

Abstract
Bacterial artificial chromosomes (BACs) and P1 artificial chromosomes (PACs), which contain large fragments of genomic DNA, have been successfully used as transgenes to create mouse models of dose-dependent diseases. They are also potentially valuable as transgenes for dominant diseases given that point mutations and/or small rearrangements can be accurately introduced. Here, we describe a new method to introduce small alterations in BACs, which results in the generation of point mutations with high frequency. The method involves homologous recombination between the original BAC and a shuttle vector providing the mutation. Each recombination step is monitored using positive and negative selection markers, which are the Kanamycin-resistance gene, the sacB gene and temperature-sensitive replication, all conferred by the shuttle plasmid. We have used this method to introduce four different point mutations and the insertion of the β-galactosidase gene in a BAC, which has subsequently been used for transgenic animal production.
INTRODUCTION
There is a growing need for high throughput techniques to determine gene function and spatial/temporal patterns of expression as more human genes are predicted or identified. This includes a requirement for the generation of animal models to examine the effects of single or multiple mutations causing dominant and/or gene dose-dependent genetic diseases (1). In order to accurately recapitulate the endogenous pattern of gene expression in an integration site-independent manner, it is necessary that all the regulatory elements of each gene are preserved. The use of large DNA molecules containing the entire genomic sequence and regulatory elements of a gene, therefore, is not only desirable but also offers a complementary approach to modification of the mouse germ line by gene targeting.
Although there have been a number of studies in which yeast artificial chromosomes (YACs) (2) have been used for this purpose, this approach is limited by the genetic instability of DNA fragments cloned into YAC vectors, the difficulty in obtaining pure intact YAC DNA with high yields and the low frequency of YAC transgene integration (3–6; our unpublished data). This has focused attention on the development of other useful systems for cloning large DNA fragments such as those of P1s bacteriophages (7), P1 artificial chromosomes (PACs) (8) and bacterial artificial chromosomes (BACs) (9). Library availability, size of insert and insert stability are attractive features of BACs and PACs and have led to their widespread use in genome sequencing experiments (10).
Several methods have been described in order to modify large clones (BACs/PACs) by homologous recombination in Escherichia coli, all of which aimed to introduce insertions or deletions (11–16). Here, we describe a new, efficient and rapid method for introducing point mutations into BAC clones in order to generate mouse models of a dominant human disease. We have used the shuttle vector pKOV, previously constructed to modify E.coli genes (17), as the basis of a new generation of vectors for modification of any genomic sequence encoded by BACs by means of a two-step procedure. The recombination was mediated by RecA, which, unlike all the previously described approaches, was expressed from a separate plasmid and not the shuttle vector. The method involves the introduction of efficient positive and negative selection markers, which results in the generation of point mutations with high frequency. Both recombination steps can be simply confirmed by PCR and/or Southern blot analysis. BACs modified by this method yield transgenic mice with high efficiency.
MATERIALS AND METHODS
Constructs
A 6.1 kb EcoRI fragment containing exons 6–9 (including introns) of mouse Fgfr1 was subcloned from a BAC containing the entire gene into pBluescript (Stratagene; pEx7-Eco). Exon 7 (including partial flanking introns) was excised from pEx7-Eco with BglII–XhoI and cloned into pBluescript (pEx7).
The Kanamycin-resistance gene (Kan) and its promoter were amplified from vector pPAC4 using primers Kan.2L: 5′-GAC CAG GTC AAG AAA TCA CAG CCG AAG C-3′ and Kan.2R: 5′-GAC CAG GTG CGT GAT CTG ATC CTT CAA CT-3′ and Pfu polymerase (Promega) according to Narayanan et al. (13). The product was cloned into pCR2.1 (Invitrogen) in two orientations. The Kan gene was excised with NotI and BamHI and cloned into the same sites of vector pKOV (http://arep.med.harvard.edu/labgc/pko3.html), in two orientations: either the same as the chloramphenicol (Cm)-resistance gene of the pKOV (pKOV-KanF) or the opposite (pKOV-KanR). Vector pKOV-Kan mentioned throughout the article refers to pKOV-KanF unless otherwise stated. Following any manipulation of vectors derived from pKOV, we reassessed the positive and negative selections by plating cells on Cm and Cm/sucrose and growing them at 30°C, as well as plating cells on Cm and growing them at 43°C.
A nucleotide polymorphism was introduced in exon 7 using PCR (18). The first PCR was performed using Pfu polymerase (Promega) and primers fgfr.21L: 5′-AGA TCT GGG AAG GGT CTA AG-3′ and fgfr.15R: 5′-CCT GCC TGC AGG ATG GGT C-3′ (carrying the mutation) on the pEx7 template. This PCR product was used as forward primer together with fgfr.21R: 5′-CTC GAG TCT GGG AGC GAG AG-3′ on template pEx7. The 920 bp product was cloned in vector pCR2.1 and sequenced. The insert was excised with BamHI and XhoI and ligated into the BamHI and SalI sites of pKOV-Kan.
Plasmid DNA preparation
All pKOV-Kan, BAC, pDF25 and co-integrant DNA plasmid extractions were performed using a modified version of the miniprep protocol by Xiang et al. (19). pKOV-Kan constructs were grown in 10 ml LB with 20 µg/ml Cm and 20 µg/ml Kan at 30°C. BAC clones (except for oocyte injection, see below) were grown in 10 ml LB with 20 µg/ml Cm at 37°C. pDF25 was grown in 10 ml LB with 20 µg/ml Cm at 30°C. Co-integrants were grown in 10 ml LB with 20 µg/ml Cm and 20 µg/ml Kan at 43°C. Cells were harvested by centrifugation, resuspended in 200 µl GTE (50 mM glucose, 25 mM Tris–HCl pH 8, 10 mM EDTA), alkaline lysed with 400 µl 0.2 M NaOH/1% SDS, precipitated with 300 µl 5 M potassium acetate and centrifuged for 15 min at 4°C. The supernatant was precipitated with 0.7 µl of isopropanol and washed with 70% ethanol. The pellet was resuspended in 50 µl TE.
BAC DNA for oocyte injection was prepared using the Qiagen plasmid midi kit (Qiagen) according to the manufacturer’s guidelines. The BAC was linearised using the P1-SceI endonuclease (NEB) and prepared for pronuclear injection as described by Chrast et al. (20). Briefly, it was separated in a 1% SeakemGold (FMC) agarose gel in 1× TAE using field inversion gel electrophoresis (FIGE, BioRad), excised, electroeluted in 0.5× TAE and dialysed against a large volume of TE (10 mM Tris, 0.25 mM EDTA, pH 7.5).
Bacterial transfections and selection of recombinants
DH10B bacterial cells containing the BAC clone were made competent using CaCl2 (21). Aliquots of 5 µl pDF25 and 1 µl shuttle plasmid miniprep DNA (~70 ng/µl) were transfected into 100 µl chemically-competent DH10B cells carrying the BAC clone. LB (1 ml) was added, the cells were incubated at 30°C for 1.5 h and then 100 µl were plated on Cm and Kan and grown at 30°C. Four to six colonies were picked separately into 1 ml LB and 100 µl were plated on Cm and Kan and grown at 43°C to select co-integrant clones. Tens to hundreds of colonies grew over a thick lawn of non-recombinant small colonies.
DH10B cells containing the co-integrant clones were made competent as above, except that they were grown at 43°C with Cm and Kan. An aliquot of 1 µl of pDF25 miniprep DNA was transfected into 10 µl of cells, 1 ml of LB was added and cells were incubated at 30°C for 1.5 h. Aliquots of 100 µl of cells were plated on Cm and allowed to resolve at 43°C overnight. Several colonies were picked and streaked on Cm/sucrose and left at 30°C overnight to select single colonies. The larger colonies were selected and re-streaked on Cm/sucrose or Cm/Kan/sucrose at 30°C and grown overnight. The colonies that only grew on Cm/sucrose were analysed further.
PCR and Southern blot analyses of co-integrant and resolved BAC clones
Integrity of co-integrant and resolved BAC clones was confirmed by Southern blot analysis. Miniprep DNA was digested with EcoRI and analysed on a 1% Seakem Gold (FMC) agarose gel using FIGE under the following conditions: run time 15 h, temperature 4°C, buffer 1× TAE, switch time ramp 0.1–0.3 s, forward voltage 180 V and reverse voltage 120 V. The DNA was transferred on GeneScreen Plus“ membrane (NEN‘ Life Science Products) and hybridised. The probe was labelled with dig-dUTP using random priming or PCR amplification (Roche).
RT–PCR
Total RNA was extracted from mouse livers using Trizol“ Reagent (Gibco BRL). One step RT–PCR was performed on 100 ng of total RNA using primers fgfr.8L: 5′-ACC TAC CAG CTT GAC GTC GTG-3′ and fgfr.6R: 5′-CAT TTC CTT GTC GGT GGT ATT AAC-3′ using Qiagen OneStep RT–PCR kit.
RESULTS
The aim of these experiments is to construct transgenic mouse models of human skeletal dysplasias induced by mutations in the fibroblast growth factor receptors (FGFRs) 1–3 (reviewed in 22,23). These mutations cause several syndromes characterised by various degrees of craniosynostosis, including Pfeiffer, Crouzon, Apert, Jackson–Weiss syndromes, achondroplasia, thanatophoric dysplasia and skeletal–skin–brain syndrome (reviewed in 23). The FGFRs consist of three extracellular immunoglobulin-like domains, a transmembrane region and a split tyrosine kinase domain (reviewed in 24). They are expressed in multiple forms as result of a complex pattern of alternative splicing (25,26). Taking into consideration the complex pattern of alternative splicing of FGFRs and the autosomal dominant mode of inheritance of craniosynostosis syndromes, we chose to create mouse models by introducing BAC transgenes harbouring specific point mutations into the mouse germ line.
A number of methods have been described to introduce rearrangements in large clones (BACs/PACs) by homologous recombination in E.coli (11–16). The first and last methods are based upon RecA-assisted recombination, while the others are RecE and RecT dependent. In pilot studies, we employed two of the previously described vectors (11,13) to introduce point mutations into a FGFR1 BAC but encountered several problems. At least in our hands, the shuttle vector pSV1.recA produced very low yields of DNA, the unique SalI cloning site placed severe limitations upon potential cloning strategies and non-homologous integrations of the shuttle vector into the BAC occurred with an undesirably high frequency.
As a result, we modified the shuttle vector pKOV, which was developed and successfully used to introduce modifications in the E.coli genome (17). pKOV exhibits temperature-dependent DNA replication: the plasmid can propagate at the permissive temperature of 30–33°C, while replication is deficient at 43°C. It also contains the Cm-resistance gene for positive and the levansucrase gene (sacB) of Bacillus amyloliquefaciens for negative selection (27). Colonies that contain the sacB gene grow very slowly on sucrose. Because the target BAC also confers Cm resistance, we introduced the Kan-resistance gene for positive selection into the shuttle vector (see Materials and Methods and Fig. 1). At the same time, we introduced a new BamHI, next to the existing SalI restriction site for insert cloning. This new vector was named pKOV-Kan.
Figure 1.

Shuttle vector pKOV-KanF. Cm, chloramphenicol-resistance gene; Kan, Kanamycin-resistance gene; RepA, temperature-sensitive replication. The cloning site contains BamHI and SalI restriction enzyme sites.
A genomic fragment of mouse Fgfr1 containing exon 7 and part of introns 6 and 7 was subcloned from a BAC containing the entire gene and a series of point mutations, including the silent polymorphism presented here, were engineered into exon 7 using PCR (Materials and Methods). Each point mutation was designed to introduce a new restriction site in order, eventually, to distinguish the expression of the endogenous gene from that of the transgene (Fig. 3A). A 920 bp fragment of mutant exon 7 and surrounding introns was cloned in the pKOV-Kan shuttle vector. The nucleotide change is located 250 bp from the 5′ end and 670 bp from the 3′ end of this fragment. The size of the fragment was selected to exceed 500 bp in total because the recombination frequency is reported to decline rapidly when the fragment is smaller than this (11). This plasmid was cotransfected into E.coli cells harbouring the BAC target together with plasmid pDF25 expressing RecA (16). The pDF25 plasmid can only propagate at the permissive temperature of 30–33°C and not at 43°C. We specifically did not introduce the RecA gene into the shuttle vector but utilised a cotransfection approach in order to emphasise the stability of the intermediate stage, the co-integrant clones, as explained below.
Figure 3.

(A) The box shows the exon sequence around the introduced nucleotide change and its translation into protein. The first line shows the original sequence. The nucleotide change, in the third line, is shown in lower case letters and the new PstI site is underlined. (B) PCR amplification of BAC-shuttle co-integrants using primers shown on the right (see Fig. 2) and digested with PstI. Clones 2, 3, 5 and 6 contained the mutation on the ‘left’ arm of the co-integrant and clones 1 and 4 on the ‘right’ arm. (C) Southern blot of the co-integrant clones, the original BAC, the shuttle vector and resolved BAC clones carrying the mutation, were digested with EcoRI and hybridised with exon 7.
The cells were first grown on Cm and Kan plates at the permissive temperature of 30°C. A few of these colonies were selected and grown on the same antibiotics at 43°C to select for recombined clones. In the first recombination event, the ‘integration’, the shuttle vector integrates in the BAC (Fig. 2). Surviving clones were analysed by PCR and Southern blot analysis (Fig. 3B and C). Homologous recombination between the shuttle vector and the BAC produced co-integrant clones with a duplication of exon 7. The mutation was present either on the ‘left’ or ‘right’ arm of the co-integrant depending on whether the recombination had occurred 3′ or 5′ of this particular nucleotide, respectively (Fig. 2). The frequency of co-integration within exon 7 varied between 75 and 85%. Two different nucleotide changes were tested, with the same size of homology arms flanking the mutation, and both showed similar recombination efficiencies. Both types of co-integrant were found in 27–33% of these colonies. Occasionally, we observed clones that contained a co-integrant and an unrecombined BAC (e.g. clone 5 in Fig. 3C). We have not detected any integration within the Cm-resistance gene, which is present in both vectors, as determined by Southern blot analysis (data not shown). When the homology 5′ of the nucleotide change was reduced to 198 bp from 250 bp the integration frequency dropped to 50%.
Figure 2.

The recombination consists of two events. 1. The integration of the shuttle vector pKOV-Kan into the BAC and 2. the resolution of the co-integrant into either the original or the mutant BAC. The position of primers used for PCR are shown. EcoRI sites on the BAC are indicated by vertical lines. EcoRI sites on the pKOV-Kan are indicated by arrows. E, EcoRI; m, mutant; n, normal nucleotide.
Co-integrants containing homologously recombined shuttle vector and BAC were allowed to resolve on Cm at 43°C and were then grown on 5% sucrose/Cm at 30°C, which selects for a second recombination event: the resolution (Fig. 2). The surviving colonies contained either the original or a BAC with the point mutation. This selection is possible because the pKOV-Kan vector contains the sacB gene, which blocks the growth of clones containing co-integrant BACs on sucrose. Single colonies were picked on Kan to verify that they survived on sucrose because they lost the pKOV-Kan vector and not because of a mutation in the sacB gene. Fifty percent of the clones that were able to grow on sucrose also grew on Kan, which indicates that the mutation rate within this gene is high. Despite this high frequency, the number of colonies that had lost the pKOV-Kan plasmid was also high. The presence of the mutation was tested by PCR, and the integrity of the resolved BAC was examined by agarose gel electrophoresis and Southern blot analysis (Fig. 3C). It is of interest that the resolution step for each co-integrant is strongly biased towards one of two possible outcomes in these experiments; each independent co-integrant only resolves into normal or mutant BAC.
During the first recombination event, the integration, the clones exhibit insert stability, as viewed by Southern blot (Fig. 3C). However, this is not the case during the second recombination event, where only 7% (9/129) were intact and the remainder lost various insert fragments (data not shown). Because the RecA plasmid also exhibits temperature-sensitive replication, it is possible that the colonies, which have undergone an aberrant second recombination event, have lost this plasmid during the selection for integration. To overcome this problem we retransfected the RecA plasmid into the co-integrants before resolving them. This time, the frequency of accurate resolution was ~97% (31/32).
We tested whether the orientation of the Kan-resistance gene relative to the Cm-resistance gene affects the recombination efficiency of the modified vector. We observed that, upon transformation into BAC cells, only the shuttle vector containing Kan- and Cm-resistance genes with the same orientation was able to produce co-integrants. We did not explore any further why the other shuttle vector did not yield any co-integrants.
A control experiment was performed where the RecA-expressing plasmid was omitted. The number of colonies surviving on Kan at 43°C was not significantly different from when the RecA plasmid was co-transfected. However, none of these colonies contained BAC shuttle vector co-integrants, as opposed to the high percentage of accurate co-integration mentioned above. A similar experiment was performed using the shuttle vector pKOV-Kan without an insert. Again, no integration of the two was detected within the Cm-resistance gene.
The mutant BAC DNA was prepared for pronucleus injection as described by Chrast et al. (20) and in the Materials and Methods. Animals were tested for transgene incorporation using PCR amplification of tail DNA and transgene expression using RT–PCR (Fig. 4). The RT–PCR primers were designed on exons flanking the mutation and the product was digested with PstI. Twenty-five percent of animals born contained the transgene. This corresponds to 2.8% of eggs that developed to two-cell embryos and were transferred. We have tried several DNA dilutions of the mutant BAC for oocyte injection. The concentration of 0.3 ng/µl gave the highest rate of embryos developing into the two-cell stage (69%).
Figure 4.
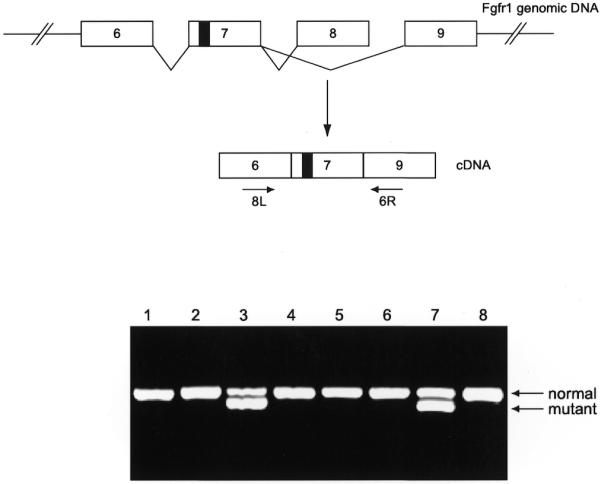
RT–PCR on liver RNA from E19 mice. Exons 8 and 9 of Fgfr1 are alternatively spliced. 8L-6R PCR product was digested with PstI, which detects the mutation (indicated by a black box in exon 7) carried by the mutant BAC. Embryos 3 and 7 were carrying and expressing the BAC transgene.
DISCUSSION
The insert stability of BAC vectors containing large DNA fragments makes them an excellent tool for use in genetics research. Establishing methods to introduce modifications in these clones is necessary for the utilisation of these clones to produce animal models of dominant diseases caused by various point mutations, insertions or deletions. Moreover, the production of animal models for gene dose-dependent diseases (e.g. Down syndrome; 6) requires the presence of a DNA polymorphism in the transgene. Often, several kilobases of coding regions are sequenced to detect a base change (28). For highly conserved genes, however, this may not be possible (e.g. Fgfr2, Twigg; A.O.Wilkie, personal communication). Instead, our method could be used to create such a polymorphism and it provides the opportunity to introduce a convenient restriction enzyme site. Several methods have been developed so far to introduce rearrangements in BACs or PACs. Here we report a method for introducing a single point mutation within the coding region of a gene carried on a BAC with high efficiency. This method consists of a homologous integration and subsequent excision of a BAC and a shuttle plasmid sharing a region of homology. The recombination events were mediated and dependent on the presence of a RecA-expressing plasmid.
Unlike all the previous approaches using RecA-mediated recombination, we chose not to include the RecA gene in the shuttle vector but to express it from a different plasmid. This method, therefore, does not result in the integration of RecA sequences in the intermediate step. With this protocol, the RecA plasmid is lost at the non-permissive temperature of 43°C. Since the BAC clones contain very large pieces of genomic DNA, it is very possible they contain repeated sequences. In the continued presence of RecA this could result in increased instability of the co-integrant clones. Moreover, this method is more flexible and can therefore be used for many different purposes including construction of in-frame LacZ fusion mutations for studying expression patterns where an excision step is not required. For example, using only the first recombination step, we have inserted the β-galactosidase gene in-frame with the Fgfr1 gene. Since the translational stop codon is provided by the β-galactosidase gene there is no need for a second recombination event.
The fragment of the gene carrying the mutation was 918 bp in length. It was constructed in order to conform to the recombination frequencies obtained with different homology lengths (11). While this manuscript was in preparation Imam et al. (16) reported a different modification system from that of Yang et al. (11), which relied on temperature sensitivity and the rpsL+ gene, and demonstrated increased recombination frequencies. Our observed frequencies for a homology length of 250 bp 5′ and 670 bp 3′ of the mutation were 75–85% in the first step and 97% in the second, when RecA carrying plasmid was reintroduced. The frequency of integration is slightly higher than reported by Imam et al. (16). During the excision step, however, the method reported here is likely to be more efficient. This is an indirect assumption we made from the lower frequency of excision (40–80%) reported in the paper for clones with >1 kb of homology from both sides of the nucleotide change (16). The difference between the two previously published methods (11,16) describing RecA-mediated recombination in E.coli and the one reported here, is that in our case the recombinase is expressed from a separate plasmid. It is possible that this contributes to the slightly increased recombination efficiencies we observed.
We observed that when each co-integrant was forced to undergo the second recombination event it resulted in either exclusively normal or mutant BAC clones. In our first three experiments we tested 23 co-integrant clones and 20 resolved BACs from each. Because unidirectional recombination was observed in all, we then screened only four resolved clones per co-integrant. The number of co-integrants that resolve towards the original BAC is similar to those that resolve to the mutant. Therefore, in these experiments there does not seem to be a predisposition towards one or the other type. It is likely that the bacterial cell has, in some way, ‘signposted’ the site of the first recombination event and performs the second one in spatial relation with the first. Although the mechanism of this phenomenon remains to be defined, it represents a very important practical point for potential users of this method.
While we only describe use of this method in introducing point mutations, we have also used it to produce an in-frame Fgfr1-lacZ fusion, explained above. This, and further adaptations of this method, can be employed to engineer complex genetic alterations in any large piece of genomic DNA cloned into BACs.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs X. W. Yang and N. Heintz for the pSV1.RecA, Dr K. Narayanan for the pGET, Drs A. J. Link and G. M. Church for the pKO3 and pKOV and Dr D. Sherratt for the pDF25. We thank Drs D. Sherratt, A. O. M. Wilkie, S. Twigg, H. D. Burns and S. Bidonde for useful discussions and suggestions and Ms Karen Faulkner and Céline Jones for technical assistance. M.D.L. is supported by an EMBO long term fellowship. This work is supported by the Wellcome Trust.
References
- 1.Magdaleno S.M. and Curran,T. (1999) Gene dosage in mice–BAC to the future. Nature Genet., 22, 319–320. [DOI] [PubMed] [Google Scholar]
- 2.Burke D.T., Carle,G.F. and Olson,M.V. (1987) Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors. Science, 236, 806–812. [DOI] [PubMed] [Google Scholar]
- 3.Green E.D., Riethman,H.C., Dutchik,J.E. and Olson,M.V. (1991) Detection and characterization of chimeric yeast artificial-chromosome clones. Genomics, 11, 658–669. [DOI] [PubMed] [Google Scholar]
- 4.Schedl A., Beermann,F., Thies,E., Montoliu,L., Kelsey,G. and Schutz,G. (1992) Transgenic mice generated by pronuclear injection of a yeast artificial chromosome. Nucleic Acids Res., 20, 3073–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manson A.L., Trezise,A.E., MacVinish,L.J., Kasschau,K.D., Birchall,N., Episkopou,V., Vassaux,G., Evans,M.J., Colledge,W.H., Cuthbert,A.W. et al. (1997) Complementation of null CF mice with a human CFTR YAC transgene. EMBO J., 16, 4238–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith D.J., Stevens,M.E., Sudanagunta,S.P., Bronson,R.T., Makhinson,M., Watabe,A.M., O’Dell,T.J., Fung,J., Weier,H.U., Cheng,J.F. et al. (1997) Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nature Genet., 16, 28–36. [DOI] [PubMed] [Google Scholar]
- 7.Sternberg N. (1990) Bacteriophage P1 cloning system for the isolation, amplification, and recovery of DNA fragments as large as 100 kilobase pairs. Proc. Natl Acad. Sci. USA, 87, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ioannou P.A., Amemiya,C.T., Garnes,J., Kroisel,P.M., Shizuya,H., Chen,C., Batzer,M.A. and de Jong,P.J. (1994) A new bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nature Genet., 6, 84–89. [DOI] [PubMed] [Google Scholar]
- 9.Shizuya H., Birren,B., Kim,U.J., Mancino,V., Slepak,T., Tachiiri,Y. and Simon,M. (1992) Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl Acad. Sci. USA, 89, 8794–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hattori M., Fujiyama,A., Taylor,T.D., Watanabe,H., Yada,T., Park,H.S., Toyoda,A., Ishii,K., Totoki,Y., Choi,D.K. et al. (2000) The DNA sequence of human chromosome 21. The chromosome 21 mapping and sequencing consortium. Nature, 405, 311–319. [DOI] [PubMed] [Google Scholar]
- 11.Yang X.W., Model,P. and Heintz,N. (1997) Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat. Biotechnol., 15, 859–865. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y., Buchholz,F., Muyrers,J.P. and Stewart,A.F. (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nature Genet., 20, 123–128. [DOI] [PubMed] [Google Scholar]
- 13.Narayanan K., Williamson,R., Zhang,Y., Stewart,A.F. and Ioannou,P.A. (1999) Efficient and precise engineering of a 200 kb beta-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther., 6, 442–447. [DOI] [PubMed] [Google Scholar]
- 14.Muyrers J.P., Zhang,Y., Testa,G. and Stewart,A.F. (1999) Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res., 27, 1555–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muyrers J.P., Zhang,Y., Benes,V., Testa,G. and Stewart,A.F. (2000) Point mutation of bacterial artificial chromosomes by ET recombination. EMBO rep., 1, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imam A.M., Patrinos,G.P., de Krom,M., Bottardi,S., Janssens,R.J., Katsantoni,E., Wai,A.W., Sherratt,D.J. and Grosveld,F.G. (2000) Modification of human beta-globin locus PAC clones by homologous recombination in Escherichia coli. Nucleic Acids Res., 28, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Link A.J., Phillips,D. and Church,G.M. (1997) Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol., 179, 6228–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene, 15, 51–59. [DOI] [PubMed] [Google Scholar]
- 19.Xiang C., Wang,H., Shiel,P., Berger,P. and Guerra,D.J. (1994) A modified alkaline lysis miniprep protocol using a single microcentrifuge tube. Biotechniques, 17, 30–32. [PubMed] [Google Scholar]
- 20.Chrast R., Scott,H.S. and Antonarakis,S.E. (1999) Linearization and purification of BAC DNA for the development of transgenic mice. Transgenic Res., 8, 147–150. [DOI] [PubMed] [Google Scholar]
- 21.Cohen S.N., Chang,A.C. and Hsu,L. (1972) Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA. Proc. Natl Acad. Sci. USA, 69, 2110–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilkie A.O., Morriss-Kay,G.M., Jones,E.Y. and Heath,J.K. (1995) Functions of fibroblast growth factors and their receptors. Curr. Biol., 5, 500–507. [DOI] [PubMed] [Google Scholar]
- 23.Wilkie A.O. (1997) Craniosynostosis: genes and mechanisms. Hum. Mol. Genet., 6, 1647–1656. [DOI] [PubMed] [Google Scholar]
- 24.Johnson D.E. and Williams,L.T. (1993) Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res., 60, 1–41. [DOI] [PubMed] [Google Scholar]
- 25.Johnson D.E., Lu,J., Chen,H., Werner,S. and Williams,L.T. (1991) The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol., 11, 4627–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Twigg S.R., Burns,H.D., Oldridge,M., Heath,J.K. and Wilkie,A.O. (1998) Conserved use of a non-canonical 5′ splice site (/GA) in alternative splicing by fibroblast growth factor receptors 1, 2 and 3. Hum. Mol. Genet., 7, 685–691. [DOI] [PubMed] [Google Scholar]
- 27.Gay P., Le Coq,D., Steinmetz,M., Berkelman,T. and Kado,C.I. (1985) Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol., 164, 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chrast R., Scott,H.S., Madani,R., Huber,L., Wolfer,D.P., Prinz,M., Aguzzi,A., Lipp,H.P. and Antonarakis,S.E. (2000) Mice trisomic for a bacterial artificial chromosome with the single-minded 2 gene (Sim2) show phenotypes similar to some of those present in the partial trisomy 16 mouse models of down syndrome. Hum. Mol. Genet., 9, 1853–1864. [DOI] [PubMed] [Google Scholar]