

Abstract
Cardiac plasmin activity is increased following myocardial ischemia. To test the hypothesis that macrophage-derived uPA is a key mediator of repair following myocardial infarction we performed myocardial infarction on mice with macrophage specific over-expression of uPA (SR-uPA mice). SR-uPA+/0 mice and wild-type littermates were sacrificed at 5 days or 4 weeks after infarction and cardiac content of macrophages, collagen, and myofibroblasts was quantified. Cardiac function and dimensions were assessed by echocardiography at baseline and at 4 weeks post-infarction. At 4 weeks after myocardial infarction, macrophage counts were increased in SR-uPA+/0 mice in the infarct (13.1 vs. 4.9 %, P < 0.001) and distant uninfarcted regions (5.9 vs. 2.4%, P < 0.001). Infarct scar was thicker in SR-uPA+/0 mice (0.54 ± 0.03mm vs. 0.45 ± 0.03mm, P <0.05) and infarct cardiac collagen content was increased (72.4 ± 3.3% vs. 63.0 ± 3.6%, P < 0.06). Functionally, these changes resulted in mildly improved fractional shortening in SR-uPA+/o mice compared to controls (24.6 ±1.68 vs. 19.8 ± 1.3% P = 0.03). At 5 days after infarction there was increased collagen content in the scar without increases in macrophages or myofibroblasts. To understand the mechanisms by which macrophage derived uPA increases collagen, cardiac fibroblasts were treated with macrophage conditioned medium or plasmin and expression of ColIα1 measured by qPCR. Conditioned media from SR-uPA+/o or plasmin-treated nontransgenic macrophages but not plasmin alone increased collagen expression in isolated cardiac fibroblasts. We hypothesize that plasmin generation in the heart in response to injury may induce activation of macrophages to a profibrotic phenotype to allow rapid formation of collagenous scar.
Keywords: remodeling, inflammation, macrophage, plasminogen, collagen
1. Introduction
Because of the heart’s unique mechanical stresses, repair of injured myocardium must be both rapid and durable [1–3]. Current data support a model in which in response to injury chemokines are immediately released to stimulate the immigration of neutrophils into the myocardium. Neutrophils then promote the influx and accumulation of bone marrow-derived monocytes that subsequently differentiate into macrophages. Early after injury these monocyte-derived macrophages produce proteineases that degrade damaged tissue and later elaborate factors that stimulate collagen deposition in the scar by fibroblasts [4]. This balance of tissue degradation and matrix deposition is critical to both preventing cardiac rupture and avoiding excess matrix deposition and resultant cardiac fibrosis.
The plasminogen activator/plasminogen system is hypothesized to have a key role in myocardial repair. Plasmin activity is increased in chronically ischemic human hearts and in animal models of acute ischemia [5–7]. Genetic absence of urokinase plasminogen activator (uPA–which converts plasminogen to the active zymogen plasmin) or plasminogen, inhibits macrophage accumulation, degradation of injured tissue, and collagen deposition following myocardial infarction [8, 9]. These findings appear to depend on plasmin activation within the cardiac extracellular matrix as mice deficient in tissue plasminogen activator (tPA–which is limited predominantly to the intravascular compartment) have preserved cardiac repair [8]. These data have led to the model that plasmin mediated degradation of injured matrix is critical for macrophage and fibroblast migration to the area of injury. In the absence of fibroblasts, a collagenous matrix cannot be formed.
Studies to elucidate the downstream mediators of plasmin-directed cardiac repair have focused on matrix degradation by selective members of the matrix metalloproteinase (MMP) family. Some MMP’s may be activated by plasmin in vivo or function in plasmin-MMP cascades [9–11]. Furthermore, a substantial literature has linked plasmin-dependent activation of MMP–2 and –9 with cardiac repair [12]. However, evidence that these two MMP’s play a significant role in matrix turnover is scant, at best, and MMP activation has been associated with both increased and decreased cardiac collagen accumulation [13–15]. Moreover, MMP’s may serve other roles in cardiac repair such as regulation of vascular integrity [16].
Because uPA is abundantly produced by macrophages–a cell at the crux of the repair process–we hypothesized that macrophage-derived uPA is a critical regulator of plasmin activation in the injured heart. Moreover, we hypothesized that macrophage-derived uPA can directly mediate fibroblast and matrix accumulation in response to cardiac injury. To test this hypothesis, we utilized two lines of transgenic mice with macrophage-specific over-expression of murine uPA. We have previously reported that the M87 line, which has very high levels of uPA expression, develops spontaneous, plasminogen-dependent cardiac collagen deposition by 15 weeks of age [17, 18]. Here we describe studies using the lower expressing M29 line, which does not develop early spontaneous fibrosis. These mice underwent myocardial infarction to determine if moderate increases in macrophage derived uPA can accelerate cardiac repair without promoting cardiac rupture. We found that not only is macrophage-derived uPA a critical determinant of matrix accumulation in the infarcted myocardium, but it also contributes to matrix accumulation in distant uninjured myocardium. In addition, contrary to previous models, macrophage derived uPA does not increase fibroblast number or MMP activation in the infarct. Rather, increased plasmin activation promotes increased numbers of macrophages that exhibit a novel profibrotic phenotype.
2. Materials and Methods
2.1 Transgenic mice
Generation of SR-uPA+/0 mice has been previously described [19]. Three lines with various levels of transgene expression were originally generated using the human scavenger receptor A promoter (SR) to target mouse uPA expression to macrophages. For these studies, we used the M29 line that has detectable but lower levels of transgene expression than the M87 line we described [19]. This line was selected because it does not develop spontaneous cardiac fibrosis. All mice were backcrossed into the C57BL/6 background greater than 8 generations.
2.2 Cell Culture
Isolation of peritoneal macrophages
Macrophages were collected by peritoneal lavage three days after injection with 3 ml of a 4% thioglycollate solution. Cells were plated and after 3 hours non-adherent cells removed. To generate macrophage-conditioned media, plating media was replaced with serum free media M199. Cells were incubated overnight and the following day cultured medium was removed and immediately used for fibroblast stimulation studies.
Isolation of cardiac fibroblasts
We cultured cardiac fibroblasts from nontransgenic mice as described by Haudek et al [20]. Hearts of nontransgenic mice were harvested and minced. The cells were dispersed using repetitive digestions with Liberase Blendzyme 4. Dispersed cells were plated and non-adherent cells washed off after 3 hours. Fibroblasts were cultured until ~80% confluent at which point the fibroblast media was replaced with conditioned media from peritoneal macrophages from SR-uPA+/0 or SR-uPA0/0 mice for 24 hours or M199 with or without plasmin (Sigma). Fibroblast mRNA was then isolated using RNAeasy (Qiagen).
2.3 Taqman Quantitative PCR
cDNA was produced using the Superscript First-Strand Synthesis System for RT-PCR (Invitrogen: cat# 11904-018) according to the manufacturer’s instructions. Primer/probe sets for ColIα1 (Mm00801666_g1) were pre-validated sets obtained from Applied Biosystems (Inventoried TaqMan® Gene Expression Assays: cat# 4331182). qPCR was performed using the Taqman® Universal PCR Mastermix from Applied Biosystems (cat# 4304437). All PCR reactions were normalized internally using either an eukaryotic 18S RNA primer set (Applied Biosystems; cat# 4319413E) or GAPDH RNA primer set as an endogenous control and run in triplicate. Quantitative Taqman PCR was performed using the Applied Biosystem 7500 Real Time PCR System and analyzed using the Applied Biosystem 7500 System sequence detection software, version 1.3.
2.4 Myocardial infarction model and echocardiography
Hearts from 35 SR-uPA+/0 and 41 SR-uPA0/0 littermates at 12–16 weeks of age were infarcted by permanent ligation surgery with a goal to infarct 20–40% of the myocardium. This procedure generates sufficient ventricular remodeling to respond to therapeutic interventions [21, 22]. Surgeons were blinded to the genotype of the mice. 8 mice were harvested at 5 days post infarction and 48 mice harvested at 4 weeks post infarction. Echocardiographic images were obtained at 48 hours and 4 weeks after infarction. The animals were lightly anesthesized using 1% isofluorane and echocardiographic images were obtained with a GE Vivid 7 system using a 13 MHz linear transducer (General Electric Healthcare) [23]. Parasternal long axis and short axis images at the mid-papillary level were obtained to acquire M-mode measurements of the left ventricular end-diastolic (LVEDD) and systolic dimensions (LVESD). Data were averaged from 3–5 cardiac cycles. Fractional shortening was calculated from the LVEDD and LVESD by using the formula: (LVEDD-LVESD)/LVEDD × 100. All measurements were made in accordance with guidelines approved by the American Society of Echocardiography and were determined by a single reader, who was blinded to the genotypes and time-points of the study.
2.5 Immunohistochemistry and morphometry
For histologic studies, hearts were obtained at 5 days or 4 weeks following infarction. Hearts were fixed in 4% paraformaldehyde, sectioned into four transverse slices of equal thickness, and paraffin-embedded. Sections (5 µm) were cut and stained with H & E for morphometry, Picrosirius red with fast green counterstain for collagen, or immunohistochemistry (IHC) was performed for macrophages (Mac 3 antibody, 1:1000, Pharmingen) or smooth muscle alpha actin (smooth muscle alpha actin, 1:50, Dako) using 3-3’diaminobenzidine chromogen (DAB) and Hematoxylin nuclear staining. Infarct size was measured by taking the areas of the infarct region and the left ventricle from cross sections of the heart from apex to the ligation area and averaged. Collagen content was measured by imaging the cross section of the heart containing the maximal infarct area and quantifying the picrosirius red-stained area using computer-assisted image analysis (Image Pro 3.0 software, Media Cybernetics). The collagen content of the uninjured septal region opposite the infarct was also measured. Macrophages were counted by taking 3–5 areas of the infarct and septum directly away from the infarct using a 100 µm2 grid at 40× objective and counting all cells with nuclei. The percent macrophages were determined by taking the total number of positive Mac 3 cells in the 100 µm2 region. Alpha-smooth muscle actin (α-SMA) positive cells were also manually counted at 10× objective in the infarct region and area away from the infarct using a 100 µm2 grid. In order to determine whether differences in ventricular remodeling were due to myocyte hypertrophy in the region away from the infarct, the diameter of the cardiomyocytes in the septum were measured using Image J program (NIH). Cardiomyocytes were measured in long axis at the level of the nucleus. All images used for analysis were photographed using CapturePro camera. All histologic quantification was performed by observers blinded to genotype.
2.6 OmniMMP assay
Hearts were harvested 2 weeks following infarction and protein extracted in 50 mM HEPES, 10 mM CaCl2, 0.05% Brij-35, 10 µM ZnCl2, pH 7.0. Protein was quantified and equivalent samples aliquoted in a 96-well plate in triplicates. OmniMMP MCA peptide (Biomol International, Plymouth Meeting, MA) was added to a final concentration of 0, 5, 10, 15, or 20 µM (100 µl total volume). Samples were excited at 328 nm and fluorescence output was read at 393 nm in 30 sec intervals for 30 min with a SpetraMax 250 plate reader (Molecular Devices, Sunnyvale, CA). Initial velocity (Vi) for each reaction was determined with using SoftmaxPro 4.6 plate reader software and was used to calculate Km and Vmax values.
2.7 Statistics
Data that are normally distributed are presented as mean ± SEM and are compared with Student’s t-test. Data that were not normally distributed are presented as median (25–75% range) and group medians were compared with the Mann-Whitney rank-sum test.
2.8 Statement of Responsibility
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
3. Results
3.1 M29 SR-uPA+/o mice do not have spontaneous cardiac fibrosis
We have previously shown that mice with high levels of macrophage-derived uPA develop spontaneous cardiac fibrosis by 15 weeks of age [17]. To examine the role of uPA in myocardial repair without the confounding variable of existing fibrosis, we used a second line of mice generated for these studies, referred to as the M29 line. Quantitative PCR of mRNA for uPA shows that M29 SR-uPA+/0 peritoneal macrophages have 100 fold increased expression of uPA compared to macrophages from non-transgenic littermates. These levels are 8 fold less than the previously reported M87 line (Figure 1A). In contrast to that seen in M87 mice, M29 SR-uPA+/0 mice have no statistically significant increase in cardiac collagen content at 12–16 weeks of age compared to nontransgenic mice (0.9±0.2 vs 0.68±0.3% picrosirius red area, P = 0.5, Figure 1 B, C, D). At baseline, there were no differences in echocardiographic measurements between SR-uPA+/0 and non-transgenic mice (Supplemental Table 1).
Figure 1. Expression of uPA and collagen content in M29 SR-uPA+/0 mice.
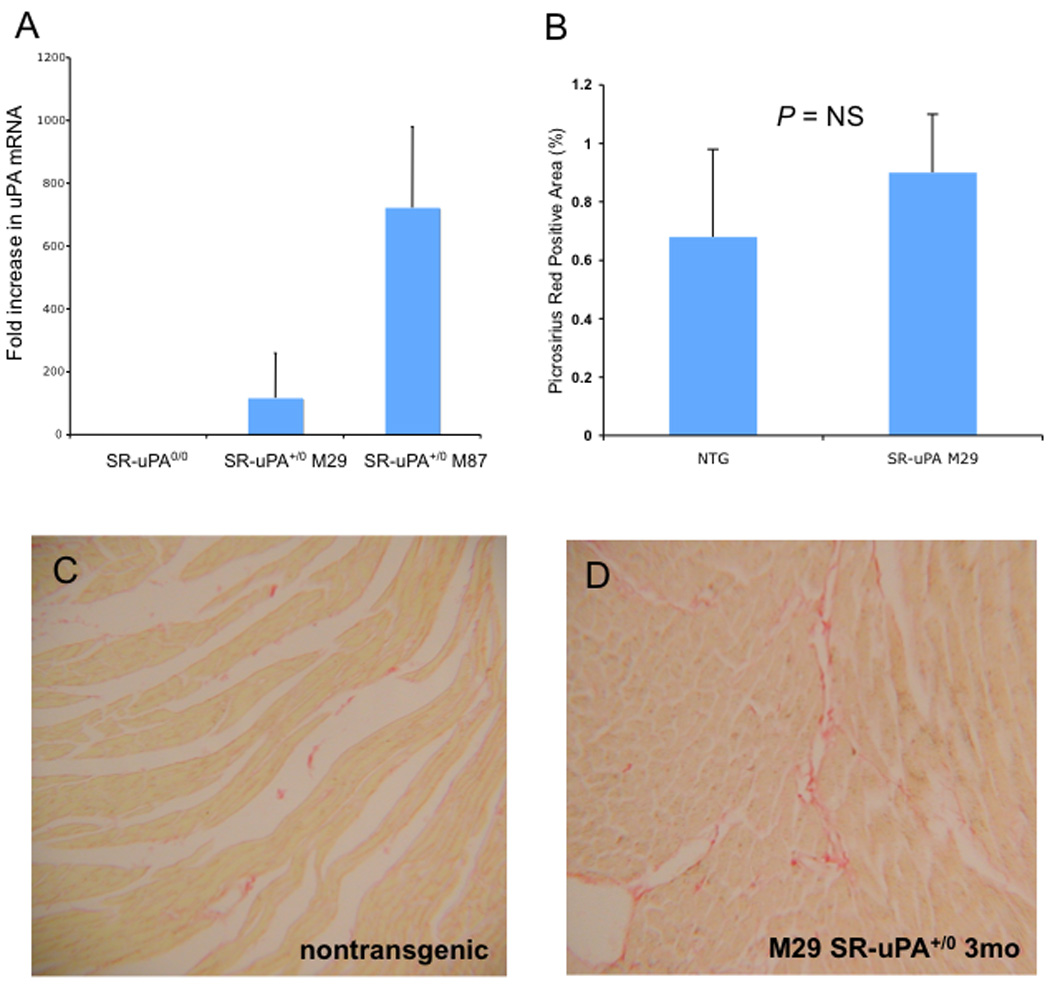
A. Quantitative Taqman PCR for uPA mRNA in isolated peritoneal macrophages from nontransgenic, SR-uPA M29 and SR-uPA M87 mice. B. Percent fibrillar collagen in uninfarcted nontransgenic and SR-uPA M29 hearts. Picrosirius red stain of uninfarcted hearts at 3 months of age from C. nontransgenic, and D. SR-uPA M29 mice.
3.2 Survival and rupture
A total of 76 animals were infarcted for this study (N=35 SR-uPA+/o and N=41 non-transgenic). There was no significant difference in survival between SR-uPA+/o and nontransgenic littermates post infarction. Mice infarcted early in the study had a high intra-operative mortality due to intermittent ventilator dysfunction. After obtaining a new ventilator, mortality was less than 15% in both groups. At 5 days, 22 of 35 SR-uPA+/o mice and 26 of 41 non-transgenic mice had survived myocardial infarction (P = 1.0 by Fisher Exact Test). Late mortality was infrequent, occurring in 2 of 28 and 1 of 33 mice aged to 4 weeks following infarction. Myocardial rupture was not seen in either group.
3.3 SR-uPA+/0 mice have thicker infarct scars
Hearts were harvested 4 weeks after myocardial infarction and morphometric analysis of histology was performed. Although there was no difference in infarct size between SR-uPA+/0 and non-transgenic mice (21.0±1.9% vs. 24.7 ± 4.2% infarcted area, respectively, p=NS), SR-uPA+/o mice had significantly thicker scars compared to nontransgenic mice (0.54 ± 0.03mm vs. 0.45 ± 0.03mm, P <0.05, Figure 2). Thicker scars were not due to increased cardiomyocyte hypertrophy. Analysis of the uninjured myocardium of infarcted mice revealed that cardiomyocytes in both the transgenic and non-transgenic mice had comparable cardiomyocyte diameter (15.1 ± 1.0 µm vs. 14.0 ± 0.6 µm, n= 15–18, P = 0.8).
Figure 2. Infarct scar thickness in SR-uPA+/0 versus nontransgenic mice post myocardial infarction.
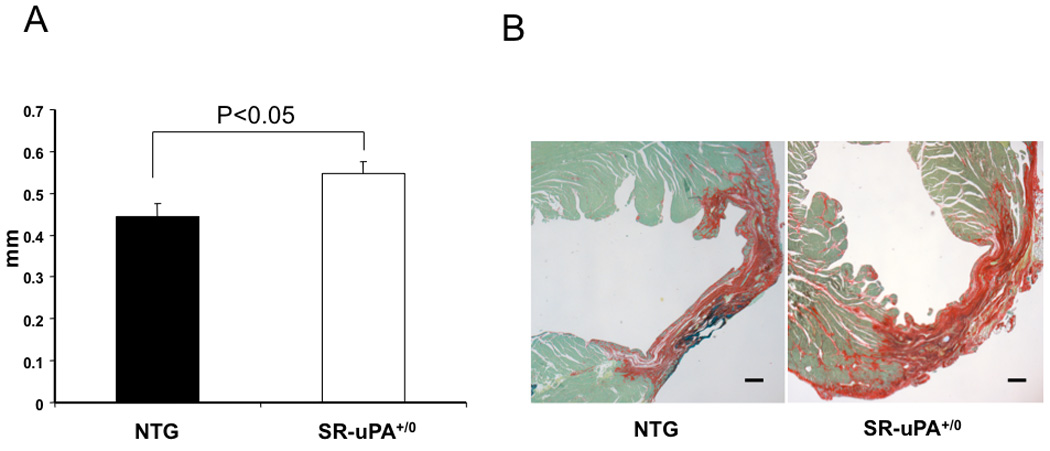
A. Infarct thickness measured on picrosirius red stained sections in non-transgenic (NTG) mice and SR-uPA+/0 mice 4 weeks after myocardial infarction (n = 11–15, P <0.05). Data are mean ± SEM. B. Picrosirius red stain of hearts four weeks after myocardial infarction. Picrosirus red staining delineates collagenous scar as red. Fast-green counterstain highlights non-collagen tissue. Scale bar = 10 µm.
3.4 SR-uPA+/0 mice have increased cardiac macrophage accumulation late after myocardial infarction
To test the hypothesis that excess macrophage-derived uPA would increase the accumulation of macrophages in infarcts, we quantified macrophages in hearts of SR-uPA+/0 and nontransgenic mice 4 weeks after myocardial infarction. Macrophages were more abundant in the infarct in the SR-uPA+/0 mice compared to nontransgenic littermates [13.1 (11.2–19.3%) vs. 4.9 (3.9–8.5%) Mac-3 positive cells, n=12–14, P < 0.001, Figure 3]. In addition, higher macrophage content was seen in the uninfarcted myocardium in SR-uPA+/0 mice compared to non-transgenic mice [5.9(4.3–8.1%) vs. 2.4(1.5–2.9%) Mac-3 positive cells, n=12, P < 0.001, Figure 3]. In five mice harvested two weeks following myocardial infarction, there was no difference in macrophage counts between groups (data not shown).
Figure 3. Macrophage content in hearts of SR-uPA+/0 versus nontransgenic mice late post myocardial infarction.
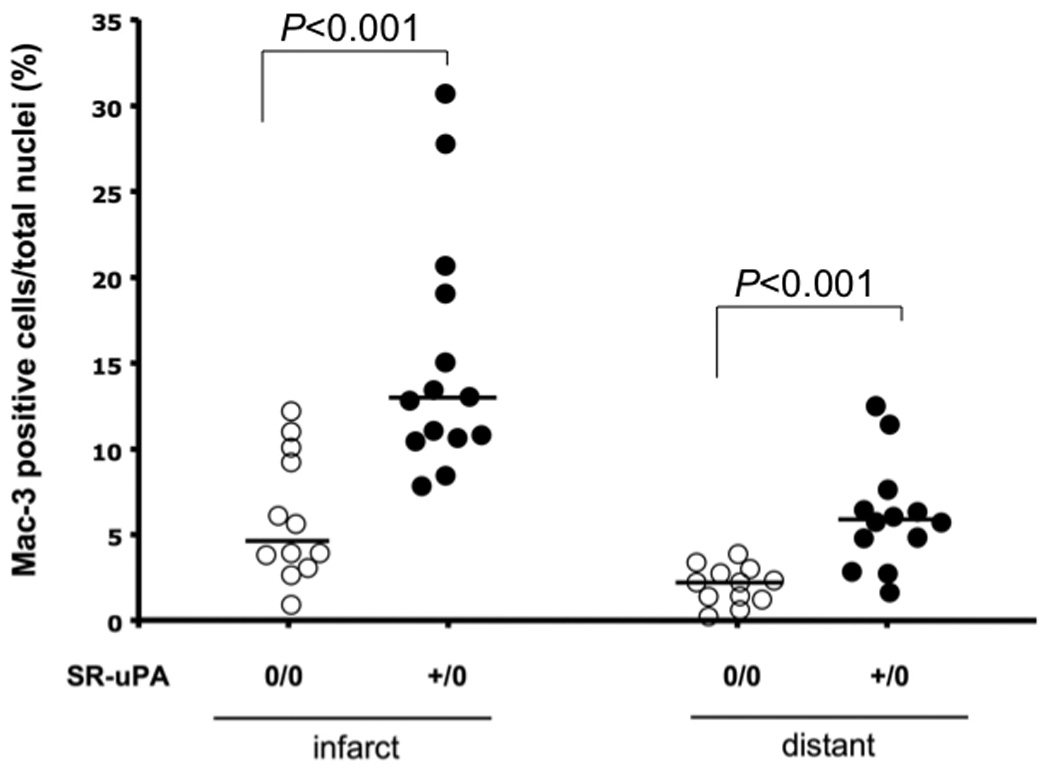
Macrophage content (Mac-3 immunostain) in the infarct region and the area distant to the infarct 4 weeks after myocardial infarction in the SR-uPA+/0 mice and non-transgenic (SR-uPA0/0) littermates (n = 12–16), horizontal lines are group medians.
3.5 SR-uPA+/0 mice have increased cardiac collagen content late after myocardial infarction
To test the hypothesis that excess macrophage derived uPA would be associated with increased collagen after injury, we quantified collagen in scar and uninjured myocardium. There was a trend towards more collagen in the infarct region of transgenic mice (72.4 ± 3.3% vs. 63.0 ± 3.6%, P < 0.06, Figure 4). In remote uninjured myocardium, SR-uPA+/o mice had a significantly higher percentage of collagen compared wild-type littermates (17.8± 2.0% vs. 8.3 ± 3.3%, P < 0.005, Figure 4) or to hearts of uninfarcted M29 mice (0.9±0.2%, P = 0.002). To determine if increased collagen was due to increased numbers of activated fibroblasts, we stained sections for α-smooth muscle actin to detect myofibroblasts. There was no difference in the percentage of α-smooth muscle actin positive cells in the infarct region (4.0 ± 0.2% vs. 4.0 ± 0.5%, n=10–13, P = 0.9) or in the uninjured septal region (3.7 ± 0.3% vs. 3.3 ± 0.2%, n=13, P = 0.4) between SR-uPA+/0 and non-transgenic mice.
Figure 4. Collagen content in SR-uPA+/0 versus nontransgenic mice late post myocardial infarction.
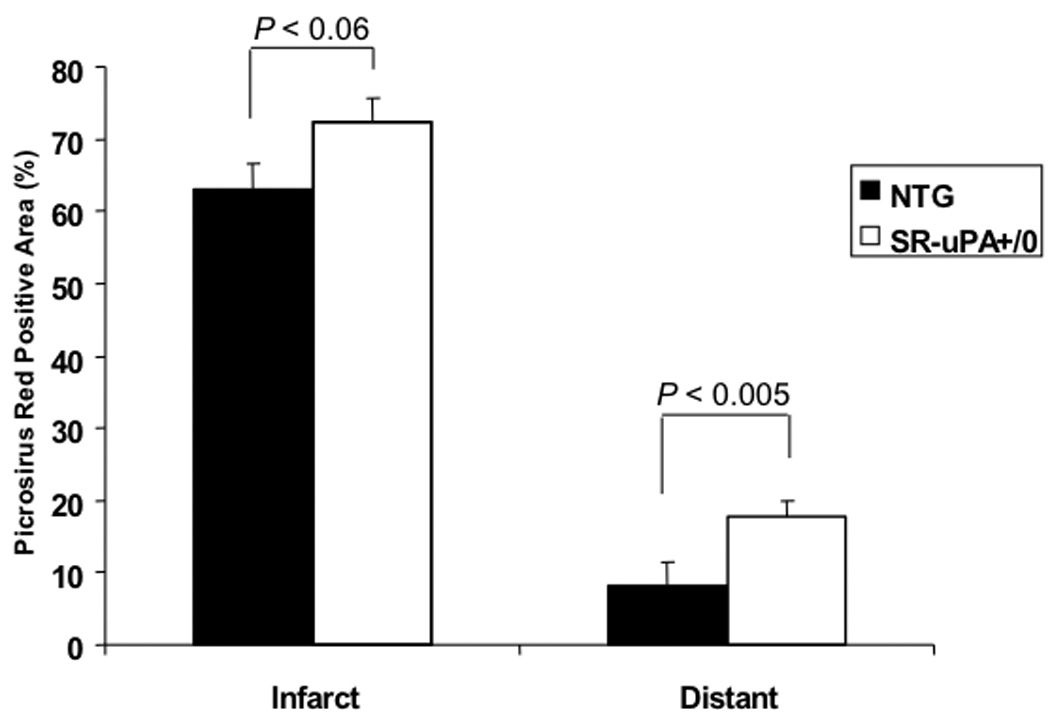
Percent fibrillar collagen content in area of infarction and uninfarcted distant myocardium measured by picrosirus red 4 weeks after myocardial infarction. There was a significant increase in collagen in the septal region away from the infarct in the SR-uPA+/0 group compared to the non-transgenic (17.8 ± 2.0% vs. 8.3±3.3%, p<0.0005) and a trend towards more collagen deposition in the infarct region (72.4 ± 3.3% vs. 63.0 ± 3.6%, p<0.06).
3.6 SR-uPA+/0 mice have improved fractional shortening late after myocardial infarction
Echocardiographic data were obtained from 13 SR-uPA+/0 and 16 nontransgenic mice at 48 hours and 15 SR-uPA+/0 and 18 nontransgenic mice 4 weeks post myocardial infarction. Echocardiography done at 48 hours post infarction showed no significant difference in fractional shortening between SR-uPA+/0 and non-transgenic mice (37.7 ± 1.2 versus 33.3 ± 2.1% fractional shortening, P = 0.1, Table 1). These data confirm comparable initial infarct sizes between the groups. At 4 weeks following myocardial infarction, echocardiographic measurements demonstrate significantly higher fractional shortening of SR-uPA+/0 mice compared to non-transgenic mice (24.6 ±1.68 vs. 19.8 ± 1.3% fractional shortening, respectively; P = 0.03, Table 1). There was a trend towards less ventricular dilatation in the transgenic group at 4 weeks in both the diastolic (0.4 ± 0.02cm vs. 0.43 ± 0.01cm, P = 0.2) and systolic measurements (0.3 ± 0.02cm vs 0.34 ± 0.01cm, P = 0.09, Table 1).
Table 1. Summary of echocardiographic data at 48 hours and 4 weeks post-infarction.
Cardiac function and remodeling post myocardial infarction. Fractional shortening, left ventricular end-diastolic (LVEDD) and left ventricular end-systolic (LVESD) dimensions measured by echocardiography at 48 hours (baseline) and 4 weeks after myocardial infarction in non-transgenic (NTG) and SR-uPA+/0 mice. Data represent mean ± SEM of n=13–16 mice at 48 hours and n=15–18 mice at 4 weeks.
| LVEDD (cm) |
LVESD (cm) |
FS(%) | HR (bpm) |
|
|---|---|---|---|---|
| 48 hours | ||||
| NTG | 0.34±0.01 | 0.23±0.01 | 33.3±2.1 | 475±52 |
| TG | 0.34±0.01 | 0.21±0.01 | 37.7±1.2 | 480±56 |
| 4 WEEKS | ||||
| NTG | 0.43±0.01 | 0.34±0.01 | 19.8±1.3 | 523±49 |
| TG | 0.40±0.02 | 0.30±0.03 | 24.6±1.7* | 474±44 |
= p<0.05
3.7 Total MMP activity is not increased after infarction in SR-uPA+/0 hearts
Because activity of MMP’s –2 and –9 is critical for macrophage accumulation in response to injury[8] and SR-uPA+/0 hearts had increased macrophages, we tested the hypothesis that MMP activity is increased in hearts of SR-uPA+/0 mice following infarction. An additional 5 mice were infarcted and the apical portion of the infarcted ventricle was flash frozen in liquid nitrogen then protein extracted for assay of total MMP activity. Uninfarcted nontransgenic hearts were used as a negative control. All infarcted hearts had increased MMP activity in comparison to uninfarcted hearts. There were no differences in MMP activity between any infarcted hearts, SR-uPA+/0 or nontransgenic (Supplemental Figure 1).
3.8 SR-uPA+/0 mice have increased collagen deposition early after infarction
It is hypothesized that increased protease activity facilitates early migration of fibroblasts to the infarction. To determine if increases in collagen in late remodeling were due to early increases in scar myofibroblast density in SR-uPA+/0 mice, we examined the characteristics of early infarct healing in SR-uPA+/0 mice. A total of 10 mice (6 SR-uPA+/0 and 4 nontransgenic) underwent LAD ligation and hearts were harvested at 5 days post infarction. One mouse in each group died prior to harvest.
Histologic evaluation showed increased collagen in infarcted myocardium of SR-uPA+/0 mice in comparison to non-transgenic mice (11.3 ± 0.05% versus 2.6 ± 0.01% picrosirius red area, Figure 5A,B) without increases in myofibroblasts (31.3 ± 8.1% versus 32.6 ± 4.0% α-smooth muscle actin positive nuclei, Figure 5C,D) or macrophages (25.2 ± 4.1% versus 26.9 ± 3.7% Mac-3 positive nuclei, Figure 5E,F). In contrast to histologic findings late after infarction, neither group had appreciable increases in macrophages, myofibroblasts or collagen in uninfarcted myocardium.
Figure 5. Collagen, myofibroblast, and macrophage content early post myocardial infarction.
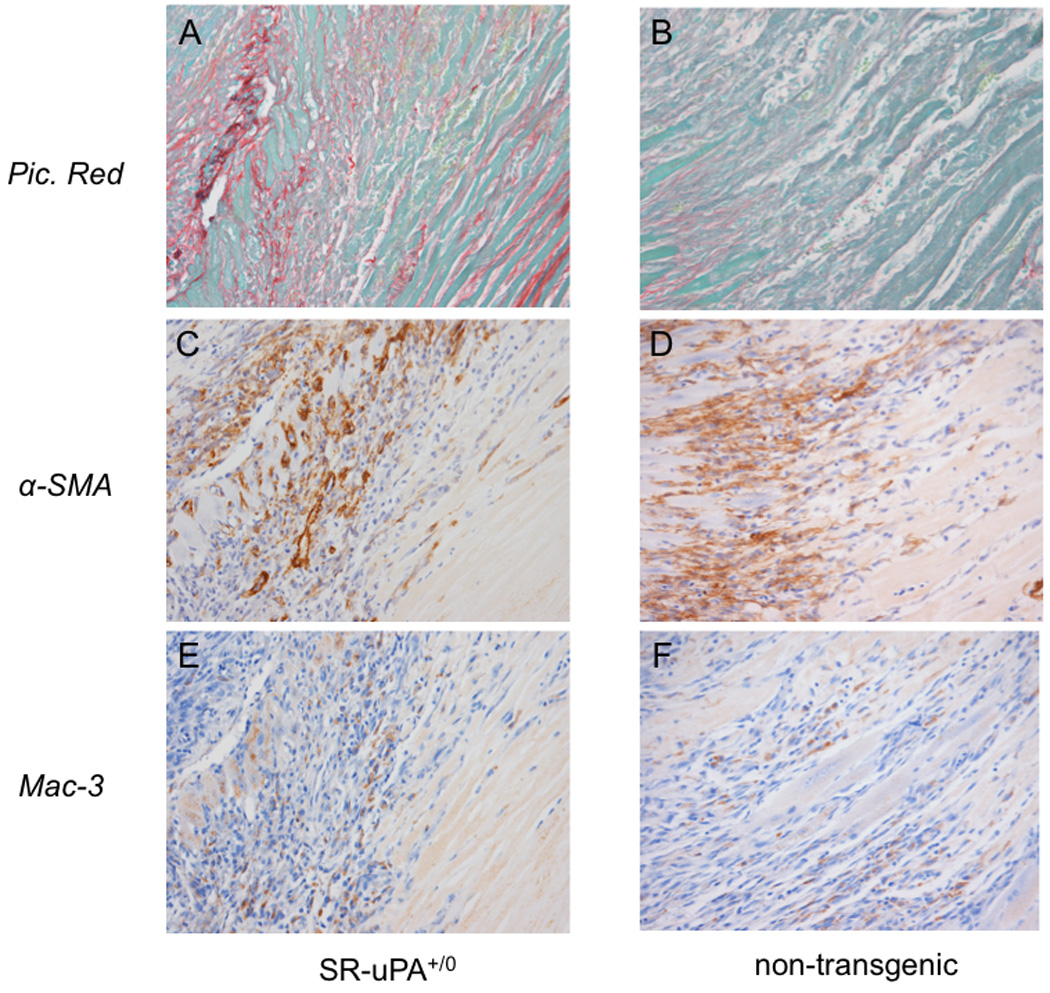
Serial sections from a SR-uPA+/0 (A, C, E) or nontransgenic (B, D, F) heart five days following myocardial infarction. Sections are stained for collagen with picrosirius red (A,B), myofibroblasts with IHC to alpha smooth muscle actin (C,D) or macrophages with IHC to Mac-3.
3.9 SR-uPA+/0 macrophages promote transcription of collagen by cardiac fibroblasts
Because hearts of SR-uPA+/0 mice had increased collagen deposition following myocardial infarction in the absence of increased numbers of myofibroblasts, we tested the hypothesis that SR-uPA+/0 macrophages can directly stimulate collagen production by isolated cardiac fibroblasts. To establish the degree to which collagen expression can be upregulated in vitro, isolated cardiac fibroblasts were treated with serum free medium (M199) alone or with TGF-β1 (20ng/ml). In addition, cardiac fibroblasts (n=4 donors) were treated with M199 conditioned by nontransgenic or SR-uPA+/0 macrophages for 18 hours. Taqman quantitative PCR was used to measure expression of the alpha 1 chain of collagen I (ColIα1), the predominant collagen in the heart. Fibroblasts treated with SR-uPA+/0 macrophage-conditioned medium had significantly more ColIα1 mRNA than did fibroblasts treated with nontransgenic macrophage conditioned medium [6.7 (4.6–8.4) fold increase, n=4 mice each genotype, P < 0.03, Figure 6B]. Moreover, the stimulation of collagen expression by SR-uPA+/o macrophage-conditioned medium was greater than the stimulation mediated by TGF-beta1, a well-established fibrogenic cytokine (1.67 ± 0.03 fold increase versus M199 alone, P < 0.03, Figure 6A).
Figure 6. Collagen production by isolated cardiac fibroblasts.
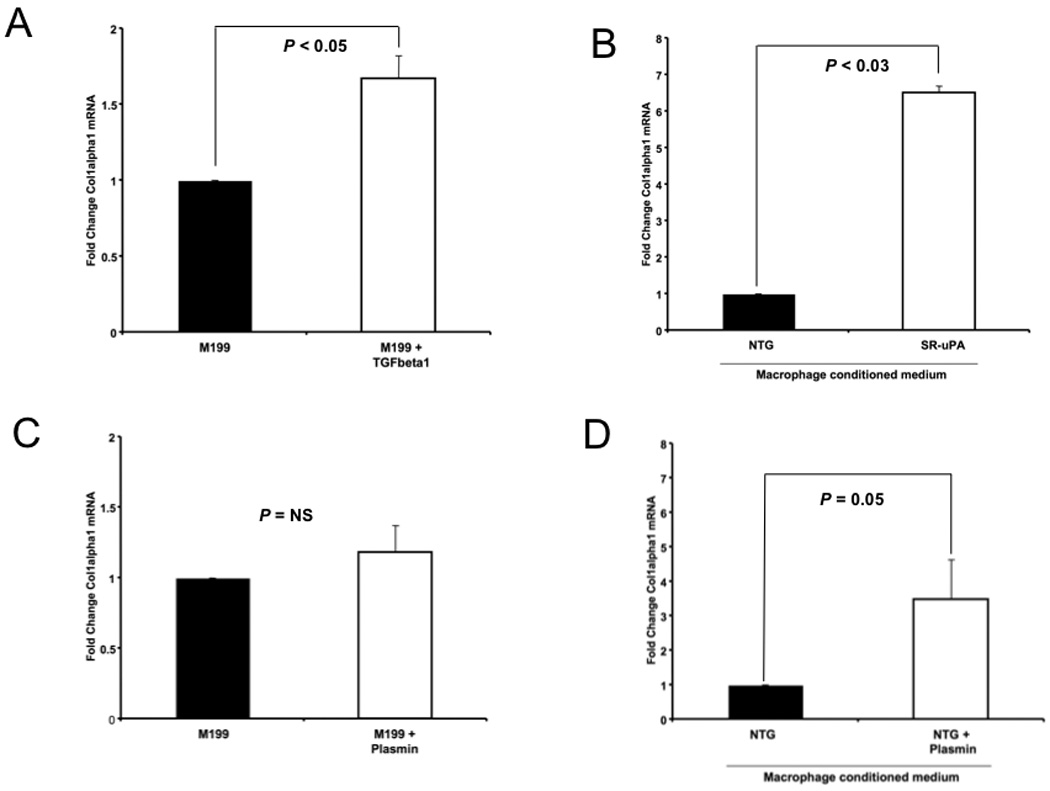
Quantitative Taqman PCR for ColIα1 mRNA from isolated cardiac fibroblasts treated with A. TGF-β1 (20ng/ml), n=4 B. conditioned media from nontransgenic (NTG) or SR-uPA+/0 macrophages, n=4 each group, C. plasmin (5nM), n=7, D. conditioned media from nontransgenic (NTG) macrophages treated with M199 alone or M199 with plasmin (5nM), n=9. Data represent mean ± S.D. of fibroblasts from individual mice treated with macrophages from different individual mice.
3.10 Plasmin stimulates fibroblast collagen transcription via a macrophage-derived factor
To test the hypothesis that increased plasmin activity alone is sufficient to promote collagen expression by cardiac fibroblasts, we first determined the plasmin activity generated by SR-uPA+/0 macrophages in the presence of plasminogen. Plasmin activity using the chromogenic substrate S-2251 was measured in conditioned medium from SR-uPA+/0 macrophages and on serial dilutions of human plasmin (Hematologic Technologies). Conditioned media from SR-uPA+/0 macrophages generated PA activity equivalent to 5.3 ± 1.9 nM of plasmin. Therefore we treated isolated cardiac fibroblasts with 5nM of plasmin in M199 overnight and measured expression of ColIα1 mRNA levels. Cardiac fibroblasts treated with plasmin had no increase in collagen expression with plasmin treatment [n = 6 fibroblast donors, median fold increase 1.1 (0.9–1.4), P = 0.4 by Mann Whitney; P = 0.8 for paired t test of ColIα1 CT normalized to 18S CT, Figure 6C]. Experiments using higher doses of plasmin to directly treat cardiac fibroblasts failed to stimulate increased transcription of collagen (data not shown).
Based on these results and our previous data showing that SR-uPA+/0 induced cardiac fibrosis is dependent on plasminogen, we hypothesized that increased plasmin activity promotes a macrophage-derived profibrotic factor [18]. To test this hypothesis we cultured wild-type murine peritoneal macrophages in M199 medium with or without plasmin (5nM) overnight, and added the resultant conditioned medium to isolated cardiac fibroblasts. Macrophage conditioned media from plasmin treated macrophages significantly increased expression of ColIα1 in cardiac fibroblasts compared to conditioned medium from untreated macrophages [n = 9 macrophage donors and 9 fibroblast donors, median fold increase 1.46 (1.1–5.8), P = 0.05 by Mann Whitney; P = 0.04 for paired t test of ColIα1 CT normalized to 18S CT, Figure 6D].
3.11 Macrophages with excess uPA do not express excess pro-inflammatory cytokines
A major characteristic of macrophages is the production of pro-inflammatory cytokines such as IL-1beta, and previous studies have implicated these cytokines with cardiac collagen accumulation in response to infarction [24, 25]. To determine if over-expression of uPA alters production of pro-inflammatory cytokines by macrophages, we measured expression of the prototypic inflammatory cytokine IL-1beta in un-stimulated and LPS-stimulated peritoneal macrophages from 5 SR-uPA+/0 and 5 littermate nontransgenic mice. There were no differences in baseline expression of IL-1beta between nontransgenic and SR-uPA+/0 macrophages (n = 5 donors per genotype, 7.2 ± 0.8 versus 7.6 ± 1.6, arbitrary units normalized to GAPDH, P = NS). In addition, macrophages from both SR-uPA+/0 and nontransgenic mice showed robust upregulation of IL-1 beta in response to LPS (2584 ± 1755 versus 1804 ± 436 fold increase in IL-1 beta expression, P = NS).
4. Discussion
We investigated the role of macrophage-derived uPA in repair following myocardial infarction. In this paper we demonstrate that macrophage-derived uPA regulates collagen deposition at several stages of cardiac repair. In SR-uPA+/0 mice, the most dramatic changes in collagen were in the healing scar early after infarction (over a 300% increase versus non-transgenic) and late after infarction in the heart distant to the site of injury (114% increase versus non-transgenic). In contrast, scar collagen late after infarction increased by 15%. These changes indicate an important role for macrophage-derived uPA in both early infarct repair and compensatory remodeling post myocardial infarction.
Macrophage accumulation is increased in SR-uPA+/0 mice concurrent with increased collagen in both injured and un-injured myocardium late after infarction. However, in early injury, macrophage derived uPA does not promote increases in macrophage density. This finding indicates that uPA may mediate macrophage mediated collagen deposition by different mechanisms in acute versus chronic injury. One potential mechanism is via macrophage specific elaboration of potent fibrogenic factors. Here we demonstrate that exposure of macrophages to excess plasmin activity, either via overexpression of uPA or direct plasmin treatment, promotes generation of a product that directly stimulates transcription of ColIα1 by isolated cardiac fibroblasts.
Plasminogen activity is increased in the heart with myocardial infarction or ischemia, and mice that are deficient in uPA or plasminogen have decreased adsorption of injured tissue [5–8]. Our data indicate that macrophages–a cell abundant in injured myocardium–can be activated by plasmin to induce collagen expression in cardiac fibroblasts. In combination, these data support a model in which plasmin generation in response to accumulation of uPA secreting macrophages is critical to both matrix degradation and collagen accumulation (Figure 7). This mechanism may–in part–underlie the heart’s ability to rapidly respond to ischemic injury without rupture.
Figure 7.
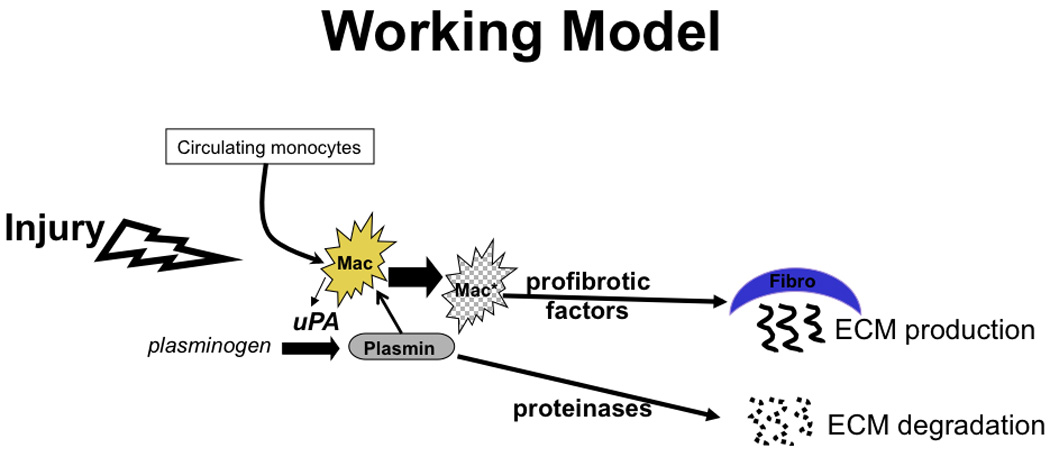
Model of the role of plasmin in macrophage mediated repair post-myocardial infarction.
Functionally, SR-uPA+/0 mice had mild improvements in fractional shortening late after myocardial infarction. Potential benefits of thicker, more compact scars in SR-uPA+/0 mice were likely limited by the increased accumulation of collagen in uninjured myocardium. In addition, increased macrophages in these areas may contribute negative inotropic stimuli, such as nitric oxide. We hypothesize that limiting up-regulation of macrophage derived uPA to the immediate post infarction period will result in more dramatic improvements in ventricular remodeling and function. Further experiments with a temporally regulated uPA transgene cassette could address this hypothesis.
Inflammation is crucial to the wound repair process following myocardial infarction, and macrophages, in particular, are essential to infarct repair. Van Amerongen et al pharmacologically depleted macrophages using clodronate--an agent that induces macrophage apoptosis--and demonstrated significantly reduced collagen deposition in cryoinjured mouse myocardium with macrophage depletion [26]. Our data support that macrophage derived products can regulate collagen deposition early after injury. Macrophages are also associated with collagen deposition in chronic cardiac repair and inhibition of macrophage accumulation is associated with decreased collagen deposition [27]. Here we show that uPA mediates both increased macrophage accumulation and collagen in areas of chronic stress. These data lead us to hypothesize that in chronic injury uPA/plasmin activates a factor that stimulates both macrophage and collagen accumulation.
The mechanisms by which macrophages promote collagen accumulation in the heart may depend on the stage of injury at which they are active. Nahrendorf et al demonstrated that Ly-6hi monocytes–the circulating precursor to macrophages–play an important role in the proteolytic function after infarction peaking during the acute phase of repair and tapering down by day 5. The Ly-6lo subset appeared later and was important for angiogenesis, myofibroblast accumulation, and collagen deposition [28]. Although, we did not differentiate the monocyte subsets in our current study, we did find differential increases in collagen deposition between time-points and locations in the heart. These data suggest that monocyte subsets may be susceptible to different macrophage activation pathways by local factors (cytokines, growth factors, wall stress) once recruited to the heart. In addition, our data support the hypothesis that increased plasmin activity is an important regulatory of these activation states. Further studies will be necessary to test this hypothesis.
The precise mechanism(s) by which macrophage-derived uPA promotes collagen deposition in the heart are as yet unknown. The lack of increase in myofibroblast numbers imply that increased cardiac collagen is due to increased collagen production by myofibroblasts in hearts of SR-uPA+/0 mice. Whether plasmin is activating a pre-formed pro-fibrotic macrophage factor via cleavage or inducing de novo production of a specific factor is unknown. Others have shown a role for the macrophage derived profibrotic factor transforming growth factor beta-1 (TGF-beta1) in collagen deposition early after infarction [29]. However, our previous data indicate that TGF-beta1 is not a proximal mediator of uPA-induced cardiac fibrosis [18]. Here we show that that conditioned media from uPA expressing macrophages can directly increase collagen production by cardiac fibroblasts. These data support our hypothesis that macrophage derived uPA–via paracrine effects on macrophages–promotes collagen production in the heart. In this study we demonstrate an in vivo dose-response between macrophage uPA expression and the rate of development of cardiac fibrosis. High levels of macrophage-derived uPA induce fibrosis at 15 weeks of age, however, lower expressing M29 SR-uPA+/0 mice do not have spontaneous cardiac fibrosis at the same time-point [18]. These data suggest a dose response between plasmin activity in the heart and production of macrophage derived profibrotic factors. In vitro studies combined with genomic and proteomic strategies will be necessary to determine the identity of this profibrotic factor.
Consistent with our previous data, we demonstrate that plasmin activity is a critical mediator of macrophage accumulation in the heart. It remains uncertain, however, by what mechanism plasmin promotes macrophage accumulation. Here we show that MMP activity is increased in the infarct in comparison to uninfarcted myocardium. However, excess macrophage derived uPA did not further increase MMP activity. These data are consistent with our previous report that high levels of macrophage-derived uPA did not increase activity of MMP-2 or -9 in the heart, and others who have questioned the role of plasmin activation of MMP’s in vivo [18, 30]. It is possible that small changes in MMP were not detectable in the number of mice studied. An alternative hypothesis is that increased plasmin generates production of a factor that homes macrophages to the myocardium. Determining if plasmin treated macrophages preferentially traffic to the heart would be critical to test this hypothesis.
Finally, our experiments point to the utility of expressing proteins in macrophages as a means to target infarct repair. Macrophages naturally home to sites of injury and survive well in the harsh infarct environment. Macrophages are well suited for protein secretion and persist throughout the process of infarct repair. Once repair is complete, the majority of macrophages die off or exit the tissue, naturally limiting the duration of transgene delivery. Therefore, delivery of therapeutic products to the infarct via genetically modified macrophages seems a promising technique to pursue.
In summary, macrophages play a key role in infarct repair and cardiac fibrosis and macrophages are regulated by the presence of uPA. Early after infarction, macrophages in SR-uPA+/0 mice are not elevated but are associated with increased scar collagen. At 4 weeks after infarction, high levels of macrophages in SR-uPA+/0 mice were associated with thicker scar and attenuated dilation of the ventricle but also with increased collagen in uninjured myocardium. This finding suggests that initially, stimulation of robust scar formation by uPA may be beneficial to prevent ventricular remodeling. However, persistent plasminogen activation may underlie the deleterious fibrotic changes seen in uninjured myocardium late after myocardial infarction. Plasminogen activation appears to be a critical link between the accumulation of macrophages and collagen production in the heart in response to injury. Understanding these mechanisms may allow the development of targeted macrophage based cell therapies to improve repair both early and late after cardiac injury.
Supplementary Material
Acknowledgements
We acknowledge the assistance of Drs. William Parks and Michael Chin with manuscript review, Teja Dyamenahalli with mouse genotyping, and Abigail Plawman with mouse husbandry. We also acknowledge the Seattle Mouse Metabolic Phenotyping Consortium for assistance in generating the 5-day myocardial infarcts.
Funding Sources: This work was supported in part by National Institute of Health grants K08 HL70941 to Dr. Stempien-Otero and P01 HL003174, R01 HL084642, R24 HL64387, U24 DK076126 to Dr. Murry. Drs. Stempien-Otero and Minami were also supported in part by the Locke Charitable Trust.
Footnotes
Disclosures: None
REFERENCES
- 1.Weisman HF, Bush DE, Mannisi JA, Weisfeldt ML, Healy B. Cellular mechanisms of myocardial infarct expansion. Circulation. 1988 Jul;78(1):186–201. doi: 10.1161/01.cir.78.1.186. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker P, Boughner DR, Kloner RA. Role of collagen in acute myocardial infarct expansion. Circulation. 1991 Nov;84(5):2123–2134. doi: 10.1161/01.cir.84.5.2123. [DOI] [PubMed] [Google Scholar]
- 3.Olivetti G, Capasso JM, Sonnenblick EH, Anversa P. Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats. Circ Res. 1990 Jul;67(1):23–34. doi: 10.1161/01.res.67.1.23. [DOI] [PubMed] [Google Scholar]
- 4.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005 Apr 29;96(8):881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 5.Tyagi S, Kumar S, Haas S, Reddy H, Voelker D, Hayden M, et al. Post-transcriptional regulation of extracellular matrix metalloproteinase in human heart end-stage failure secondary to ischemic cardiomyopathy. J Mol Cell Cardiol. 1996;28:1415–1428. doi: 10.1006/jmcc.1996.0132. [DOI] [PubMed] [Google Scholar]
- 6.Gaertner R, Jacob MP, Prunier F, Angles-Cano E, Mercadier JJ, Michel JB. The plasminogen-MMP system is more activated in the scar than in viable myocardium 3 months post-MI in the rat. J Mol Cell Cardiol. 2005 Jan;38(1):193–204. doi: 10.1016/j.yjmcc.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Knoepfler PS, Bloor CM, Carroll SM. Urokinase plasminogen activator activity is increased in the myocardium during coronary artery occlusion. J Mol Cell Cardiol. 1995 Jun;27(6):1317–1324. doi: 10.1016/s0022-2828(05)82394-6. [DOI] [PubMed] [Google Scholar]
- 8.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but imparis therapeutic angiogenesis and causes cardiac failure. Nature Medicine. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 9.Creemers E, Cleutjens J, Smits J, Heymans S, Moons L, Collen D, et al. Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am J Pathol. 2000 Jun;156(6):1865–1873. doi: 10.1016/S0002-9440(10)65060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999 May 7;274(19):13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 11.Monea S, Lehti K, Keski-Oja J, Mignatti P. Plasmin activates pro-matrix metalloproteinase-2 with a membrane-type 1 matrix metalloproteinase-dependent mechanism. J Cell Physiol. 2002 Aug;192(2):160–170. doi: 10.1002/jcp.10126. [DOI] [PubMed] [Google Scholar]
- 12.Vanhoutte D, Schellings M, Pinto Y, Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res. 2006 Feb 15;69(3):604–613. doi: 10.1016/j.cardiores.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004 Aug;4(8):617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 14.Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, et al. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension. 2006 Apr;47(4):711–717. doi: 10.1161/01.HYP.0000208840.30778.00. [DOI] [PubMed] [Google Scholar]
- 15.Van Linthout S, Seeland U, Riad A, Eckhardt O, Hohl M, Dhayat N, et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol. 2008 Jul;103(4):319–327. doi: 10.1007/s00395-008-0715-2. [DOI] [PubMed] [Google Scholar]
- 16.Fert-Bober J, Leon H, Sawicka J, Basran RS, Devon RM, Schulz R, et al. Inhibiting matrix metalloproteinase-2 reduces protein release into coronary effluent from isolated rat hearts during ischemia-reperfusion. Basic Res Cardiol. 2008 Sep;103(5):431–443. doi: 10.1007/s00395-008-0727-y. [DOI] [PubMed] [Google Scholar]
- 17.Moriwaki H, Stempien-Otero A, Kremen M, Cozen AE, Dichek DA. Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice. Circ Res. 2004 Sep 17;95(6):637–644. doi: 10.1161/01.RES.0000141427.61023.f4. [DOI] [PubMed] [Google Scholar]
- 18.Stempien-Otero A, Plawman A, Meznarich J, Dyamenahalli T, Otsuka G, Dichek DA. Mechanisms of cardiac fibrosis induced by urokinase plasminogen activator. J Biol Chem. 2006 Jun 2;281(22):15345–15351. doi: 10.1074/jbc.M512818200. [DOI] [PubMed] [Google Scholar]
- 19.Cozen AE, Moriwaki H, Kremen M, DeYoung MB, Dichek HL, Slezicki KI, et al. Macrophage-targeted overexpression of urokinase causes accelerated atherosclerosis, coronary artery occlusions, and premature death. Circulation. 2004 May 4;109(17):2129–2135. doi: 10.1161/01.CIR.0000127369.24127.03. [DOI] [PubMed] [Google Scholar]
- 20.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006 Nov 28;103(48):18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossow CF, Minami E, Chase EG, Murry CE, Santana LF. NFATc3-induced reductions in voltage-gated K+ currents after myocardial infarction. Circ Res. 2004 May 28;94(10):1340–1350. doi: 10.1161/01.RES.0000128406.08418.34. [DOI] [PubMed] [Google Scholar]
- 22.Reinecke H, Murry CE. Cell grafting for cardiac repair. Methods Mol Biol. 2003;219:97–112. doi: 10.1385/1-59259-350-x:97. [DOI] [PubMed] [Google Scholar]
- 23.Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006 Jul;12(7):787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murtuza B, Suzuki K, Bou-Gharios G, Beauchamp JR, Smolenski RT, Partridge TA, et al. Transplantation of skeletal myoblasts secreting an IL-1 inhibitor modulates adverse remodeling in infarcted murine myocardium. Proc Natl Acad Sci U S A. 2004 Mar 23;101(12):4216–4221. doi: 10.1073/pnas.0306205101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008 Jul;173(1):57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007 Mar;170(3):818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007 Feb 6;115(5):584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 28.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007 Nov 26;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frantz S, Hu K, Adamek A, Wolf J, Sallam A, Maier SK, et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. 2008 Sep;103(5):485–492. doi: 10.1007/s00395-008-0739-7. [DOI] [PubMed] [Google Scholar]
- 30.Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007 Oct;26(8):587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.