

Abstract
In this paper we describe CB513 a non-redundant dataset, suitable for development of algorithms for prediction of secondary protein structure. A program was made in Borland Delphi for transforming data from our dataset to make it suitable for learning of neural network for prediction of secondary protein structure implemented in MATLAB Neural-Network Toolbox. Learning (training and testing) of neural network is researched with different sizes of windows, different number of neurons in the hidden layer and different number of training epochs, while using dataset CB513.
1. PROTEIN STRUCTURE PREDICTION PROBLEM
1. 1. Introduction
Current technology for three-dimensional protein structure prediction relies on using unknown sequences of proteins and overlaying them with homologous proteins with known structure. Such practice is faulty as it does not account for proteins which do not have a homologous protein in their database or current technology is not yet able to identify existing homologous proteins. Most of today’s algorithms which are employed in secondary protein structure prediction rely heavily on known three-dimensional structures. Using those algorithms certain parameters are then imposed upon unknown sequences. Such methods rely on available data for their predictions. Earlier algorithms for prediction of secondary protein structures reported high success, but these studies were based on small quantities of data which was mainly derived during training sessions. For example, Lim [1] claimed 70% Q3 prediction success of 25 proteins; Garnier [2] achieved 63% success for 26 proteins; while Qian and Sejnowski [3] reported 64.3% prediction success in 19 proteins. Using such a different protein pool for training and testing of algorithms produces a challenge for objective evaluation of such algorithms. Rost and Sander [4] tested a method in prediction of proteins in which they did not use the same proteins for the formation of their algorithm. They achieved prediction success better than 70%. Prediction success of Lim’s study [1] was reduced by 14% down to 56%. Cross-validation of methods of testing whereby initial proteins are removed from the training pool of proteins yields more realistic predictions.
1.2. Problem with objective testing of methods in prediction of secondary protein structure
For a protein sequence with an unknown 3D structure which is similar to a protein with a known 3D structure, the best prediction model for protein’s secondary structure is the alignment of sequences using algorithms derived from dynamic programming [5]. Methods for predicting secondary structures are employed when similar sequences are not available. Testing of exactness of a prediction on a training dataset yields unrealistically high success rates. An ideal approach is to test a desired sequence of a protein whose sequence is not included in the testing set and has no similarities.
Today there are around 500 different proteins with non-similar sequences with known 3D structures which can be used in assessing the accuracy of technology for prediction of secondary structure of proteins. However, many of today’s prediction models are based on a set of 126 protein chains used by Rost and Sander [6]. Cuff J A and Barton G J developed a new non-redundant set of 396 protein domains which include proteins from RS126 protein chains [7]. In our research we used developed algorithm [8] using CB513 data set [7] and expanded application interface API-EPE2 with better performances in comparing to API-EPE [8].
2. EXTRACTION, PREPARING, AND ENCODING OF DATA SAMPLES
The concept of algorithm for predicting secondary protein structure is shown in Fig. 1. The implementation of this algorithm is provided with two software packages. The first one is API_EPE2 (APplication Interface used for Extracting, Preparing and Encoding), and second is MATLAB&NN Toolbox.
Figure 1.

Neural network can learn general rules of association between primary sequence of protein chains and corresponding secondary structures elements
The purpose of API_EPE2 software is to extract, prepare, and encode data examples from CB513 data set taken from link: http://www.paraschorpa.com/project/evoca_prot/index.php. CB513 data set consists of 513 files with extension .all. These files contain information about protein sequences, but, for our algorithm, only information about primary and secondary structures is essential.
The first step in using our application software is to process all CB513 files and separate primary sequences and corresponding secondary structures. The result of this step is saved as file IzdvCB513.txt which consists of 513 non-homologous protein sequences and corresponding secondary structures. An example of this data is:
MVLSEGEWQLVLHVWAKVEADVAGHGQDILI…
CCCCHHHHHHHHHHHHHHGGGHHHHHHHHHH…
MNIFEMLRIDEGLRLKIYKDTEGYYTIGIGH…
CCHHHHHHHHHCCEEEEEECTTSCEEEETTE…
The second step is to encode protein sequences and corresponding secondary structures from 1-letter amino acid codes into numeric codes (Table 1) and result is saved as file KodCB513.txt. We also encode DSSP classes [9] into numeric codes (Table 2).
Table 1.
| Amin acids | Amino acids (referred in our language) | 1-letter symbol | Code |
|---|---|---|---|
| Alanine | Alanin | A | 01 |
| Cysteine | Cistein | C | 02 |
| Aspartate | Asparaginska kiselina | D | 03 |
| Glutamate | Glutaminska kiselina | E | 04 |
| Phenylalanine | Fenilanin | F | 05 |
| Glycine | Glicin | G | 06 |
| Histidine | Histidin | H | 07 |
| Isolecine | Izoleucin | I | 08 |
| Lysine | Lizin | K | 09 |
| Leucine | Leucin | L | 10 |
| Methionine | Metionin | M | 11 |
| Asparagine | Asparagin | N | 12 |
| Proline | Proline | P | 13 |
| Glutamine | Glutamin | Q | 14 |
| Arginine | Arginin | R | 15 |
| Serine | Serin | S | 16 |
| Threonine | Treonin | T | 17 |
| Valine | Valin | V | 18 |
| Tryptophan | Triptofan | W | 19 |
| Threonine | Tirozin | Y | 20 |
Table 2.
| DSSP classes | Structures used in our algorithm | Code |
|---|---|---|
| H,G | Helix | 01 |
| E | Strand | 02 |
| B, I, S, T, C, L | Other structures | 03 |
After that, we separate primary sequences from secondary structures and result is file for primary protein sequences PrimCB513.txt:
11 18 10 16 04 06 06 19 14 10 07 18 19 01 09 18 04 01 03 18 01…
11 12 08 05 04 11 10 15 08 03 04 06 10 15 10 09 08 20 09 03 17…
and file for secondary structures SekCB513.txt is:
3 03 03 03 01 01 01 01 01 01 01 01 01 01 01 01 01 01 01 01 01…
03 03 01 01 01 01 01 01 01 01 01 03 03 02 02 02 02 02 02 03 03 …
3. NEURAL NETWORK BASED ALGORITHM FOR SECONDARY STRUCTURE PREDICTION
3.1. Determination of neural network training sets batch matrices
Data files PrimCB513.txt and SekCb513.txt are used for creation of two matrices; the first one for input samples and second one for output samples. The samples in these two matrices are used for training of neural network. To make batch matrix of patterns from the amino acids sequences (stored in the file PrimCB513.txt) we use two functions implemented in MATLAB. The first function returns a vector which contains sequences of amino acids separated by number 0. For example, if file PrimCB513.txt contains three amino acids sequences in rows:
11 12 08 05 04 11 10 15
01 01 09 16
06 18 18 17 09 03 04 01 04 09 10 05
the first function (prepare_data) produces vector:
11 12 8 5 4 11 10 15 0 1 1 9 16 0 6 18 18 17 9 3 4 1 4 9 10 5
The second function (prepare_pattern) returns pattern batch matrix using one window. A window is a short segment of a complete protein string. In the middle of it there is an amino acid for which we want to predict secondary structure. Each column of matrix created by this function corresponds to one window in protein string (Figure 3).
Figure 3.
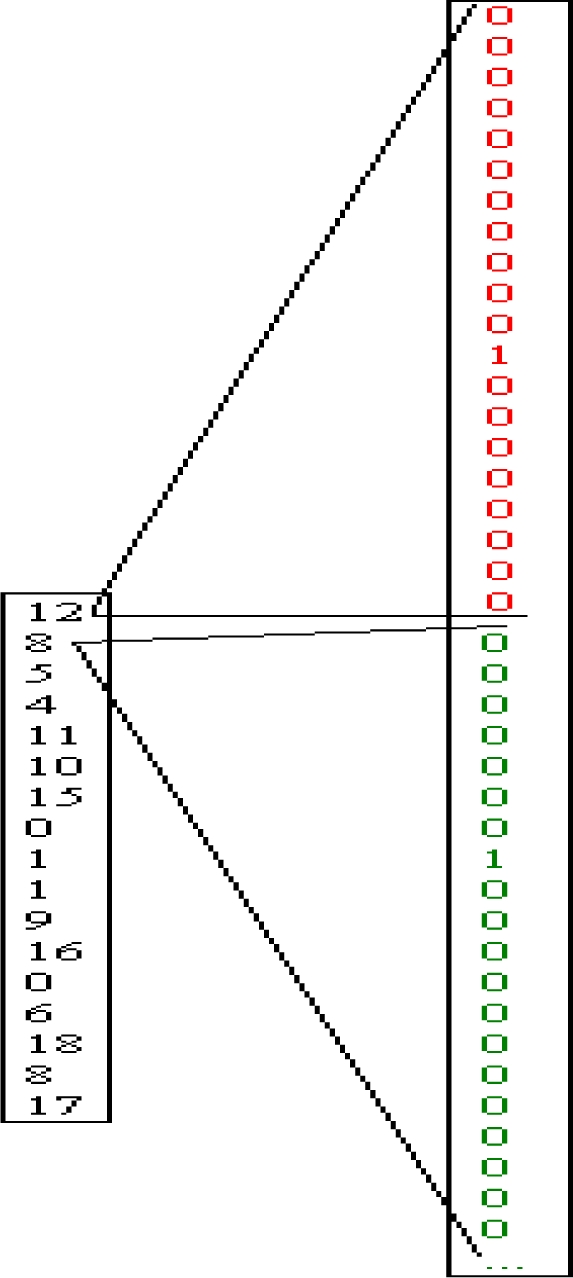
Transforming 1-number code into 20-number code
This window moves through protein, 1 amino acid at a time. Our prediction is made for central amino acid and if we encounter 0 (spacer between two proteins) at that position of window, than function prepare_pattern doesn't permit for that window to be placed into the pattern matrix. Then, with this function we transform 1-number code of amino acid into 20 number code. As you can see from the Figure 3, number 12 is transformed into 000000000001 00000000 and number 8 into 000000010000-00000000. Our matrix will require a neural network with 20 x17 input nodes (17 is length of the window) which have values 0 or 1.
To make target matrix from the secondary structure (stored in the file SekCB513.txt), we use two functions implemented in MATLAB. The first function (prepare_data) returns a vector which contains only three types of numbers (1,2,3) and also 0-numbers corresponding to null-numbers at the input vector produced with function prepare_data (PrimCB513.txt). Second function prepare_target produces output matrix using the length of the window which is the same as the length of the input vector string. That function eliminates those secondary structures from the output vector which correspond to amino acids for which we don't make any prediction (the first eight and the last eight amino acids) in the input vector. As all elements in target vector must have values between 0 and 1 (activation function at output layer is logsig), function transforms 1-number code of secondary structure into 3-number code and generates target matrix whose column vectors denote secondary structure patterns: helix, strand, and other structures. For example, if secondary structure sequence is:
11231111
1132
112122211111
the function prepare_data produces the following string:
1123111101132011212221111.
The function prepare_target produces from the previous string the following target matrix:
1 1 0 0 1 1 0 1
0 0 0 1 0 0 1 0
0 0 1 0 0 0 0 0
3.2. Design, learning, and performance evaluation of neural networks for different size of windows
Prediction of secondary structure was implemented using non-linear neural network with three layers based on feed-forward supervised learning and backpropagation error algorithm [8][10]. Default values for window size, number of neurons in hidden layer and number of training epochs are presented in Table 3.
Table 3.
| Algorithm parameters | Initial values |
|---|---|
| Window sizes | 13 |
| Number of neurons in hidden layer | 5 |
| Number of training epochs | 250 |
Instruction creatennw is used for designing neural networks with different sizes of windows (11, 13, 15, 17, and 19); instruction createCB513w is used for creation of training set, and training of neural network is started with instruction trainw. After that, we evaluated performances of neural networks using testing sets stored in files PrimStrTestB.txt and SekStrTestB.txt. Results are shown in Table 4.
Table 4.
| training/testing sets | window size (11–19) | Evaluated results | ||
|---|---|---|---|---|
| Q3% (training set) | Q3% (test set) | |||
| 1 | trainsetw11/testsetw11 | 11 | 62,2150 | 60,9910 |
| 2 | trainsetw13/testsetw13 | 13 | 62,4055 | 61,1011 |
| 3 | trainsetw15/testsetw15 | 15 | 57,0756 | 54,7338 |
| 4 | trainsetw17/testsetw17 | 17 | 62,5809 | 61,7053 |
| 5 | trainsetw19/testsetw19 | 19 | 63,5312 | 62,5160 |
| Average value Q3 | 61,5616 | 60,2094 | ||
3.3. Design, learning and performance evaluation of neural networks for different number of neurons in hidden layer
For this purpose we use the following instructions:
creatennhn (to design neural networks with different number of neurons in hidden layer),
trainhn (to train neural networks), and
accuracyhn (for performance evaluation). Results are shown in Table 5.
Table 5.
| Number of neurons in hidden layer (1–13) | Test | ||
|---|---|---|---|
| Q3% (training set) | Q3% (testset) | ||
| 1 | 2 | 44.1607 | 41.9736 |
| 2 | 3 | 63.2946 | 61.5593 |
| 3 | 4 | 63.2101 | 62.2373 |
| 4 | 5 | 63.5312 | 62.5160 |
| 5 | 6 | 62.8499 | 61.4689 |
| 6 | 7 | 63.6632 | 62.1544 |
| 7 | 8 | 63.6323 | 62.3352 |
| 8 | 9 | 63.6168 | 62.1695 |
| 9 | 10 | 63.4052 | 61.8531 |
| 10 | 11 | 63.5086 | 62.1544 |
| 11 | 12 | 63.4111 | 62.0866 |
| 12 | 13 | 63.7809 | 62.3051 |
| Average value Q3 | 61,8387 | 60,4011 | |
3.4. Design, learning and performance evaluation of neural networks for different number of epochs
At the end we used five neurons in hidden layer, window size 19 and different number of epochs using instructions traine. Results are shown in Table 6.
Table 6.
| Number of neurons in hidden layer (1–13) | Test | ||
|---|---|---|---|
| Q3% (training set) | Q3% (test set) | ||
| 1 | 100 | 44.5602 | 43.0508 |
| 2 | 150 | 57.8345 | 56.1582 |
| 3 | 200 | 63.0461 | 61.9962 |
| 4 | 250 | 63.5312 | 62.5160 |
| 5 | 300 | 63.5229 | 62.3879 |
| 6 | 500 | 63.5550 | 62.4633 |
| 7 | 1000 | 63.7357 | 62.3879 |
| 8 | 2000 | 64.3885 | 62.5838 |
| 9 | 3000 | 64.7369 | 62.8173 |
| 10 | 4000 | 64.8546 | 62.8776 |
| 11 | 5000 | 64.8914 | 62.8399 |
| 12 | 10000 | 64.5740 | 61.9736 |
| Average value Q3 | 61.9539 | 60.3377 | |
3.5. Performance evaluation of neural networks using nonhomologus data test
CB513, data set in which there is no sequence similarity between protein sequences, was divided into two sets:
413 training protein sequences (stored in PrimTrain413.txt data file and SekTrain413 data file), and
100 test protein sequences (stored in PrimTest100.txt and SekTest100.txt data file).
In MATLAB a software package nonhomtest was created with the following functions:
design of input data (samples) matrix using PrimTrain413.txt data file, and design of output data matrix using SekTrain413 data file,
design of neural network based on 5 neurons in hidden layer, and using window size 19,
training of designed neural network in time interval of 4000 epochs,
design of input data (samples) matrix using PrimTest100.txt data file, and design of output data matrix using SekTest100 data file, and
accuracy evaluation of our neural networks using.
Accuracy of a neural network prediction evaluated with non-homologous test set is 62.7253.
4. CONCLUSION
In this paper we have demonstrated the power of the artificial neural networks in extracting information from the protein structure database and in predicting secondary structural features from sequence alone. For design of neural network for prediction of secondary protein structure we used the modified algorithm API_EPE [8]. In that work [8] neural network was evaluated with protein set without considering existence of similarity between protein sequences. In this study, we used training set (CB513) in which there are no sequence similarities between protein sequences. But, in test set we did not consider homology between protein sequences and that test set was taken from PDBFIND2 data base. Because of that we cannot assure that results of predictions 62.8776 and 63.6261 [8] are fully objective. The solution in this study is based on dividing CB513 non-homologous data set into two subsets. The first one consists of 413 nonhomologous protein sequences and was used for training of our neural network. The second subset consists of 100 non-homologous protein sequences and was used for test of our neural networks. Accuracy of prediction for these two subsets is 62.7253, and we can assert that this is objective. The achieved exactness is relatively smaller compared to the study which used neural network training and test sets without verification of protein homology.
Figure 2.

Design of input pattern matrix taken from the string of encoded protein sequences
REFERENCES
- [1].Lim VI. Algorithms for prediction of α helices and β structural regions in globular proteins. J. Mol. Biology. 1974;88:873–894. doi: 10.1016/0022-2836(74)90405-7. [DOI] [PubMed] [Google Scholar]
- [2].Garnier J, Osguthorpe DJ, Robson B. Analysis and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biology. 1978;120:97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- [3].Qian N, Sejnowski T.Predicting the secondary structure of globular proteins using neural network models J. Mol. Biology 1988202865–884.Available from: http://brahms.cpmc.columbia.edu/publications/protein.pdf [DOI] [PubMed] [Google Scholar]
- [4].Rost B, Sander C. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biology. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- [5].Campbell AM, Heyer LJ. Discovering Genomics, Proteomics and Bioinformatics. CHSL Press, Benjamin Cummings; San Francisco: 2003. [Google Scholar]
- [6].Rost BR, Sander C, Schneider R. Redefining the goals of protein secondary structure prediction. J. Mol. Biology. 1994;235:13–26. doi: 10.1016/s0022-2836(05)80007-5. [DOI] [PubMed] [Google Scholar]
- [7].Cuff JA, Barton GJ.Evaluation and Improvement of Multiple Sequence Methods for Protein Secondary Structure Prediction, PROTEINS: Structure, Function, and Genetics 199934508–519.Available from: http://binf.gmu.edu/vaisman/csi731/pr99-cuff.pdf [DOI] [PubMed] [Google Scholar]
- [8].Avdagic Z, Purisevic E. Proccedings: Cybernetics and Systems. Vol. 1. Vienna: Austrian Society for Cybernetic Study; 2006. Feed-Forward Neural Network for Protein Structure Prediction; p. 198. [Google Scholar]
- [9].David W. Mount, Bioinformatics: Sequence and Genome Analysis. New York: CHSL Press; 2001. p. 13. [Google Scholar]
- [10].Holbrook SR, Muskal SM, Kim SH. Predicting Protein Structural Features with Artificial Neural Networks. In: Hunter L, editor. Artificial Intelligence and Molecular Biology. Massachusetts: The MIT Press, Massachusetts Institute of Technology; 1993. p. 161. [Google Scholar]