

Abstract
Cancer progression is an abnormal form of tissue repair characterized by chronic inflammation. IκB kinase-β (IKKβ) required for nuclear factor-κB (NF-κB) activation plays a critical role in this process. Using EOC cells isolated from malignant ovarian cancer ascites and solid tumors, we identified IKKβ as a major factor promoting a functional TLR–MyD88–NF-κB pathway that confers to EOC cell the capacity to constitutively secrete proinflammatory/protumor cytokines and therefore promoting tumor progression and chemoresistance. Furthermore, we describe for the first time the identification of the microRNA hsa-miR-199a as a regulator of IKKβ expression. Our study describes the property of ovarian cancer cells to enhance the inflammatory microenvironment as a result of the expression of an active IKKβ pathway. Identification of these markers in patients' tumor samples may facilitate the adequate selection of treatment and open new venues for the development of effective therapy for chemoresistant ovarian cancers.
Keywords: inflammation, ovarian cancer, IKKβ, MyD88, chemoresistance
Introduction
Epithelial ovarian cancer (EOC) is the fifth leading cause of cancer-related deaths in women in the United States and the leading cause of gynecologic cancer-related deaths (Schwartz, 2002; Jemal et al., 2007). One major limitation in the treatment of EOC is the development of cross-resistance to a wide range of chemotherapeutic agents. Although 80–90% of patients initially respond to first-line chemotherapy such as carboplatin and paclitaxel, less than 10–15% will remain in remission (Jemal et al., 2006). Treatment advances have led to improved 5-year survival, approaching 45%, but not in overall survival.
Recent studies have demonstrated a potential link between inflammation, cancer progression and chemoresistance (Karin et al., 2002; Balkwill and Coussens, 2004; Chen et al., 2007). Nuclear factor-κB (NF-κB) is one of the key transcription factors in proinflammatory response, and numerous evidence has been reported that links NF-κB activation and cancer development (Chen et al., 2007). Cytokines and chemokines, such as interleukin (IL)-6, IL-8, tumor necrosis factor (TNF-α), macrophage chemotactic protein 1 (MCP-1), growth-regulated protein α (GRO-α) and migration inhibitory factor (MIF) produced at the microenvironment by immune cells through NF-κB activation, are thought to drive the neoplastic process (Karin et al., 2002). However, the contribution of cancer cells themselves (especially non-leukemic cancer cells) in the maintenance of a proinflammatory environment that promotes cancer growth is largely overlooked and is sometimes considered passive, even though numerous studies have shown that NF-κB, and its activator IκB kinases (IKKs) are constitutively active in most cancer cells (Shishodia and Aggarwal, 2004; Chen et al., 2007).
The IKK complex is the direct upstream activator of NF-κB. The canonical IKK complex consists of three major subunits, IKKα, IKKβ and IKKγ. By phosphorylating the inhibitor of NF-κB α (IκBα), activated IKKs promote the proteosome-mediated degradation of IκBα and nuclear translocation of NF-κB. Although IKKα is more important in cell differentiation, lymphoid organogenesis and the regulation of adaptive immunity (Hacker and Karin, 2006; Liu et al., 2006), IKKβ is crucial for the production of proinflammatory cytokines that are related to cell survival and cell proliferation (Greten et al., 2004; Hu et al., 2004). IKKβ has been found highly active in many different types of cancer including breast cancer, pancreatic cancer, thyroidal C-cells carcinoma, melanoma and acute myeloid leukemia (Ludwig et al., 2001; Romieu-Mourez et al., 2001; Tamatani et al., 2001; Yang and Richmond, 2001; Baumgartner et al., 2002; Li et al., 2004). IKKβ deletion in mouse enterocytes decreased the incidence of colitis-associated tumor by 75% (Greten et al., 2004). In addition, Hu et al. (2004) previously reported that the breast cancer cell line 453 stably expressing IKKβ has a higher proliferation rate than its IKKβ-negative counterpart.
Furthermore, tumor growth could be considered as a form of abnormal compensatory proliferation in response to injury and tissue damage. This tissue repair process has been reported to depend on Toll-like receptor 4 (TLR4)-MyD88 signaling (Fukata et al., 2005). TLR4-MyD88 signaling is important to maintain intestinal epithelial homeostasis in response to gut injury, and both TLR4- and MyD88-knockout mice displayed impaired compensatory proliferation and increased apoptosis (Rakoff-Nahoum et al., 2004; Fukata et al., 2005; Pull et al., 2005). Similarly, in a mouse model of acute lung injury, hyaluronan released from injured cells protected epithelial cells from apoptosis through the TLR2/4–MyD88–NF-κB pathway (Jiang et al., 2005). Moreover, Medzhitov and colleagues found that spontaneous intestinal tumorigenesis was significantly decreased in MyD88-knockout mice (Rakoff-Nahoum and Medzhitov, 2007). These animal studies strongly suggest that MyD88 expression and associated molecules may play an important role in tumor progression; however, until now this information has not been confirmed in human cases.
Recently, we described the ubiquitous expression of TLR4 in EOC cells and showed that ligation of TLR4 by lipopolysaccharide (LPS)- or paclitaxel-induced cell proliferation and enhanced cytokine/chemokine production (Kelly et al., 2006). However, this effect was limited to a group of EOC cells expressing the TLR adaptor protein MyD88 (Kelly et al., 2006). Constitutive production of cytokines and growth factors are mechanisms that can suppress cancer cell apoptosis and therefore inhibit the efficacy of chemotherapy. Therefore, understanding the intracellular pathways mediating this unique tumor-enhancing characteristic is critical for developing adequate therapies for recurrent and chemoresistant cancer cells.
In this study, we identified IKKβ and miR-199a as crucial molecules expressed by EOC cells, which promotes a proinflammatory environment. Using EOC cells isolated from malignant ovarian cancer ascites and solid tumors, we identified IKKβ as a major factor promoting a functional TLR–MyD88–NF-κB pathway that confers to EOC cell the capacity to constitutively secrete proinflammatory/protumor cytokines and therefore promoting tumor progression and chemoresistance. Our findings demonstrate two possible differential stages in tumor development with unique characteristics in cytokine production, NF-κB regulation and chemo-response.
Results
Type I EOC cells constitutively secrete protumor cytokines
Previously, we identified the differential expression of MyD88 in EOC cells, which correlated with their in vitro response to LPS and paclitaxel (Kelly et al., 2006). Furthermore, we showed that ligation of TLR4 by LPS or paclitaxel in MyD88-positive (Type I EOC cells) but not MyD88-negative cells (Type II EOC cells) enhanced cytokine/chemokine production (Kelly et al., 2006). As the tumor microenvironment is determined, in great part, by the factors produced by the tumor cells, our first objective was to characterize the cytokine profile for each of these two cell types. Type I EOC cells (MyD88-positive) are characterized by constitutive secretion of proinflammatory cytokines and chemokines including IL-6, IL-8, MCP-1, MIP-1α, Regulated on Activation, Normal T-cell Expressed and Secreted (RANTES), GRO-α, granulocyte monocyte-colony-stimulating factor (GM-CSF) and MIF (Figure 1). Conversely, none of these cytokines, with the exception of MIF, were detected in Type II EOC cells (Figure 1).
Figure 1.
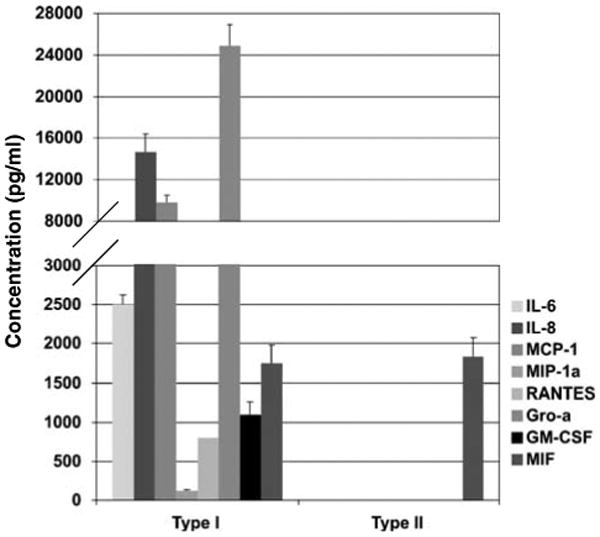
Differential expression of cytokines between Type I and II epithelial ovarian cancer (EOC) cells. Cytokine production was determined in the supernatant from EOC cells. Type I EOC cells expresses significant levels of inflammatory cytokines. No inflammatory cytokines were detected in Type II EOC cells in any condition. Representative figure of 20 evaluated EOC cells. Each sample was performed in triplicates.
Differential patterns of NF-κB activity in EOC cells
It is well accepted that NF-κB has a central role in the inflammatory response by inducing the production of proinflammatory cytokines (Karin et al., 2002). Therefore, our next objective was to determine the expression and function of the transcription factor NF-κB and its possible relationship with the continuous cytokine production we observed in Type I EOC cells. We monitored the level of NF-κB activity in both Type I and II EOC cells using a luciferase reporter system (Leung et al., 2006). As shown in Figure 2a, Type I EOC cells have constitutive NF-κB activity characterized by cyclic changes during a 12-h time course. In contrast, in Type II EOC cells, NF-κB activity remained constant during the same time period (Figure 2a). Further differences in NF-κB activity were observed when EOC cells were stimulated with either LPS or paclitaxel (both of which activate NF-κB in a MyD88-dependent pathway). LPS and paclitaxel enhanced NF-κB activity in Type I but not Type II EOC cells, confirming that MyD88 expression is necessary for the effects of these two compounds (Figure 2b). These data suggest that constitutive NF-κB activity in Type I EOC cells may be responsible for the constitutive production of proin-flammatory cytokines, and the induced NF-κB activation may account for the enhanced cytokine production after LPS or paclitaxel stimulation. Indeed, treatment of Type I EOC cells with a specific NF-κB inhibitor, Eriocalyxin B (Leung et al., 2006), suppressed the production and secretion of cytokines in Type I EOC cells. As show in Figure 2c, the secretion of two NF-κB-dependent cytokines, represented by IL-6 and MCP-1, are significantly inhibited following Eriocalyxin B treatment (Figure 2c). Similar effects were observed for GRO-α, GM-CSF and RANTES (data not shown).
Figure 2.
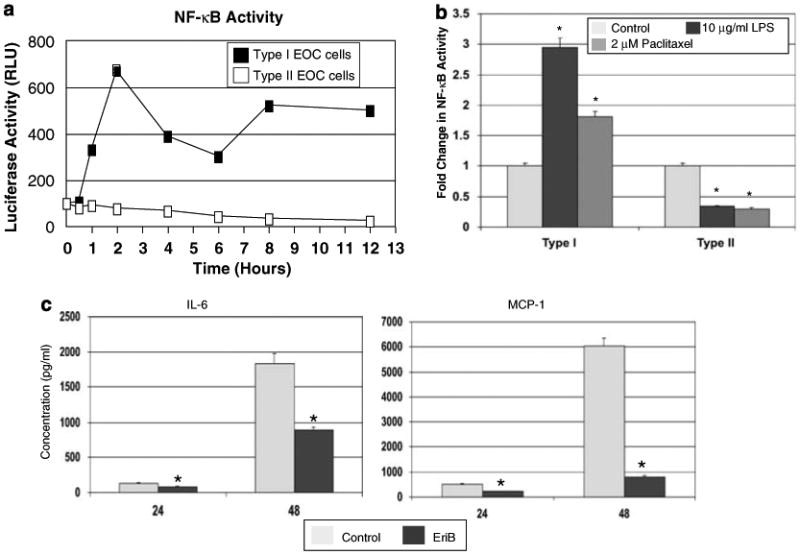
Determination of nuclear factor-κB (NF-κB) activity in ovarian cancer cells transfected with a luciferase reporter construct containing two κB-binding sites. (a) Endogenous cyclic NF-κB activity was observed in Type I but not Type II epithelial ovarian cancer (EOC) cells during a period of 12 h. (b) Treatment with lipopolysaccharide (LPS) and paclitaxel induced NF-κB activity in Type I but not in Type II EOC cells, as represented by data at 12 h after treatment. *P<0.05. (c) Treatment of Type I EOC cells with 2 μM EriB, a NF-κB inhibitor, blocks the constitutive cytokine production, indicating its NF-κB dependence. EriB, Eriocalyxin B. *P<0.05. Representative experiment of two Type I and II EOC cell lines. Similar results were observed with other cells of the same type. n=3 per sample per time point.
Differential expression and regulation of the inhibitor of NF-κB α (IκBα) in EOC cells
We hypothesized that the constitutive NF-κB cyclic activity observed in Type I EOC cells may be the result of specific regulatory elements upstream of the pathway. Thus, we examined the expression of IκBα, a major inhibitor of NF-κB. Type I EOC cells have low levels of IκBα, whereas high levels of expression was observed in Type II EOC cells (Figure 3a). Interestingly, this pattern of expression was inversely correlated with MyD88 expression (Figure 3a). Furthermore, evaluation of IκBα levels over a period of 12 h showed a similar cyclic pattern as observed for NF-κB in Type I but not Type II EOC cells (data not shown). These data demonstrate a correlation between the levels of IκBα and NF-κB activity, suggesting that the differential regulation of NF-κB activity in Type I and II EOC cells may be due to upstream regulators of IκBα.
Figure 3.
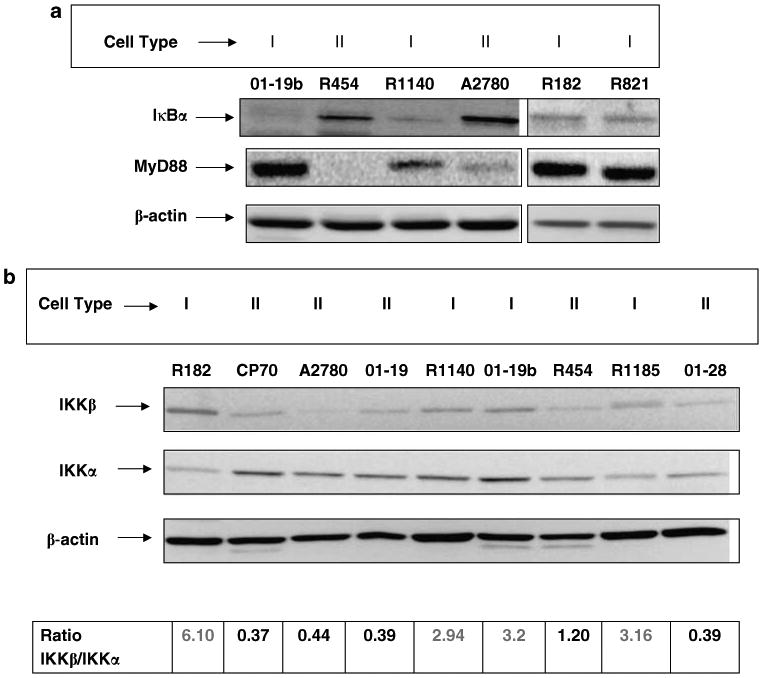
Differential expression of IκBα and IKK subunits between Type I and II epithelial ovarian cancer (EOC) cells. (a) Western blot analysis for IκBα expression in ovarian cancer cells. Note the lack of IκBα expression in Type I EOC cells compared to Type II. Low IκBα expression corresponds to high MyD88 expression levels. (b) Differential expression of IKKα and IKKβ in Type I and II EOC cells. Type I EOC cells are characterized by a high IKKβ/IKKα ratio. Representative figure of four Type I and five Type II EOC cells out of 40 cancers evaluated in five independent experiments. Each experiment was performed in triplicates.
The degradation of IκBα depends on the phosphorylation of the IKK (Rothwarf and Karin, 1999; Greten and Karin, 2004). The IKK complex has two catalytic subunits IKKα and IKKβ, and an essential regulatory subunit IKKγ/NEMO (Mercurio et al., 1999). Differential expression of IKK subunits has been associated with differential cytokine production (Luo et al., 2005). Therefore, we hypothesized that the difference we observed in Type I and II EOC cells may be related to the expression of IKK subunits. Thus, the levels of IKKα and IKKβ were determined by western blot analysis. We found that the ratio of IKKβ/IKKα was significantly higher in Type I than Type II cells (Figure 3b), suggesting that this differential expression of IKKα and IKKβ may explain the observed difference in NF-κB and IκBα activity.
Ectopic expression of IKKβ induce cytokine production in Type II EOC cells
Deletion of the IKKβ subunit has been shown to inhibit inflammatory response (Li et al., 1999; Li and Verma, 2002). As Type II EOC cells express low levels of IKKβ, we hypothesized that the ectopic expression of IKKβ in these cells will increase the ratio of IKKβ/IKKα and may induce Type II EOC cells to produce proinflammatory cytokines. Indeed, the ectopic overexpression of a constitutively active form of IKKβ (pCMV2-IKK2 S177E S181E) (Mercurio et al., 1999) in Type II EOC cells resulted in a significant decrease in the expression of IκBα (Figure 4a). The changes in IκBα levels inversely correlated with the IKKβ expression levels (Figure 4a). Furthermore, IKKβ overexpression in Type II cells led to the production of high levels of proinflammatory cytokines similar to that we saw in Type I cells (Figure 4b). Interestingly, we also observed increased MyD88 expression in these transfectants (Figure 4a), which may be the result of NF-κB activation, as MyD88 is also a target of NF-κB (Harroch et al., 1995).
Figure 4.
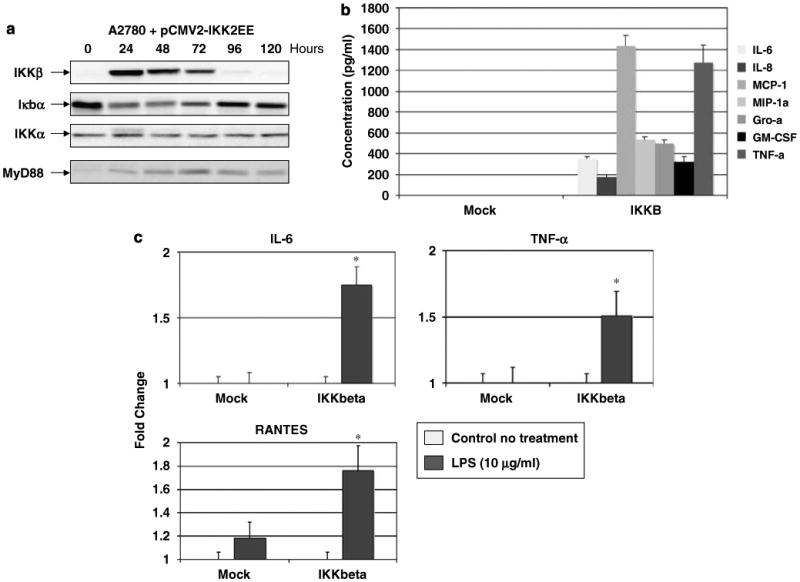
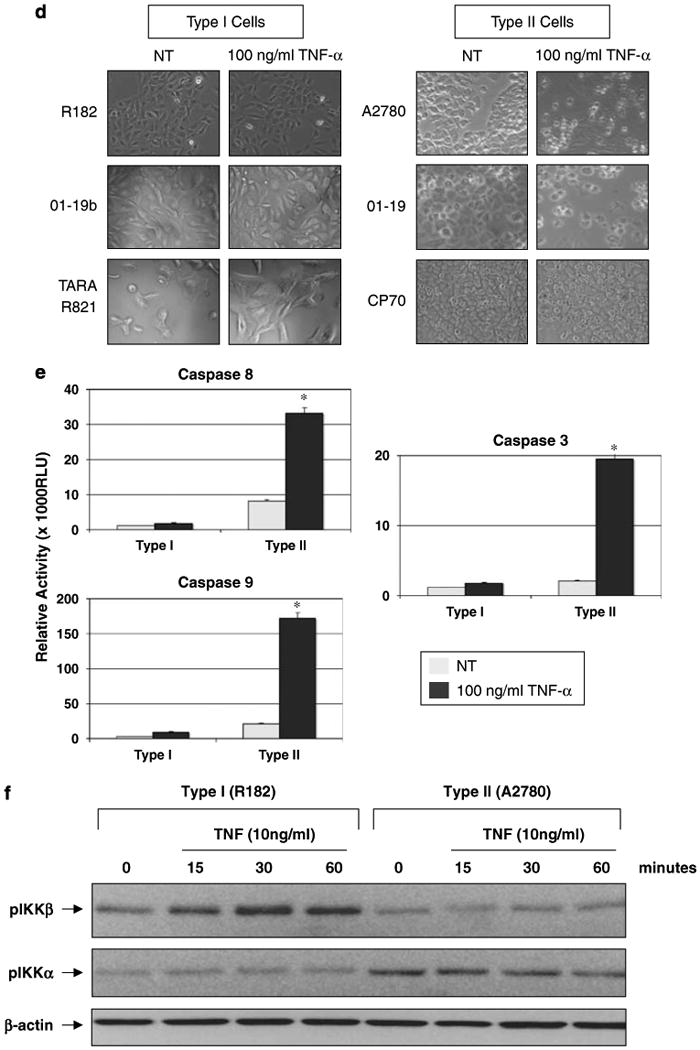
Effect of ectopic overexpression of IKKβ in Type II epithelial ovarian cancer (EOC) cells. (a) Transient overexpression of a constitutively active form of IKKβ (IKKβ S177E S181E) in Type II EOC cells induced a significant decrease in IκBα expression and increase in MyD88 expression. No change in IKKα expression was observed. (b) Overexpression of IKKβ S177E S181E in Type II EOC cells promotes cytokine production. (c) Transient overexpression of WT IKKβ in a Type II MyD88-positive stable transfectant cell line restored the functionality of the TLR4 pathway, as determined by lipopolysaccharide (LPS)-induced cytokine production. *P<0.05. pCMV2-IKKEE, plasmid expressing a constitutively active form of IKKβ (IKKβ S177E S181E). MOCK, mock transfection with the empty plasmid pCMV2. Representative figure of an experiment using A2780 cells. Similar results were obtained with two additional Type II EOC cells. (d) Differential effect of TNF-α on ovarian cancer cells. Treatment with TNF-α, 100 ng/ml, (24 h) induce cell death in Type II but not in Type I ovarian cancer cells. Six representative cell lines NT, nontreatment. (e) Induction of caspase activity by TNF-α 100 ng/ml, (24 h) in Type II EOC cells. Representative experiment of at least eight cell lines. Each experiment was performed in triplicates. *P<0.0001. (f) TNF-α treatment induces the phosphorylation of IKKβ but not IKKα in Type I EOC cells. No changes are observed in Type II cells. Representative experiment performed with five Type I and five Type II cell lines.
IKKβ overexpression restores TLR4 response in Type II EOC cells
We then evaluated whether the expression of IKKβ in Type II EOC cells could mimic the TLR4 response observed in Type I cells. Thus, Type II A2780 cells stably transfected with MyD88 (A2780MyD88+) were transiently transfected with a plasmid expressing the wild-type IKKβ gene. After 24 h of transfection, cells were treated with LPS, and cytokine production was determined in both supernatant and cell lysate. As shown in Figure 4c, only cells transfected with IKKβ have increased levels of cytokine secretion following LPS treatment. Similar results were seen in cell lysate samples (data not shown). These results confirm the important role of IKKβ in cytokine production following TLR4 ligation.
Differential response to tumor necrosis factor-α
Tumor necrosis factor-α is a proinflammatory cytokine with divergent effects on cancer cells; in some cases TNF-α induce tumor cell death (Aggarwal, 2003) and in other cases TNF-α may act as tumor promoter through activation of NF-κB (Aggarwal, 2003; Barnhart and Peter, 2003; Chen et al., 2007). The induction of NF-κB activity by TNF-α has been shown to be dependent on the expression of IKKβ and the absence of IKKβ is associated with increased susceptibility to TNF-α-induce apoptosis (Li et al., 1999). As Type I and II EOC cells have a differential expression of IKKβ, we evaluated the effect of TNF-α on these two cell types. Although Type I cells were resistant to high-dose TNF-α-induced cell death (Figure 4d), Type II cells showed a significant decrease in cell viability and underwent apoptosis as shown by increasing activity of caspases 8, 9 and 3 (Figures 4d and e), which are the main mediators of TNF-α-induced apoptosis.
Subsequently, we evaluated whether low-dose TNF-α could activate the NF-κB pathway in Type I EOC cells by determining IKKα/β phosphorylation levels. As shown in Figure 4f, treatment with TNF-α (10 ng/ml) induced a time-dependent increase in p-IKKβ in Type I cells but not in Type II EOC cells. No changes were observed on p-IKKα in either type of cells (Figure 4f).
hsa-miR-199a regulates IKKβ expression in EOC cells
Evaluation of IKKβ mRNA showed similar levels in both cell types (Figure 5a), suggestive of a post-transcriptional regulation. As microRNAs (miRNAs) are noncoding forms of RNAs involved in post-transcriptional gene regulation, we hypothesized that the differential expression of IKKβ protein may be due to miRNA regulation. To examine the potential involvement of miRNAs in the regulation of EOC cell phenotype, we analysed the expression of 316 miRNAs in Type I and II EOC cells using miRNA microarray chips (Invitrogen, Carlsbad, CA, USA) (Supplementary Figure 1). Eighteen miRNAs were identified to be significantly differentially expressed between the two cell types (Figure 5b). Three miRNAs are highly expressed in Type II, whereas 15 are downregulated, compared to Type I. Using PicTar (http://www.pictar.bio.nyu.edu/), an algorithm for the identification of microRNA targets, we found that the 3′-UTR of IKKβ mRNA contains three putative target sequences for hsa-miR-199a, which is one of the three miRNAs upregulated in Type II EOC cells (Figure 5c). To validate the results from the miRNA microarray, we performed NCode SYBR GreenER miRNA qRT–PCR analysis for hsa-miR-199a. Using total RNA from six samples in a blind manner, high levels of hsa-miR-199a expression was detected only in Type II EOC cells (Figure 5d).
Figure 5.
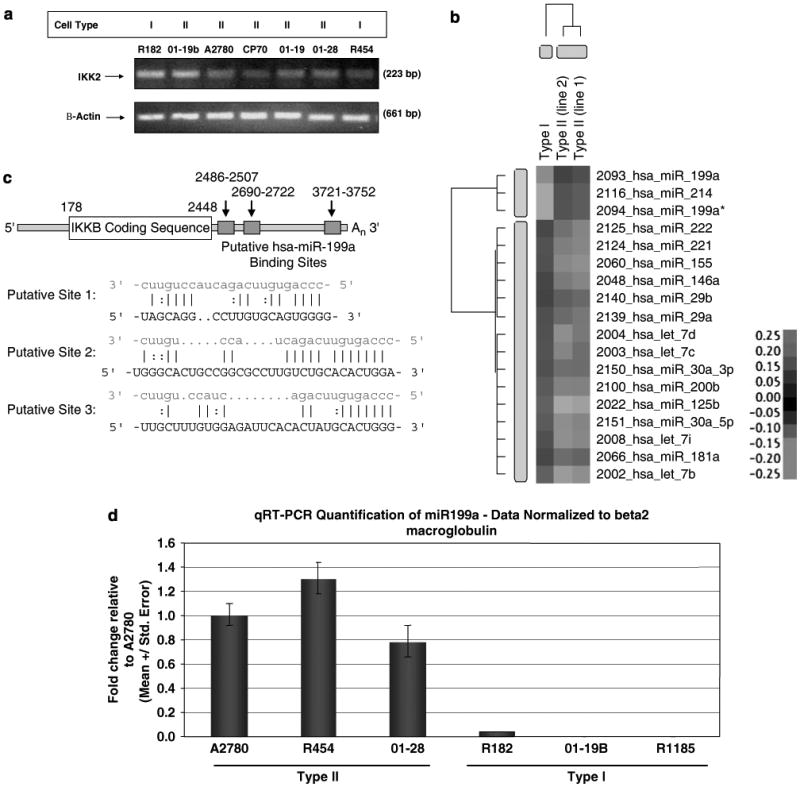
MicroRNAs (miRNA) profiles in Type I and II epithelial ovarian cancer (EOC) cells, and their relationship to IKKβ expression. (a) Type I and II cells have similar levels of IKKβ mRNA, as determined by reverse transcription (RT)–PCR. (b) miRNA profiling of one Type I and two Type II cell lines by Invitrogen NCODE miRNA microarray. (c) Panel of differentially expressed miRNA in Type I and II cells. Note the similarity of miRNA expression between the Type II cell lines and their similar differences in relation to Type I. Red indicates miRNA upregulated in Type II versus Type I; green indicates miRNA downregulated in Type II versus Type I. (d) Three putative hsa-miR-199a-binding sites within the 3′-UTR region of the IKKB mRNA, as predicted by Pictar (http://www.pictar.bio.nyu.edu/, and the algorithm ‘PicTar predictions in vertebrates, flies and nematodes’ was selected). Red sequences, hsa-miR-199a; black sequences, putative binding sites on the 3′-UTR of IKKB mRNA. (e) qRT–PCR quantification of hsa-miR-199a in ovarian cancer cells. Data normalized to β-2 macroglobulin. Note the high expression levels of hsa-miR-199a in Type II cells compared to Type I cells.
To conclusively confirm the role of hsa-miR-199a in the regulation of IKKβ, we performed loss or gain of function studies. Transient transfection of the pre-miRNA of hsa-miR-199a into Type I cells led to a significant decrease in IKKβ expression, whereas transient transfection of the anti-miRNA of hsa-miR199a into Type II cells resulted in the expression of IKKβ (Figure 6a). We further proved that the downregulation of IKKβ expression by hsa-miR-199a depends directly on the 3′-UTR of IKKβ mRNA with a luciferase reporter system (Figure 6b). Using a construct that highly expresses luciferase, we added the IKKβ 3′-UTR after the luciferase gene (pmir-RIKK2-3u-1). Afterwards, the construct was transfected into Type I EOC cells (endogenous low levels of hsa-miR-199a), and divided in three groups. Group 1 was transfected with hsa-miR-199a; group 2 received a nonspecific negative control miRNA (miR-NC-no. 1) and group 3 received mock transfection control (only transfection reagent). hsa-miR-199a transfection resulted in significant suppression of luciferase activity compared to mock transfection, whereas negative control miRNA (miR-NC-no. 1) led to no change in the luciferase activity (Figure 6b). Mutation of the hsa-miR-199a-binding sites in the IKKβ 3′-UTR completely abolished the inhibitory effect of hsa-miR-199a on luciferase activity (data not shown). These results further confirm a direct inhibition of hsa-miR-199a on IKKβ mRNA translation through its 3′-UTR.
Figure 6.
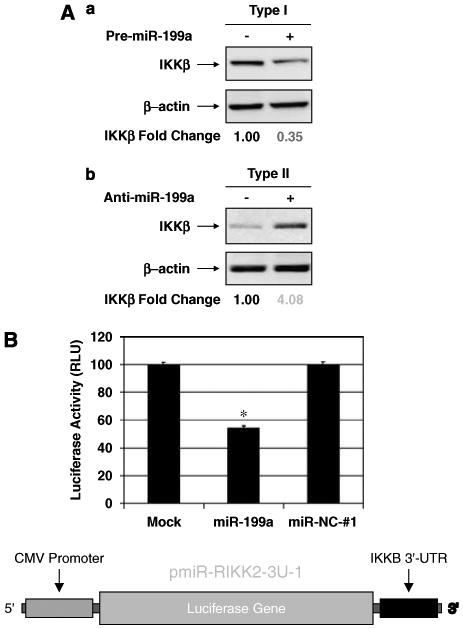
Regulation of IKKβ expression by hsa-miR-199a. (A) (a) Transient transfection of Type I cells with Pre-miR-199a inhibits IKKβ expression. (b) Transient transfection of Type II cells with anti-miR-199a induced IKKβ expression. (B) miR-199a suppressed the IKKB 3′-UTR Luciferase reporter activity compared to mock transfection, whereas the negative control miRNA (miR-NC no. 1) did not result in any changes. Diagram of the Luciferase reporter plasmid to study the function of the 3′-UTR of IKKB mRNA. The reporter consists of a Luciferase gene with the IKKB 3′-UTR driven by a cytomegalovirus promoter. *P<0.001.
Discussion
We describe for the first time the differential expression of IKKβ in EOC cells isolated from patients with ovarian cancer. We demonstrate that the expression of IKKβ in these cancer cells is associated with unique characteristics related to NF-κB activation, TNF-α response, cytokine production and miRNA profiles that correlate to chemoresponse. We further identified hsa-miR-199a as a regulator of IKKβ expression.
There is compelling evidence that inflammation promotes cancer progression and NF-κB provides the link between these two processes. In fact, recent data directly implicate NF-κB activation as the key component in inflammation-based cancer progression (Pikarsky et al., 2004). The present results indicate that, in addition to the immune cells, tumor cells may also actively contribute to the inflammatory process that will enhance the repair process and promote cell growth. However, this capacity is not intrinsic to all the cancer cells, but to a selected population that has acquired a functional TLR4–MyD88–NF-κB signaling pathway (Figure 7).
Figure 7.

Model of Type I and II EOC cells. Type I epithelial ovarian cancer (EOC) cells have high levels of IKKβ expression due to low hsa-miR-199a; therefore, when stimulated, nuclear factor-κB (NF-κB) activation leads to cytokine production, cell proliferation and induction of antiapoptotic proteins. In Type II cells expression of IKKβ is low due to high hsa-miR-199a expression levels, therefore an incomplete TLR4–MyD88–NF-κB pathway cannot respond to ligands, resulting in no cytokine production and chemosensitivity.
Interestingly, we found that in addition to MyD88, as we previously reported, high levels of IKKβ are also responsible for the phenotype of Type I EOC cells. IKKβ is one of the catalytic subunits of the IKK complex that has been shown to be crucial for the NF-κB-mediated production of proinflammatory cytokines that are related to cell survival and cell proliferation (Greten et al., 2004; Hu et al., 2004), and has been found highly active in many types of cancer (Ludwig et al., 2001; Romieu-Mourez et al., 2001; Tamatani et al., 2001; Yang and Richmond, 2001; Baumgartner et al., 2002; Li et al., 2004). Consistent with these findings, we observed a correlation between IKKβ expression levels, NK-κB activity and cytokine production. All these cytokines and chemokines produced by Type I EOC cells had been shown to be related to cell proliferation and chemoresistance (Coussens and Werb, 2002; Duan et al., 2002; Balkwill, 2004; Nakanishi and Toi, 2005).
Moreover, the expression of IKKβ has been associated with the differential response to TNF-α (Pikarsky et al., 2004). Although in some cells, TNF-α is able to induce apoptosis in a caspse-dependent manner; in other cells TNF-α induces NF-κB activation through IKKβ (Li et al., 1999). Type I EOC cells which express high levels of IKKβ are resistant to TNF-α-induced apoptosis and are characterized by increase in pIKKβ following TNF-α treatment. This is not the case for Type II EOC cells, which lack IKKβ and therefore are sensitive to TNF-α-induced apoptosis. In these cells, TNF-α activates the classical caspase-dependent apoptotic pathway, which involves caspases 8, 9 and 3.
As outlined above, the expression of IKKβ has been associated with cytokine production and inflammation (Maeda et al., 2003, 2005), a characteristic that we found in Type I EOC cells. This role of IKKβ was confirmed in our studies where transient overexpression of IKKβ in Type II cells restored both the cytokine production and MyD88 expression; therefore, confirming that IKKβ plays a very important role in the capacity of Type I cells to promote a proinflammatory environment that could lead to enhanced tumor growth, tumor recurrence and possible chemoresistance.
A major question is then, what regulates the differential expression of IKKβ in these two types of EOC cells. One of the differentially expressed miRNA was hsa-miR-199a. Indeed, hsa-miR-199a expression was significantly higher in Type II EOC cells compared with that of Type I and its expression was associated with inhibition of IKKβ expression. The characteristics of the three putative target sequences for hsa-miR-199a and mRNA levels for IKKβ observed in ovarian cancer cells suggest a translation-inhibitory effect of hsa-miR-199a rather than induction of RNA degradation.
Our findings demonstrate for the first time a functional role for hsa-miR-199a as direct inhibitor of IKKβ expression. The loss of the inhibitory effect of hsa-miR-199a on IKKβ expression in Type I cells may represent an evolutionary fine-tuning during the development of chemoresistant cells. miRNAs may provide invaluable targets to treat chemoresistant cancers.
The data presented in this study suggest that distinct characteristics of Type I and II EOC cells may lead to differential responses to stimulatory signals, including those coming from TLRs. Thus, in Type I cells, due to the high IKKβ and low hsa-miR-199a expression, TLR and TNF-α stimulation leads to NF-κB activation and cytokine production, which then may enhance the cells' resistance to cytotoxic drugs such as paclitaxel and TNF-α. In contrast, this response is not observed in Type II cells, which have high levels of hsa-miR-199a and therefore have low levels of IKKβ (Figure 7).
In conclusion, we describe for the first that hsa-miR-199a regulates IKKβ expression and this differential expression has significant effect on the type of response of the cancer cells to stimuli. Through their IKKβ expression, Type I EOC cells have the capacity to promote a protumor environment, which may have implications in tissue repair, chemoresistance and tumor progression. Identification of these markers in patients' tumor samples may facilitate the adequate selection of treatment, and open new venues for the development of effective therapy for chemoresistant ovarian cancers.
Materials and methods
Patients and samples
Malignant ovarian ascites samples were collected from stage III/IV ovarian cancer patients. Tumor samples were collected from surgery under sterile conditions; one aliquot was processed for cell preparation and a second aliquot snap frozen in liquid nitrogen for additional use. All patients signed consent forms, and the use of patient samples was approved under Yale University's Human Investigations Committee (HIC no. 10425).
Cell lines and culture conditions
A total of 20 primary EOC cells were used in this study in addition to the human EOC cell lines SKOV3, A2780 and CP70 (gifts from Dr TC Hamilton). Primary EOC cells were isolated from malignant ovarian ascites or ovarian tumors and cultured as previously described (Kamsteeg et al., 2003; Flick et al., 2004). EOC cells were grown in RPMI plus 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA, USA) at 37 °C in a 5% CO2 atmosphere. Purity of the EOC cells was 100% as determined by immunostaining for cytokeratin antigen.
Reagents
Lipopolysaccharides isolated from Escherichia coli (0111:B4), paclitaxel and rabbit anti-human β-actin antibody were purchased from Sigma (St Louis, MO, USA). Rabbit anti-human MyD88 antibody was purchased from eBioscience (San Diego, CA, USA). Mouse anti-human IκBα antibody (no. 4814) and rabbit anti-human IKKα (no. 2682) or IKKβ (no. 2684) or phospho-IKKα (Ser 180)/IKKβ (Ser 181) (no. 2681) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
Protein preparation
Protein extraction was done as previously described (Kamsteeg et al., 2003). Briefly, cell pellets were lysed on ice in 1 × phosphate-buffered saline with 1% NP40, 0.1% SDS and freshly added 20 μl/ml protease inhibitor cocktail (Sigma Chemical, St Louis, MO, USA) and 2mM phenylmethylsulfo-nyl fluoride (Sigma Chemical). Protein concentration was determined by BCA Protein Assay (Pierce Biotechnology, Rockford, IL, USA), and proteins were stored at −40 °C until further use.
SDS–PAGE and western blots
A quantity of 20 μg of each protein sample was denatured in sample buffer and subjected to 12% SDS–polyacrylamide gel electrophoresis (PAGE) as previously described (Kamsteeg et al., 2003). The following antibodies were used: rabbit anti-human MyD88 (1:1,000), mouse anti-human IκBα (1:1,000), rabbit anti-human IKKα (1:1,000), rabbit anti-human IKKβ (1:2,000) and rabbit anti-human β-actin (1:10,000). Specific protein bands were visualized using the enhanced chemiluminescence assay (Pierce Biotechnology).
NF-κB activity
Nuclear factor-κB activity was measured using a luciferase reporter construct, pBII-LUC containing two κB sites before a Fos essential promoter (a gift from Dr S Ghosh, Yale University). Cells were transiently transfected with pBII-LUC using the FuGENE 6 Transfection Reagent (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer's instructions. Luciferase activity was measured using the Luciferase Assay System (Promega, Madison, WI, USA) according to the manufacturer's protocol. Briefly, 10 μg of each protein sample in a total volume of 20 μl was mixed with 100 μl of the luciferase assay reagent, and luminescence was measured using TD 20/20 Luminometer (Turner Designs, Sunnyvale, CA, USA). Relative activity was calculated on the basis of readings measured from samples after subtracting blank values, and normalized to β-galactosidase activity expressed by a co-transfected control plasmid. Each sample was performed in triplicates.
Cytokine profiling
Cytokine profiling was performed from protein extracts or culture supernatants using the Luminex 200 system (Luminex Co., Austin, TX, USA) according to the manufacturer's instructions. In summary, 50 μl standard or sample was added to the wells of 96-well plates, and 25 μl microparticle mixture was added to each well. The plates were incubated at room temperature on an orbital shaker (500 r.p.m.) for 2 h. The plates were then washed 3 times with Beadlyte Cell Signaling Assay Buffer (Upstate, Charlottesville, VA, USA) and the microparticles were resuspended in 75 μl Assay Buffer. A quantity of 25 μl of biotinylated detection Ab was added to each well, and after 1.5 h of incubation at room temperature (500 r.p.m.), the plates were washed 3 times with Assay Buffer, and the microparticles were resuspended in 75 μl Assay Buffer. Streptavadin-PE/Assay buffer mixture (1:20) was prepare and 25μl were added per well. After 0.5 h of incubation at room temperature (500 r.p.m.), the plates were washed 3 times with Assay Buffer and resuspended in 125 μl Assay Buffer. The plates were then read by the Luminex 200 Multiplex Analyzer. The final readings were normalized against total cell number of each sample and each sample was performed in triplicates.
IKKβ transfection
The plasmid construct overexpressing a constitutively active form of IKKβ (pCMV-IKK2 S177E S181E) was obtained from Dr A Rao through www.addgene.com (Plasmid no. 11105). Transient transfection of Type II cells with pCMV-IKK2 S177E S181E, or the empty vector pCMV was carried out using the FuGENE 6 Transfection Reagent (Roche Applied Science) following the manufacturer's instructions.
RNA isolation and reverse transcription–PCR
Total RNA was isolated using the RNeasy Mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Reverse transcription was performed on 2 μg of total RNA using the First Strand cDNA Synthesis kit (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer's instructions. The primers for IKKB: forward, 5′-ACTTGGCGCCCAATGACCT-3′; reverse, 5′-CTCTGTTCTCCTTGCTGCA-3′. The primers for ACTB: forward, 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′; reverse, 5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′. Thirty cycles of PCR were performed at 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. The size of PCR products was 223 bp (IKKB) and 661 bp (ACTB), respectively.
miRNA microarray
Analysis with miRNA microarrays each containing 316 analytes were carried out using the NCode Multi-Species miRNA Microarray Kit (Invitrogen) according to the manufacturer's protocol. In summary, miRNA were isolated from total RNA and Poly(A)-tailed followed by ligation of a specific capture sequence through an Oligo(dT) bridge. The tagged miRNA were purified and hybridized to the microarray slides overnight. The slides were then washed and hybridized with Alexa Fluor3/Alexa Fluor5 Capture Reagents. The microarray slides were scanned and quantitated using a GenePix 4000B microarray scanner (Molecular Devices, Sunnyvale, CA, USA).
miRNA qRT–PCR
Quantitative reverse transcription (qRT)–PCR was performed to detect the levels of hsa-miR-199a in three Type I and three Type II EOC cell lines by NCode SYBRGreenER miRNA qRT–PCR Analysis (Invitrogen) according to the manufacturer's protocol as summarized below. A qRT–PCR forward primer for hsa-miR-199a (5′-CCCAGTGTTCAGACTACCTGTTC-3′) was designed and synthesized by Invitrogen. A polyadenylation reaction was performed on 500 ng of each total RNA sample using the NCode miRNA First-Strand cDNA Synthesis Kit (Invitrogen). Quadruplicate qPCR reactions for each cDNA were performed for hsa-miR-199a as well as for the housekeeping control genes β-2-microglobulin (B2 M, RefSeq NM_004048.2, forward primer 5′-CCGTGGCCTTAGCTGTGCTC, reverse primer 5′-TCCATTCTCTGCTGGATGACG) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, RefSeq NM_002046.3, forward primer 5′-CGCTGAGTACGTCGTGGAGTC, reverse primer 5′-GCAGGAGGCATTGCTGATGA) using the NCode SYBR GreenER miRNA qRT–PCR Kit (Invitrogen) and a 7900HT qPCR machine (Applied Biosystems, Foster City, CA, USA). The data are shown as fold change compared to sample A2780 after normalization to either housekeeping gene.
Statistical analysis
Data were presented as mean±s.d. (O'Dwyer et al., 1994). Statistical significance (P<0.05) was determined using oneway ANOVA with the Bonferonni correction.
Supplementary Material
Acknowledgments
This study was supported in part by funds from the Discovery to Cure Program, Nicholas Brady and Sands Foundation. We thank the support of Drs Mark Landers, Amy Cumeo and Heather White from Invitrogen on the micro-RNA studies. We thank Dr Yung-Chi Cheng from Pharmacology Department, Yale University for providing Eriocalyxin B. We also thank Dr Sankar Ghosh from Immunology Department, Yale University for sharing pBIIX-LUC.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431:405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- Barnhart BC, Peter ME. The TNF receptor 1: a split personality complex. Cell. 2003;114:148–150. doi: 10.1016/s0092-8674(03)00561-0. [DOI] [PubMed] [Google Scholar]
- Baumgartner B, Weber M, Quirling M, Fischer C, Page S, Adam M, et al. Increased IkappaB kinase activity is associated with activated NF-kappaB in acute myeloid blasts. Leukemia. 2002;16:2062–2071. doi: 10.1038/sj.leu.2402641. [DOI] [PubMed] [Google Scholar]
- Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the Toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Lamendola DE, Penson RT, Kronish KM, Seiden MV. Overexpression of IL-6 but not IL-8 increases paclitaxel resistance of U-2OS human osteosarcoma cells. Cytokine. 2002;17:234–242. doi: 10.1006/cyto.2001.1008. [DOI] [PubMed] [Google Scholar]
- Flick MB, O'Malley D, Rutherford T, Rodov S, Kamsteeg M, Hao XY, et al. Apoptosis-based evaluation of chemosensitivity in ovarian cancer patients. J Soc Gynecol Investig. 2004;11:252–259. doi: 10.1016/j.jsgi.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–G1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206:193–199. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Harroch S, Gothelf Y, Revel M, Chebath J. 5′ upstream sequences of MyD88, an IL-6 primary response gene in M1 cells: detection of functional IRF-1 and Stat factors binding sites. Nucleic Acids Res. 1995;23:3539–3546. doi: 10.1093/nar/23.17.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- Kamsteeg M, Rutherford T, Sapi E, Hanczaruk B, Shahabi S, Flick M, et al. Phenoxodiol—an isoflavone analog—induces apoptosis in chemoresistant ovarian cancer cells. Oncogene. 2003;22:2611–2620. doi: 10.1038/sj.onc.1206422. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66:3859–3868. doi: 10.1158/0008-5472.CAN-05-3948. [DOI] [PubMed] [Google Scholar]
- Leung CH, Grill SP, Lam W, Gao W, Sun HD, Cheng YC. Eriocalyxin B inhibits nuclear factor-kappaB activation by interfering with the binding of both p65 and p50 to the response element in a noncompetitive manner. Mol Pharmacol. 2006;70:1946–1955. doi: 10.1124/mol.106.028480. [DOI] [PubMed] [Google Scholar]
- Li L, Aggarwal BB, Shishodia S, Abbruzzese J, Kurzrock R. Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer. 2004;101:2351–2362. doi: 10.1002/cncr.20605. [DOI] [PubMed] [Google Scholar]
- Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Liu B, Park E, Zhu F, Bustos T, Liu J, Shen J, et al. A critical role for I kappaB kinase alpha in the development of human and mouse squamous cell carcinomas. Proc Natl Acad Sci USA. 2006;103:17202–17207. doi: 10.1073/pnas.0604481103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig L, Kessler H, Wagner M, Hoang-Vu C, Dralle H, Adler G, et al. Nuclear factor-kappaB is constitutively active in C-cell carcinoma and required for RET-induced transformation. Cancer Res. 2001;61:4526–4535. [PubMed] [Google Scholar]
- Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death—a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- O'Dwyer PJ, Moyer JD, Suffness M, Harrison SD, Jr, Cysyk R, Hamilton TC, et al. Antitumor activity and biochemical effects of aphidicolin glycinate (NSC 303812) alone and in combination with cisplatin in vivo. Cancer Res. 1994;54:724–729. [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Traish AM, Mercurio F, Sonenshein GE. Roles of IKK kinases and protein kinase CK2 in activation of nuclear factor-kappaB in breast cancer. Cancer Res. 2001;61:3810–3818. [PubMed] [Google Scholar]
- Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;1999:RE1. doi: 10.1126/stke.1999.5.re1. [DOI] [PubMed] [Google Scholar]
- Schwartz PE. Current diagnosis and treatment modalities for ovarian cancer. Cancer Treat Res. 2002;107:99–118. doi: 10.1007/978-1-4757-3587-1_4. [DOI] [PubMed] [Google Scholar]
- Shishodia S, Aggarwal BB. Nuclear factor-kappaB: a friend or a foe in cancer? Biochem Pharmacol. 2004;68:1071–1080. doi: 10.1016/j.bcp.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Tamatani T, Azuma M, Aota K, Yamashita T, Bando T, Sato M. Enhanced IkappaB kinase activity is responsible for the augmented activity of NF-kappaB in human head and neck carcinoma cells. Cancer Lett. 2001;171:165–172. doi: 10.1016/s0304-3835(01)00611-5. [DOI] [PubMed] [Google Scholar]
- Yang J, Richmond A. Constitutive IkappaB kinase activity correlates with nuclear factor-kappaB activation in human melanoma cells. Cancer Res. 2001;61:4901–4909. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.