

Abstract
The annotation of gene/gene products with information on associated diseases is useful as an aid to clinical diagnosis and drug discovery. Several supervised and unsupervised methods exist that automate the association of genes with diseases, but relatively little work has been done to map protein sequence data to disease terminologies. This paper augments an existing open-disease terminology, the Disease Ontology (DO), and uses it for automated annotation of Swissprot records. In addition to the inherent benefits of mapping data to a rich ontology, we demonstrate a gain of 36.1% in gene-disease associations compared to that in DO. Further, we measure disease similarity by exploiting the co-occurrence of annotation among proteins and the hierarchical structure of DO. This makes it possible to find related diseases or signs, with the potential to find previously unknown relationships.
INTRODUCTION
Associating genes with diseases and finding commonality between seemingly dissimilar disorders is an active area of research as it can lead to a better understanding of disease and reduced time and expenditure in developing effective drugs and treatment. Several methods have been developed to associate genes/gene products with diseases using microarray data, orthology, annotation of genes and protein interactions, and medical literature [1][2].
Swissprot (SP) [3] has 2554 proteins (March 1st, 2009) that have been manually annotated with disease names. These entries are a mixture of phrases explicitly referring to disease names and additional sentences that imply associated diseases. Mapping this accurate information to standard terminologies or ontologies would aid the automation of research on gene-disease relationships. A recent study published by Mottaz et al [4] used a template-based approach to link SP proteins to Medical Subject Headings (MeSH) [5] by mining the disease information in protein records. OMIM data has been mapped to MeSH to infer similarity between genes based on phenotypes [6]. Metadata from tissue microarray data has been mapped to the National Cancer Institute Thesaurus (NCI-T) and the Systematized Nomenclature of Medicine-Clinical Terms (SNOMED-CT) for classification of tumor samples [7]. Though MeSH has broad coverage on a variety of subjects, it has several missing terms and lacks detail in the disease section. For example, ‘Hypothalamic Neoplasms’ is not present in MeSH and the term Asthma has only ‘Asthma, Exercise-Induced’ and ‘Status Asthmaticus’ as child terms. As a result, MeSH-based approaches may have limited recall and any similarity metric that exploits its hierarchical nature can lack specificity. While SNOMED-CT is rich in clinical terms, its availability is restricted. The International Classification for Diseases (ICD) classifies epidemiological diseases but lacks clinical details.
It is important to have vocabulary that not only gives disease sub-classification, but also signs related to them [8]. For example, consider disease text line 5 for protein P21333 in SP: “Defects in FLNA are the cause of frontometaphyseal dysplasia (FMD) [MIM:305620]. FMD is a congenital bone disease characterized by supraorbital hyperostosis, deafness and digital anomalies.” Although ‘frontometaphyseal dysplasia’ is the disease, the knowledge that the defects are congenital musculoskeletal anomalies, deafness and hyperostosis can be potentially useful in knowledge bases and expert systems.
The Disease Ontology [9] developed at Northwestern University is part of the Open Biomedical Ontologies and has 12082 terms (ver3.0, August 2009) compared to 4323 in MeSH 2009 (Disease section). Most of the terms have been obtained from ICD, NCI, MeSH, CSP (Complications Screening Program) and MTH (UMLS Metathesaurus).
Major efforts in text mining have focused on the recognition of genes/proteins and protein interactions in publications [10]; relatively few tools map free text to disease terminologies. NLM’s MetaMap [11] is a widely used tool to map free text to controlled vocabularies in UMLS.
It is useful to discern similarity between diseases as this can lead to a better understanding of underlying common pathophysiology. This can impact diagnosis, prognosis and treatment in the clinic as drugs/procedures used to treat a particular disease may be effective for similar diseases. A few methods have been developed to compute disease similarity; one uses phenotypes to find disease relationships [12] while the other finds consistently co-occurring diseases in Medicare patient data [13].
In this paper, we use MetaMap to map the disease annotation of Swissprot protein entries to DO terms. We then estimate the similarity between diseases using co-annotation and the DO semantic hierarchy.
METHODS
Disease Terms
Based on the semantic types defined in UMLS, a term was designated as Disease-Related if it was any of the following 7 semantic types: ‘Disease or Syndrome,’ ‘Neoplastic Process,’ ‘Mental or Behavioral Dysfunction,’ ‘Acquired Abnormality,’ ‘Pathologic Function,’ ‘Anatomical Abnormality’ and ‘Congenital Abnormality.’ ‘Sign or Symptom’ though sometimes synonymous with disease names was not considered as it includes several non-specific terms like pain and fever which can confound mapping.
Augmenting DO vocabulary
DO consists of 12082 terms corresponding to 14,392 Concept User Identifiers (CUIs). Although DO has a large vocabulary, several terms lack synonyms and some are rather abstract (e.g., Reproduction Disease). In order to augment DO, we exploited the semantics of the internal hierarchy of multiple source vocabularies in UMLS - ICD10, SNOMed-CT, MeSH, MTH and NCI-T. For a given DO term, its CUI was used to extract all Atom Unique Identifiers (AUIs). These were designated as Preferred Terms. The AUIs obtained were recursively used as query terms to find additional AUIs having an IS-A relationship with the query terms. The AUIs thus extracted were labeled as Secondary terms of the DO term used to initiate the query. To avoid redundancy, the iteration was terminated whenever the extracted Secondary term matched the Primary term of an existing DOID. For example, A3124434, A2891225 and A3413463 (Fig. 1) are AUIs of DOID:10081 (Syphilitic encephalitis) extracted from MRCONSO. Using these terms, Secondary Terms are iteratively extracted from MRHIER, except for ‘A2982782’ which is a Preferred Term of another DO Identifier, ‘Syphilitic Parkinsonism.’ Thus, several Secondary terms were subsumptively mapped to each Primary DO identifier to create an augmented version of DO. To strike a balance between a rich mapping to DO and excessive abstraction, the iteration for each Preferred DO identifier was limited to a pragmatic maximum of 4 levels (based on a sample of mappings).
Figure 1.

Adding Terms to DO using UMLS
Mapping disease lines to DO
The SP database disease-annotated subset consists of 2554 proteins with 3936 disease descriptor lines. The OMIM titles and alternative titles were appended to the disease line wherever an OMIM ID was mentioned. MetaMap 2009AA was used to map the SP text entries to DO. The SP text was used to search the entire UMLS and wherever the CUI was represented in augmented DO, the corresponding DO identifier was used to annotate the SP disease line.
Entrez Gene identifiers corresponding to SP identifiers were used to add disease information based on DO-GeneRIF mappings. Thus, the final annotation was based on the cumulative annotation from SP, OMIM and GeneRIF.
Metric to compute disease similarity
Given a disease term, the subset of genes annotated with it was extracted and the associated DO terms were used to find similar diseases. This was done by finding over-represented extracted DO terms using the hypergeometric distribution and the Benjamini-Hochberg correction for multiple tests. The hypergeometric distribution tends to give less importance to abstract upper level terms and is based on an assumption of independence. The results can therefore be skewed for terms along the hierarchy and for rare terms which yield spuriously high scores. To account for random or rare occurrences, a similarity metric called BV [14] that is based on both co-annotation and hierarchy (Equations 1 and 2) was used. A p-value was calculated for similarity scores using 100,000 randomly generated pairs of diseases.
Given DO terms A & B, n(A) = number of genes annotated with A, n(A ∩ B) = number of genes annotated with both A and B, and N = total genes, similarity is given by
| (1) |
The value obtained is normalized by the average of the maximum scores for A and B, and multiplied by the average suprisal of the terms as follows.
| (2) |
Max_sim(A,i) is the maximum similarity score for DO terms A and ‘i.’ Sup(A) is the surprisal of A.
RESULTS
Comparison between DO and MeSH
The MeSH ‘Disease’ section contains 4323 terms related to various UMLS semantic types of which 3944 are ‘disease related’ (see above). These correspond to 6954 Concept User Identifiers (CUIs). In comparison, DO consists of 12082 terms. Further, the augmentation of DO using UMLS resulted in increasing the coverage to 33,085 CUIs, an increase of 2.5 fold. There were only 615 MeSH terms which did not have a corresponding CUI in the augmented DO; 84.4% MeSH disease-related terms were represented in DO.
To evaluate the efficiency of mapping, the Mottaz benchmark set of 200 records was used. These records were originally randomly chosen and manually annotated by experts using MeSH. A record consists of Protein ID, Line Number and Disease Text. The same records were parsed by MetaMap and annotated with MeSH terms. Recall and precision were calculated on a per record basis. If any term mapped by MetaMap matched the curated terms, then the record was scored as a match. It was noticed that some terms from the disease text were missed by experts. To prevent underestimation of precision, the mapped terms were manually checked by DD, the 2nd author of the paper.
The template-based extraction method in Mottaz et al required only 1 term to be extracted per record and had a precision of 86% and recall of 64% resulting in an F-score of 0.73. MetaMap (SP-MeSH mapping) performs better by achieving an average cardinality of 3.05 per correctly called record with a better recall and overall performance (Maximum Recall=89%; Precision=72%; F-score=0.81). Mappings of the 200 records to DO achieved an average cardinality of 3.06 with better F-scores (Maximum Recall=90%; Precision=76%; F-score=0.83) compared to SP-MeSH mappings (Fig 2).
Figure 2.

Evaluation of automated annotation of 200 records with MeSH and DO terms
A cutoff score of 700 was used for automated annotation of all SP entries having disease lines as it had the highest F-score of 0.827. Out of a total of 2554 proteins annotated with disease text, MetaMap was able to assign annotation to 2303 (90.4%).
The relative contributions of GeneRIF, OMIM and Swissprot annotation to disease–gene association were assessed. The existing GeneRIF annotation in DO (4376 genes annotated with 2638 DO terms) corresponds to 20183 gene-disease associations. SP records alone, devoid of any OMIM references, yielded 1191 gene-disease associations between 485 genes and 325 diseases. SP records that included information derived from OMIM contributed to the annotation of 923 Entrez genes with 564 DO terms (an additional 7293 gene-disease associations) (Fig. 3). Thus, the use of information embedded in SP records resulted in an increase of 36.1% over pre-existing annotation derived from GeneRIF and a 22.5% increase in DO terms used for annotation. Exploiting both SwissProt and GeneRIF annotation resulted in associating 3202 diseases with 5290 Entrez genes.
Figure 3.
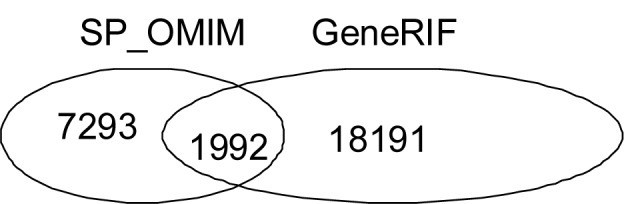
Disease-Gene associations from SP-OMIM and GeneRIF
Table I lists pairs of diseases which do not occur in the same hierarchy as the query disease. The p-value based on the hypergeometric distribution (corrected for multiple testing (BH)) was essentially zero for the associations shown. ‘Sbv’ is the p-value based on BV scores that range from 0 to 8.6 with a mean of 0.1 and standard deviation of 0.5.
Table I.
Diseases showing an association with a query disease in protein annotation space
| Query Disease | Associated Diseases | Sbv |
|---|---|---|
| Hypertension (260 genes) DOID:10763 | Diabetes Mellitus | 0.03 |
| Obesity | 0.03 | |
| Heart Diseases | 0.03 | |
| Coronary heart disease (144 genes) DOID:3393 | Hyperlipidemia | 0.03 |
| Diabetes Mellitus | 0.03 | |
| Obesity | 0.03 | |
| Obesity (151 genes) DOID:9970 | Diabetes Mellitus | 0.03 |
| Lipid Disorder | 0.03 | |
| Polycystic Ovary Syndrome | 0.05 | |
| Diabetes Mellitus (407 genes) DOID:9351 | Autoimmune Disease | 0.02 |
| Obesity | 0.05 | |
| Myocardial Ischemia | 0.03 | |
| Hearing problem (189 genes) DOID:12226 | Vision Disorders | 0.03 |
| Cranial nerve diseases | 0.02 | |
| Pneumonia (81 genes) DOID:552 | Influenza | 0.02 |
| Lung Diseases, Interstitial | 0.01 | |
| Pulmonary Fibrosis | 0.01 | |
| Dermatitis (151 genes) DOID:2723 | Skin Diseases, Eczematous | 0.01 |
| Skin Diseases, Genetic | 0.01 | |
| Asthma | 0.02 | |
| Parasitic Disorders | 0.05 | |
| Multiple Sclerosis (96 genes) DOID:2377 | Retroviridae Infections | 0.02 |
| Gastrointestinal Infection | 0.02 |
DISCUSSION
Automating the annotation of genes with standardized disease information
Controlled terminologies have been a prominent part of medical informatics for several decades. It is only recently that interest has grown in linking clinical data to biological functions of genes based on ontologies. Significant gains in understanding gene-disease relationships can be made by the knowledge integration facilitated by linking multiple ontologies [15]. A bottleneck that currently exists is the fact that the majority of useful annotation is in the form of free-text. The first part of the paper represents an effort to automate the conversion of free-text disease-specific information in SP to terms based on the Disease Ontology by using MetaMap together with upper ontological mappings in UMLS. While the level of accuracy falls short of manual annotation, it is important to note that this is a fully automated approach. It resulted in an increase of 36.1% in gene-disease annotation compared to that maintained by the DO community. Some roles of genes might be missed by an expert during the creation of a reference standard with controlled vocabulary terms, thus introducing false negatives. In turn, when this is used as the basis for evaluation of an automated method, it can lead to a spuriously lower precision if some of the missing terms are picked up. To avoid underestimation of precision, the automated annotations based on MeSH and DO were manually checked by DD in addition to comparison with the existing annotations.
MeSH is deficient in detailed nomenclature of diseases. For example, consider the text entry “Defects in TGM1 are a cause of non-bullous congenital ichthyosiform erythroderma. Clinical features are milder than in lamellar ichthyoses. Patients suffer from palmoplantar keratoderma, often with painful fissures, digital contractures, and loss of pulp volume.” The closest MeSH term is ‘lamellar ichthyoses.’ In contrast, DO includes both ‘palmoplantar keratoderma’ and ‘lamellar ichthyoses.’ Although DO is particularly rich in highly specific terms, it is a work in progress as i) synonyms are minimal, ii) there are several obsolete terms and iii) there are a large number of terms in a ‘temp holding’ category. There is no placeholder for general terms like ‘Ataxia’ or ‘Short Stature;’ only specific versions like ‘Cerebellar Ataxia’ and ‘Pituitary Dwarfism’ exist. This limits the use of DO in both molecular or clinical record annotation when an association is detected but an exact cause is yet to be ascertained.
While MetaMap is an effective tool to map text onto biomedical vocabularies, polysemy, anomalous lexical variations, negations and sub-partitioning of disease term phrases resulted in false positives. For example, text ‘Deficiency’ was mapped to Malnutrition, ‘Exhibit’ to Exhibitionism, and ‘Pyruvate Dehydrogenase Complex Deficiency Disease’ to Malnutrition and Protein Deficiency. Although MetaMap 2009AA addresses negation, we found that it fails in some cases, especially when the negation occurs in the latter half of a phrase. A DO thesaurus was constructed using Datafile Builder tool of MMTx and SP text mapped to it. Although this achieved slightly better recall than MetaMap, its precision was lower (data not shown). These limitations can be potentially overcome by incorporating advanced text mining techniques [10].
Assessing disease similarity in protein space
Table I shows a representative sample of the gene annotation based inter-disease associations that are statistically significant. Trivial associations like rheumatism and soft tissue diseases that occur in the same hierarchy are not reported. Nor are obvious matches for synonyms and variants shown here. Table I shows several well known associations between diseases. For example, Obesity, Diabetes Mellitus, Hypertension and Heart Disease are known to be linked, which is borne out by the self-consistent nature of the first few rows in the table. Interestingly, the association shown for multiple sclerosis resonates with theories on viral infections (not just Epstein-Barr) triggering the auto-immune response. Common KEGG pathways based on gene-enrichment analysis are cytokine-cytokine receptor interaction, T cell receptor signaling pathway, Toll-like receptor signaling pathway and cell adhesion molecules (CAMs). While most of these associations are common knowledge, these are based on complete automation and based on the accurate extraction of standard vocabulary from free-text. Though only a relatively small fraction of genes is currently annotated with disease-specific information, subsequent growth is likely to lead to the identification of new associations.
Many metrics take the information content in the nearest common ancestor to estimate similarity between terms; this approach can underestimate the similarity between related yet distant terms if the hierarchy is flawed. This is a problem in DO, which has close to 3654 terms under ‘temp holding.’ It is therefore important to use co-annotation relative frequency along with ontological hierarchy for a more accurate estimate of similarity.
An important area to consider is the overlap between the concepts of disease, signs, phenotype and perhaps even symptoms. At times a single sign or symptom is considered pathognomic and hence synonymous with a specific disease, while at the other extreme there are syndromes with a complex and variable presentation. It behooves the knowledge representation community to extend annotation to not just the name of a disease but to a more detailed description. This will allow make maximum use of the collective knowledge in both clinic and lab.
CONCLUSION
We have demonstrated an automated approach to map high quality annotation to the Disease Ontology and report an increase of 36.1% in gene-disease relationships. Interesting relationships between diseases can be found with better accuracy with metrics that exploit ontology as well as co-annotation. For seemingly dissimilar diseases found to be similar, it would be interesting to see if similarity exists in gene functionality, eventually resulting in knowledge that impacts diagnosis, prognosis and treatment.
Acknowledgments
We would like to thank Anais Mottaz for clarifications related to her publication, Jim Mork for his feedback on MetaMap and Arcady Mushegian for comments on the manuscript.
REFERENCES
- 1.Hristovski D, et al. “Using literature-based discovery to identify disease candidate genes,”. Int J Med Inform. 2005 Mar;74:289–98. doi: 10.1016/j.ijmedinf.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 2.Oti M, Brunner HG. “The modular nature of genetic diseases,”. Clin Genet. 2007 Jan;71:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- 3.“The Universal Protein Resource (UniProt) 2009,”. Nucleic Acids Res. 2009 Jan;37:D169–74. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mottaz A, et al. “Mapping proteins to disease terminologies: from UniProt to MeSH,”. BMC Bioinformatics. 2008;9(Suppl 5):S3. doi: 10.1186/1471-2105-9-S5-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson SJ, et al. “The MeSH translation maintenance system: structure, interface design, and implementation,”. Stud Health Technol Inform. 2004;107:67–9. [PubMed] [Google Scholar]
- 6.van Driel MA, et al. “A text-mining analysis of the human phenome,”. Eur J Hum Genet. 2006 May;14:535–42. doi: 10.1038/sj.ejhg.5201585. [DOI] [PubMed] [Google Scholar]
- 7.Shah NH, et al. “Annotation and query of tissue microarray data using the NCI Thesaurus,”. BMC Bioinformatics. 2007;8:296. doi: 10.1186/1471-2105-8-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almeida F, et al. “Controlled health thesaurus for the CDC web redesign project,”. AMIA Annu Symp Proc. 2003:777. [PMC free article] [PubMed] [Google Scholar]
- 9.Osborne JD, et al. “Annotating the human genome with Disease Ontology,”. BMC Genomics. 2009;10(Suppl 1):S6. doi: 10.1186/1471-2164-10-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jensen LJ, et al. “Literature mining for the biologist: from information retrieval to biological discovery,”. Nat Rev Genet. 2006 Feb;7:119–29. doi: 10.1038/nrg1768. [DOI] [PubMed] [Google Scholar]
- 11.Aronson AR. “Effective mapping of biomedical text to the UMLS Metathesaurus: the MetaMap program,”. Proc AMIA Symp. 2001:17–21. [PMC free article] [PubMed] [Google Scholar]
- 12.Rzhetsky A, et al. “Probing genetic overlap among complex human phenotypes,”. Proc Natl Acad Sci U S A. 2007 Jul 10;104:11694–9. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hidalgo CA, et al. “A dynamic network approach for the study of human phenotypes,”. PLoS Comput Biol. 2009 Apr;5:e1000353. doi: 10.1371/journal.pcbi.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathur S, Dinakarpandian D. “A New Metric to Measure Gene Product Similarity,”. IEEE International Conference on Bioinformatics and Biomedicine; 2007. pp. 333–338. [Google Scholar]
- 15.Burgun A, Bodenreider O. “Accessing and integrating data and knowledge for biomedical research,”. Yearb Med Inform. 2008:91–101. [PMC free article] [PubMed] [Google Scholar]