanik-Sypniewska T, Turski WA. Micromolar concentration of kynurenic acid in rat small intestine. Amino Acids. 2008;35:503–505. doi: 10.1007/s00726-007-0631-z. [DOI] [PubMed] [Google Scholar]
- Lane JR, Henderson D, Powney B, Wise A, Rees S, Daniels D, et al. Antibodies that identify only the active conformation of G(i) family G protein alpha subunits. FASEB J. 2008;22:1924–1932. doi: 10.1096/fj.07-100388. [DOI] [PubMed] [Google Scholar]
- van der Lee MM, Blomenrohr M, van der Doelen AA, Wat JW, Smits N, Hanson BJ, et al. Pharmacological characterization of receptor redistribution and beta-arrestin recruitment assays for the cannabinoid receptor 1. J Biomol Screen. 2009;14:811–823. doi: 10.1177/1087057109337937. [DOI] [PubMed] [Google Scholar]
- Leonard J, Chu ZL, Unett DJ, Gatlin JE, Gaidarov I, Qui J, et al. GPR35 and modulators thereof for the treatment of metabolic-related disorders. 2005. World Intellectual Property Organization Patent WO 2005/119252 A2.
- Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem. 2009;284:2811–2822. doi: 10.1074/jbc.M806409200. [DOI] [PubMed] [Google Scholar]
- Lorenz WW, McCann RO, Longiaru M, Cormier MJ. Isolation and expression of a cDNA encoding Renilla reniformis luciferase. Proc Natl Acad Sci USA. 1991;88:4438–4442. doi: 10.1073/pnas.88.10.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundstrom K. An overview on GPCRs and drug discovery: structure-based drug design and structural biology on GPCRs. Methods Mol Biol. 2009;552:51–66. doi: 10.1007/978-1-60327-317-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. Principles: extending the utility of [35S]GTP gamma S binding assays. Trends Pharmacol Sci. 2003;24:87–90. doi: 10.1016/s0165-6147(02)00027-5. [DOI] [PubMed] [Google Scholar]
- Milligan G, Rees S. Chimaeric G alpha proteins: their potential use in drug discovery. Trends Pharmacol Sci. 1999;20:118–124. doi: 10.1016/s0165-6147(99)01320-6. [DOI] [PubMed] [Google Scholar]
- Milligan G, Stoddart LA, Smith NJ. Agonism and allosterism: the pharmacology of the free fatty acid receptors FFA2 and FFA3. Br J Pharmacol. 2009;158:146–153. doi: 10.1111/j.1476-5381.2009.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Nevalainen T, Irving AJ. GPR55, a lysophosphatidylinositol receptor with cannabinoid sensitivity? Curr Top Med Chem. 2010;10:799–813. doi: 10.2174/156802610791164229. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- O'Dowd BF, Nguyen T, Marchese A, Cheng R, Lynch KR, Heng HH, et al. Discovery of three novel G-protein-coupled receptor genes. Genomics. 1998;47:310–313. doi: 10.1006/geno.1998.5095. [DOI] [PubMed] [Google Scholar]
- Ohshiro H, Tonai-Kachi H, Ichikawa K. GPR35 is a functional receptor in rat dorsal root ganglion neurons. Biochem Biophys Res Commun. 2008;365:344–348. doi: 10.1016/j.bbrc.2007.10.197. [DOI] [PubMed] [Google Scholar]
- Okumura S, Baba H, Kumada T, Nanmoku K, Nakajima H, Nakane Y, et al. Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer Sci. 2004;95:131–135. doi: 10.1111/j.1349-7006.2004.tb03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluszkiewicz P, Zgrajka W, Saran T, Schabowski J, Piedra JL, Fedkiv O, et al. High concentration of kynurenic acid in bile and pancreatic juice. Amino Acids. 2009;37:637–641. doi: 10.1007/s00726-008-0183-x. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Dalrymple MB, Dromey JR, Eidne KA. Monitoring interactions between G-protein-coupled receptors and beta-arrestins. Biochem Soc Trans. 2007;35:764–766. doi: 10.1042/BST0350764. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Stoddart LA, Devine NM, Jenkins L, Milligan G. The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J Biol Chem. 2009;284:17527–17539. doi: 10.1074/jbc.M109.012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Jenkins L, Brown AJ, Milligan G. Conserved polar residues in transmembrane domains V, VI, and VII of free fatty acid receptor 2 and free fatty acid receptor 3 are required for the binding and function of short chain fatty acids. J Biol Chem. 2008a;283:32913–32924. doi: 10.1074/jbc.M805601200. [DOI] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Milligan G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol Rev. 2008b;60:405–417. doi: 10.1124/pr.108.00802. [DOI] [PubMed] [Google Scholar]
- Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Tonai-Kachi H, Shinjo K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 2006;580:5003–5008. doi: 10.1016/j.febslet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Tunaru S, Lattig J, Kero J, Krause G, Offermanns S. Characterization of determinants of ligand binding to the nicotinic acid receptor GPR109A (HM74A/PUMA-G) Mol Pharmacol. 2005;68:1271–1280. doi: 10.1124/mol.105.015750. [DOI] [PubMed] [Google Scholar]
- Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem. 2006;281:22021–22028. doi: 10.1074/jbc.M603503200. [DOI] [PubMed] [Google Scholar]
- Williams C, Hill SJ. GPCR signaling: understanding the pathway to successful drug discovery. Methods Mol Biol. 2009;552:39–50. doi: 10.1007/978-1-60327-317-6_3. [DOI] [PubMed] [Google Scholar]
- Worzfeld T, Wettschureck N, Offermanns S. G(12)/G(13)-mediated signalling in mammalian physiology and disease. Trends Pharmacol Sci. 2008;29:582–589. doi: 10.1016/j.tips.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Xiao SH, Reagan JD, Lee PH, Fu A, Schwandner R, Zhao X, et al. High throughput screening for orphan and liganded GPCRs. Comb Chem High Throughput Screen. 2008;11:195–215. doi: 10.2174/138620708783877762. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, et al. G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology. 2010;86:1–5. doi: 10.1159/000314164. [DOI] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.