

Abstract
A major burden in the treatment of ovarian cancer is the high percentage of recurrence and chemoresistance. Cancer stem cells (CSCs) provide a reservoir of cells that can self-renew, maintain the tumor by generating differentiated cells (non-stem cells) which make up the bulk of the tumor and may be the primary source of recurrence. We describe the characterization of human ovarian cancer stem cells (OCSCs). These cells have a distinctive genetic profile that confers them with the capacity to recapitulate the original tumor, proliferate with chemotherapy, and promote recurrence. CSC identified in cells isolated form ascites and solid tumors are characterized by: CD44+, MyD88+, constitutive NFκB activity and cytokine and chemokine production, high capacity for repair, chemoresistance to conventional chemotherapies, resistance to TNFα-mediated apoptosis, capacity to form spheroids in suspension, and ability to recapitulate in vivo the original tumor.
Chemotherapy eliminates the bulk of the tumor but it leaves a core of cancer cells with high capacity for repair and renewal. The molecular properties identified in these cells may explain some of the unique characteristics of CSCs that control self-renewal and drive metastasis. The identification and cloning of human OCSCs can aid in the development of better therapeutic approaches for ovarian cancer patients.
Keywords: ovarian cancer stem cells, NFκB, toll like receptors, MyD88, ovarian cancer, chemoresistance, inflammation
Introduction
Epithelial ovarian cancer (EOC) is the fourth leading cause of cancer-related deaths in women in the United States and the leading cause of gynecologic cancer deaths. Approximately 80% of patients with primary disease respond to surgery and chemotherapy,1 however, the number of responders decreases to ∼15% for recurrent cancers.2-4 Individuals who succumb to advanced-stage ovarian cancer inevitably become refractory to chemotherapy, resulting in disease progression and death.
Cancers often arise from normal tissues in the skin, gut and reproductive organs (ovary, endometrium, breast) where constant proliferation is required to ensure a continuous supply of newly differentiated cells. Replacement of the mature cells is accomplished by a highly orchestrated process in which a relatively small population of self-renewing adult stem cells gives rise to progenitor cells, which undergo limited rounds of mitotic division prior to terminal differentiation.5 In the cancer tissue, this population of long-lived cells with extraordinary expansion potential has been called tumor-initiating cells or cancer stem cells (CSCs).
Current evidence suggests that CSCs are the putative mediators of chemotherapy resistance and tumor progression.6,7 It is thought that CSCs are able to survive conventional treatments, which usually target fast dividing cells, and give rise to recurrent tumors that are more chemo-resistant and more aggressive.8,9
CSCs are characterized by: (i) capacity to self-renew; (ii) ability to give rise to a heterogeneous progeny of cells; and (iii) ability to modulate and balance its own differentiation and self-renewal capacities according to genetic constraints and environmental stimuli.5,10
CSCs were originally identified in leukemia,11,12 and more recently in solid tumors.10,13-18 In ovarian cancer, Bapat et al.19 reported the identification of cells with the characteristics of CSCs. These cells were obtained from a single clone, which arise from a culture isolated from malignant ovarian cancer ascites.
We report the molecular characterization of ovarian cancer stem cells (OCSC). These cells are CD44-positive (CD44+), as recently described,20 and are present in primary and metastatic tumors, as well as in malignant ascites. OCSCs are characterized by a unique genetic profile that confers the distinctive characteristics of cancer stem cells in terms of their tumorigenicity, resistance to chemotherapy, differentiation potential both in vitro and in vivo, and the capacity to promote a pro-inflammatory microenvironment.
Results
Prevalence of CD44+ cells in EOC tumors and correlation with patient survival
CD44 has been extensively reported as a marker that can enrich for CSCs in both hematologic and solid tumors.21 Recently, its specific correlation with ovarian CSCs has been established.20 Our first objective was to determine the prevalence of CD44+ cells in paraffin-embedded EOC tumor sections, which were obtained prior to the initiation of chemotherapy. Immunohistochemical staining showed positive reactivity in all of the 147 patient samples tested. The percentage and intra-tumoral distribution of the CD44+ cells, however, varied between patients. CD44+ cells were observed either as clusters or as single cells surrounded by CD44- cells (Fig. 1A) or as clusters invading blood vessels (Fig. 1B).
Figure 1.
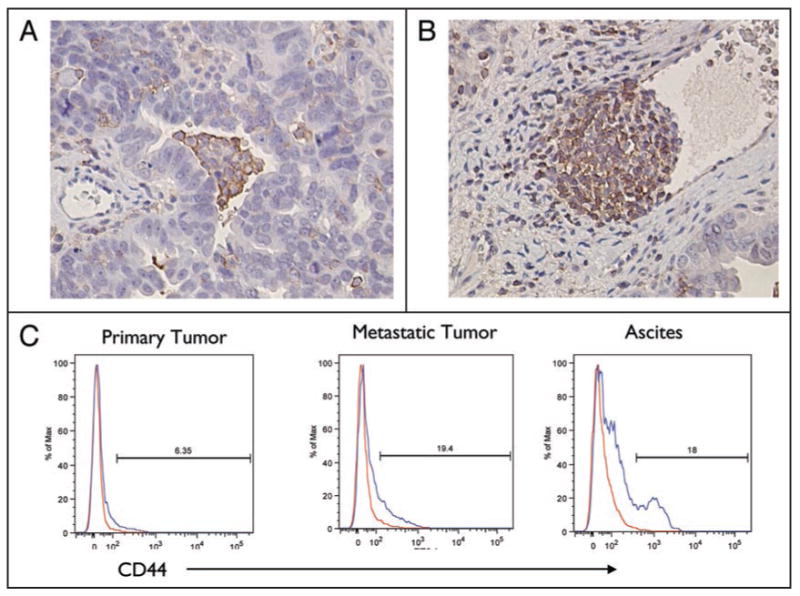
Localization of CD44+ cells in EOC tissue sections. (A and B) Five μm paraffin sections of EOC tumors were evaluated for CD44 expression by IHC. Immunoreactive cells were identified in the tumors as cluster cells surrounded by negative cells (A) or invading blood vessels (B). (C) Flow cytometry analysis for the expression of CD44 in EOC cells. Representative figure of cells isolated from primary tumor, metastatic tumor and ascites of the same patient analyzed for CD44 content. (Representative figure from 30 patients).
We also compared the percentage of CD44+ cells in individual patients when primary and/or metastatic tumor tissue and ascites samples were available. Flow cytometry analysis of cells obtained from patients with newly diagnosed ovarian cancer showed that CD44+ cells were found in both primary and metastatic sites, as well as in ascites. A higher percentage of CD44+ were observed in metastasis and ascites. Figure 1C shows a representative patient with 6.3%, 19% and 18% CD44+ cells in primary tumor, metastatic tumor and ascites, respectively.
CD44+ EOC cells form self-renewing spheroids in vitro and differentiated tumors in vivo
CD44 was reported to be a cell surface marker present in the population of EOC cells that formed spheroids in vitro.20 These spheroids were obtained from cultures grown under special conditions that selected for cells with self-renewing potential. Our next objective was to determine if CD44+ cells could enrich for cells with “stemness” potential directly from patient samples and without prior selection. Patients' ascites contain cell clusters that usually remain non-adherent in culture (Fig. 2A). CD44+ cells isolated from ascites and incubated in a suspension system consisting of a glass tube in continuous rotation or in bacteriologic dishes to prevent adherence formed clusters in 48 h (Fig. 2B and C) and compact spheroid in 4 days (Fig. 2D–F). Flow cytometry analysis revealed that after 72 h of spheroid formation, 91% of the cells remained CD44+ (Fig. 2G and H). This percentage of CD44+ cells was maintained for over 20 passages suggesting that they have the capacity to self-renew. On the other hand CD44-cells do not form spheroids and do not grow under glass suspension conditions.
Figure 2.

CD44+ EOC cells form compact spheroids in vitro. (A) EOC cells freshly isolated from malignant ascites are characterized by both single cell suspension and cell clusters. (B and C) Isolated clones of CD44+ EOC cells incubated in glass in continuous rotation form cell clusters within 24–48 h, compact spheroids (D and E), and tumor-like bodies in 4 days (F and G). Flow cytometry analysis for CD44 on spheroids (H).
We then evaluated whether CD44+ EOC cells could form tumors in a xenograft mouse model that recapitulates the original tumor phenotype. Thus, CD44+ from either ascites or tumor tissue were sorted and established as separate cultures as described in Material and Methods. 1 × 106 CD44+ EOC cells were suspended in a Matrigel™ mixture (1:1) and injected subcutaneously (s.c.) in NCR nude mice. Once tumors were established, cells were isolated and evaluated for CD44 expression by flow cytometry. Figure 3A shows a representative result from a mouse xenograft. An injection of pure CD44+ EOC cells is able to form tumors in 6–8 weeks characterized by ∼10% CD44+ and ∼90% CD44- cells (inset a and d). Additional passages in mice of isolated pure CD44+ cells resulted in similar tumor characteristics suggesting its differentiation in vivo (inset b, c and e). Histology of the resulting tumors was observed between passages (inset f and g), and have a striking morphological and histological similarity to the original patient tumor, from which the CD44+ cells were isolated (Fig. 3B, inset a–d).
Figure 3.

Tumorigenic potential of CD44+ EOC cells in NCr nude mice. (A) CD44+ cells were sorted and injected s.c. in Matrigel™ to NCR Nude mice (a and b). Resulting tumor was analyzed for CD44 using flow cytometry (c). CD44+ cells from the resulting tumor were again sorted and injected in another mouse and re-analyzed for CD44 (d and e). Note that both tumors have similar phenotype in terms of CD44 positivity and histology as demonstrated by the H&E staining (f and g). (B) Morphological and histological similarities between patient tumor (a and c) and the resulting mice tumor following injection of isolated CD44+ EOC cells (b and d). (C) Tumorigenic potential of CD44+ spheroids in NCR nude mice. CD44+ EOC cells in compact spheroids were injected s.c. in NCR nude mice without Matrigel™ (a and b) and the resulting tumors were analyzed for CD44 by flow cytometry (c). Note that the resulting tumor had similar morphology (d) as that obtained from monolayer cells shown in Figure 3A (inset f and g). (D) Spheroids were injected s.c. (insert a); inspection of tumors obtained six days after injection revealed hypervascularity (insert b); magnification of b shows small blood vessels and capillaries infiltrating the tumor (insert c); histological analysis of the tumor showed presence of numerous blood vessels (insert d). (E) Carcinomatosis resulting from CD44+ EOC spheroids in NCR nude mice. (a) seven to ten days after injection of spheroids, metastatic sites can be observed; white arrows show the tumor sites. (b) Twenty one days after injection. Note the polypoid morphology of tumors in the abdominal cavity.
To test for the tumorigenic potential of the spheroids obtained from CD44+ cells, two systems of implantation were implemented, the classic s.c. model, without the use of Matrigel™, and the intra-peritoneal (i.p.) model, which better resembles EOC in humans. Spheroids (containing ∼1 × 106 CD44+ cells) were injected s.c. in a total volume of 100 μl and i.p. in a total volume of 200 μl. Twenty days after injection, the s.c. tumors had a volume of 6 cm3. Flow cytometry analysis revealed that CD44+ cells represent only 10–15% of the total tumor cell population (Fig. 3C, inset a–d) suggesting its differentiation in vivo. Moreover, the tumors were characterized by high vascularity, which was evident as early as 6 days post-inoculation. High neovascularization was evidenced in both macroscopic (Fig. 3D, inset b and c) as well as microscopic (Fig. 3D, inset d) analysis.
In the i.p. model, tumors were found in the mesenterium, peritoneum, liver, pancreas, stomach, and spleen (Fig. 3E, inset a). Twenty-one days post spheroid injection, a disseminated carcinomatosis infiltrating the organs was observed (Fig. 3E, inset b). In addition, these animals developed malignant ascites containing cells which could be isolated and further propagated (data not shown).
Differential gene expression profile between CD44+ and CD44- EOC cells
Once we confirmed the presence of these two cell populations in our EOC samples and demonstrated the self-renewing and stemness properties of CD44+ cells in our system, our next objective was to further characterize its molecular phenotype. Thus, we determined the global gene expression of CD44+ EOC cells isolated from three individual patients and compared its profile with the CD44- cell population. Ten percent of the 24,000 evaluated genes were differentially expressed between the two cell populations. Figure 4A shows the heat map, which identifies the top 25 differentially expressed genes. Gene ontology analysis revealed that the differentially expressed genes belong to gene families associated with: (1) control of cell death and apoptosis; (2) signal transduction; (3) transcription regulation; and (4) control of cell differentiation, to name a few (Suppl. Table 1). The differential expression is reported as differences between CD44 negative vs. CD44 positive EOC cells.
Figure 4.

Differential Gene expression between CD44+ and CD44- EOC cells. (A) Dendogram depicting the differential gene expression between CD44 positive and CD44 negative EOC cells. The figure shows the top 25 genes differentially expressed between the two groups. Note the remarkable similarity observed between the cells of the same group. (See Suppl. Table 1 for list of genes). (B) Western blot analysis for CD44, CK-18 and β-catenin expression in representative CD44+ and CD44- EOC cells. Figure is a representative experiment for the validation of microarray results. OCSC = ovarian cancer stem cells. (C) Differential MyD88 expression between CD44+ and CD44- EOC cells. Note that MyD88 is expressed only in CD44+ cells while TLR4 is ubiquitously expressed. Representative figure of at least 20 patients. (D) CD44+ and CD44- EOC cells were either left untreated or treated with 10 μg/ml LPS for 48 h and cytokine/chemokine levels measured in supernatant using xMAP technology. Note the constitutive cytokine production in CD44+ but not CD44- EOC cells and the increase in cytokines levels following LPS treatment. (Complete list of the cytokines produced by CD44+ cells is included in Table 1). (E) CD44+ and CD44- EOC cells were transfected with a plasmid containing the firefly luciferase gene under the control of two NFκB binding sites. Luciferase activity was measured during a 12 h-period. Note the cyclic NFκB activity observed in CD44+ cells but lacking in CD44- cells. Representative study from at least 6 cell lines.
The results from the gene array established the optimal separation between the two cell populations (154-fold difference in expression between the CD44- and CD44+ cells); which correlates with the Flow cytometry findings. This was further confirmed at the protein level by western blot analysis (Fig. 4B).
CK18 is another gene differentially expressed (27-fold higher in the CD44+ cell population) and indeed, evaluation of CK18 by western blot analysis confirmed the high levels of CK18 expression in CD44+ cells but not in CD44- cells (Fig. 4B).
Similarly, β-catenin, an important component of the WNT pathway was found, in the gene array, to be higher in CD44+ cells (2.6 fold). β-catenin expression was also detected only in CD44+ cells but not in CD44- EOC cells (Fig. 4B).
CD44+ EOC cells have a functional TLR4/MyD88 pathway
Our next objective was to characterize the functionality of some of the differentially expressed genes and their potential role in cancer development.
Toll Like Receptors (TLRs), especially the TLR4 signaling pathway, have a critical role in the control of infection, tissue renewal and repair22,23 and may be associated with tumor formation.24 Families of genes involved in the TLR pathway are also shown to be differentially expressed between CD44+ and CD44- EOC cells. One of these genes is Myeloid Differentiation Factor 88 (MyD88; 10-fold upregulation in CD44+ cells). MyD88 is a critical component of the TLRs pathway and its activation leads to downstream activation of the NFκB signaling pathway.25 Thus, we evaluated whether CD44+ EOC cells express TLR4 and MyD88. TLR4 expression was observed in all EOC cells tested, however, as shown in the gene array, the signaling adaptor protein MyD88 was expressed only in CD44+ EOC cells, suggesting that the TLR4 pathway may be functional only in the CD44+ cell population (Fig. 4C).
We then treated CD44+ and CD44- cultures with the TLR4 ligand, LPS and evaluated the changes in their secreted cytokine profile. A significant increase in IL-6, IL-8, MCP-1 and GROα was observed only in CD44+ and not CD44- EOC cells (Fig. 4D), confirming the functionality of the TLR4-MyD88 pathway in only the CD44+ cell population.
Interestingly, when CD44+ cells were treated with paclitaxel, a known ligand for TLR4,26 we saw a significant increase in NFκB activity (Suppl. Fig. 2A). This response was not observed in the CD44- cells, which instead undergo apoptosis following paclitaxel treatment (Suppl. Fig. 2B). Similarly, TNFα treatment not only enhanced NFκB activity and promoted cytokine production in CD44+ EOC cells (Suppl. Fig. 3A and B), but also induced apoptosis in CD44- cells as demonstrated by a significant increase in the levels of active caspase-3 (Suppl. Fig. 3C). All of this data suggests that MyD88 expression might play a critical (crucial) role on the differentiation process and response to environmental factors.
CD44+ EOC cells are characterized by constitutive NFκB activity and constitutive cytokine production
The process of tissue repair is accomplished by production of cytokines, chemokines and growth factors, which enhance cell proliferation, regulate immune response and promote neovascularization. Interestingly, genes associated with inflammation are also highly expressed in CD44+ cells, including Interleukin-1betaβ (IL-1β; 52 fold) Interleukin-8 (IL-8; 160 fold), and Interleukin-6 (IL-6; 33 fold). We then examined the cytokine/chemokine profile of the cell culture supernatants from CD44+ and CD44- EOC cells. We found significant levels of IL-1ββ, IL-8 and IL-6 in supernatants from CD44+ EOC cells but not in CD44- cells (Suppl. Table 2), confirming the findings from the array.
In addition we observed high levels of MCP-1, MIP-1α, RANTES, GROα, GM-CSF, G-CSF and MIF. Conversely, none of these cytokines, with the exception of MIF, were detected in CD44- cells (Suppl. Table 2).
Since the NFκB pathway has been shown to play a central role in the regulation of cytokine production, particularly those involved in inflammation,27 we examined NFκB activity in the ovarian cancer cells. Using a luciferase reporter system inducible by active NFκB28 we found that CD44+ EOC cells have constitutive NFκB activation characterized by cyclic changes during a 12-hour time course. In contrast, in CD44- EOC cells, NFκB activity remained constant during the same time period (Fig. 4E). The constitutive NFκB activity observed in CD44+ cells may explain the high levels of pro-inflammatory cytokines produced by CD44+ cells.
CD44+ EOC cells are chemo-resistant
The family of genes that regulate cell death processes, including apoptosis, is one of the sets of genes identified to be differentially expressed between CD44+ and CD44- EOC cells. It is well accepted that most chemotherapeutic agents induce cancer cell death by activating the apoptotic pathway and that chemoresistance is a result of a cancer cell's resistance to activate apoptosis. Indeed, chemoresistance is one of the major obstacles in the treatment of EOC patients. Therefore, we determined how CD44+ EOC cells will respond to therapeutic doses of Paclitaxel and Carboplatin, and compared this response to that of CD44- cells. CD44+ and CD44- cells were sorted from either dissociated tumor tissue or ascites from patients and treated with different doses of the drugs for 24 h. As shown in Figure 5A, CD44- cells were sensitive to treatment. However, CD44+ cells obtained from the same patient were resistant and even proliferated in the presence of the drugs. Similar results were obtained with all the samples evaluated (Suppl. Fig. 1).
Figure 5.
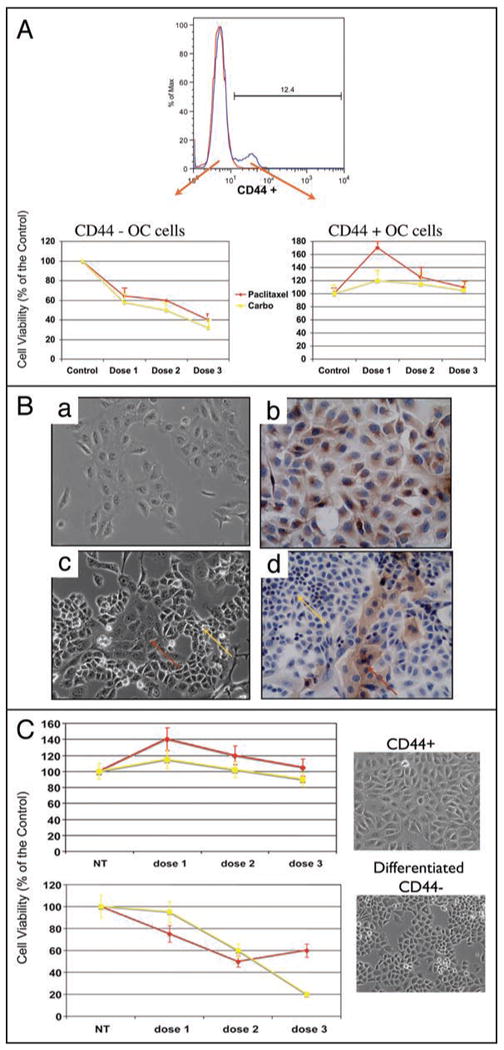
CD44+ cells are chemoresistant to Paclitaxel and Carboplatin. (A) CD44+ and CD44- cell populations were sorted from the same sample and treated in vitro with Paclitaxel (0.2, 2, 20 μM) and Carboplatin (50, 100, 200 ug/ml) for 24 h. Note the increase in viability in CD44+ cells in concentrations that induce cell death in CD44- cells. Representative figure of 30 patients evaluated. (B) CD44+/MyD88+ EOC cells differentiate into CD44-/MyD88- cells in vitro. CD44+ cells (a) were passed in low density until a change in morphology was observed. The heterogeneous culture could be observed after 6 passages (c). Inserts (b and d) are IHC for MyD88 of (a) and (c), respectively. (C) Differential response to carboplatin and paclitaxel between CD44+ EOC cells and the differentiated CD44- EOC cells. The culture was treated in vitro with Paclitaxel (dose 1 = 0.2 μM, dose 2 = 2 μM and dose 3 = 20 μM) or Carboplatin (dose 1 = 50 μug/ml, dose 2 = 100 ug/ml and dose 3 = 200 ug/ml) for 24 hours before cell viability was tested. NT = no treatment control. Note that the differentiated culture became sensitive to the treatment of these drugs.
In vitro differentiation of CD44+ EOC change their response to chemotherapy
Results from the in vivo studies suggest that CD44- cells originate from the CD44+ cells. To further test this hypothesis, we evaluated whether we could replicate the in vivo differentiation using an in vitro system. Pure CD44+ EOC cells obtained from either dissociated tumor tissue or ascites from patients (Fig. 5B, inset a and b) were passed several times in low concentrations until we could see a significant change in cell morphology. After about six passages in a span of 6 weeks, the culture was comprised of a heterogeneous cell population (Fig. 5B, inset c and d). While the original/pure CD44+ cells have a large cytoplasm (Fig. 5B, insert a), characteristic of cells with low replicative rate, that expresses MyD88 (Fig. 5B, inset b), the new cell population had a much smaller cytoplasm and was MyD88-negative (Fig. 5B, inset c and d). Flow cytometry analysis of the differentiated cultures showed that the cell population was composed of less than 10% CD44+ cells (data not shown). Furthermore, as observed with the cells isolated from the patients, the differentiated CD44- cells responded to carboplatin and paclitaxel treatment while CD44+ cells reminded resistant (Fig. 5C).
Discussion
We demonstrated that OCSCs have a unique molecular profile that may explain their capacity to recapitulate the original tumor, promote a pro-inflammatory microenvironment, and proliferate with chemotherapy. In addition, we report a distinctive phenotype characterized by CD44+/MyD88+, constitutive NFκB activity, cytokine production and pan-chemo-resistance.
CSCs are defined as cells within the tumor that possess the capacity to self-renew and to cause the heterogeneous lineage of cancer cells that comprise the whole tumor.5,29 CSCs have been postulated as the potential source of tumor formation as well as of recurrence and chemoresistance.5,30 It is therefore important to identify and characterize these cells in order to develop new ways of diagnosis and therapy.
Recent publications have identified CD44 as a potential marker for the identification of cancer stem cells in breast and ovarian cancer.17,20 Indeed, we demonstrate that CD44 is able to enrich for cells that can recapitulate the morphology of the original tumor; and give rise to a tumor with both CD44+ and CD44- cells, suggesting that they can differentiate and self-renew. The process of differentiation was also observed in vitro wherein 100% of CD44+ cells seeded at very low density eventually gave rise to cultures that looked morphologically different. In addition, whereas the original CD44+ cell culture doubles every 36 h, the new CD44- cell culture doubles every 16 h. More importantly, while CD44+ cells (slow dividing cells) are chemoresistant, the new CD44- cell cultures (fast dividing cells) became responsive to chemotherapy. This model represents a simple but unique system to study the process of CSC differentiation. It is possible that this model mimics a “repair condition” where the continuous passaging in low density becomes a signal for repair/proliferation, and therefore, accelerates the process of cell division and induces differentiation into faster-dividing cells.
We also tested the capacity of CD44+ EOC cells to grow independently of surface attachment, another characteristic of CSCs.31,32 CD44+ (but not CD44-), EOC cells can form spheroids and structures that resemble solid tumors. A striking property of these cells is their capacity to form tumors in vivo, independent of MatriGel™ support, reminiscent of carcinomatosis found in patients with ovarian cancer.
The genetic profile of the CSCs that we identified in this study is associated with some of the hallmarks of cancers including inflammation, and resistance to apoptosis.
A novel characteristic observed in the CD44+, but not in CD44- EOC cells, is the constitutive NFκB activity and secretion of pro-inflammatory cytokines. NFκB regulates numerous genes that function in diverse processes, including inflammatory responses, immune system development, apoptosis and tissue development. Tumor initiation and progression largely depend on the production of pro-inflammatory cytokines which serve as growth factors for the neoplastic cells.33,34 The cyclic activity of NFκB in the CD44+ EOC cells may ensure the continuous production of cytokines such as the observed IL-6, MCP-1 and IL-1, which can create a pro-inflammatory environment that might enhance resistance to apoptosis, boost proliferation and trigger differentiation.33 Furthermore, the observed chemokine, IL-8, is an angiogenic factor and can, therefore, induce the formation of new blood vessels.35
Tumors, like normal tissues, have the ability of compensatory proliferation in response to injury and tissue damage (including necrosis and apoptosis). This tissue repair process has been reported to depend on TLR4-MyD88 signaling.36 TLR4-MyD88 signaling play a critical role to maintain intestinal epithelial homeostasis in response to gut injury, and both TLR4 and MyD88 knockout mice displayed impaired compensatory proliferation and increased apoptosis.22,23,36 In addition, it has been suggested that TLR signaling, mainly that of TLR4, is involved in sterile inflammatory responses induced by products of tissue damage.24,37 CD44+ EOC cells express TLR4 and MyD88 and respond to TLR-4 ligands by activating NFκB, suggesting that the TLR4 pathway may play a critical in the process of repair/differentiation triggered by the cancer stem cells. Indeed, the presence of necrotic centers in tumors has been associated with poor prognosis due to the presence of immune infiltrates, which creates a pro-inflammatory environment. Therefore, it is reasonable to hypothesize that the damage induced by chemotherapeutic drugs could also initiate a compensatory repair process, which includes all the cellular and molecular characteristics associated with sterile inflammatory response (Fig. 6A and B).
Figure 6.

Model of tumor composition and response. (A) Ovarian cancer tumors are made of heterogeneous population consisting of cancer stem cells (CD44+, MyD88+, IKKββ) progenitor cells (CD44-, MyD88+, IKKβ) and the bulk of the tumor made by fast dividing cells (CD44-, MyD88-, IKKβ). (B) Upon chemotherapy the fast dividing cells are eliminated leaving the stem cells, which will repair and recreate the tumor as a recurrence.
The specific gene expression profile observed in the CD44+ EOC cells revealed that they represent defined cell population with distinct biologic characteristics. In addition to the gene profile studies, we have also performed miRNA arrays and likewise found a specific miRNA profile that distinguishes CD44+ from CD44- EOC cells (Alvero et al. in preparation). These differences may be associated with some of the unique characteristics observed in CSCs. For example, we recently reported that miR199-A regulates IKKβ and therefore the NFκB pathway. Indeed we found that CD44+ EOC cells have very low levels of miR-199A compared to CD44- cells.38
In summary, we have further confirmed the stemness potential of CD44+ EOC cells and report its specific molecular signature. The establishment of these cells in culture can aid in the further understanding of CSC biology and possibly the development of better therapeutic approaches for ovarian cancer patients.
Material and Methods
Patients and samples
Malignant ovarian ascites samples were obtained from stage III/IV ovarian cancer patients. Tumor samples were collected at the time of surgery under sterile conditions; one aliquot was processed for cell preparation and a second aliquot snap frozen in liquid nitrogen for additional use. All patients signed consent forms, and the use of patient samples was approved under Yale University's Human Investigations Committee (HIC #0606001587).
Cell lines and culture conditions
Primary EOC cells were isolated either from malignant ovarian ascites or from ovarian tumors and cultured as previously described (Flick et al. 2004; Kamsteeg et al. 2003). EOC cells were grown in RPMI plus 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA) at 37°C in a 5% CO2 atmosphere. Purity of the EOC cells was 100% as determined by immunostaining for cytokeratin antigen.
After isolation from ascites or tumors, OCSCs were maintained as low growing rate cultures and kept at minimum 80% confluence. The cells were grown in 199/155 media plus 4 ng/ml EGF and 5% serum.
NFκB activity
NFκB activity was measured with a luciferase reporter construct, pBII-LUC containing two B sites before a Fos essential promoter (a gift from Dr. S. Ghosh, Yale University). Cells were transiently transfected with pBII-LUC using the FuGENE 6 Transfection Reagent (Roche Applied Science, Indianapolis, IN) following the manufacturer's instructions. Luciferase activity was measured using the Luciferase Assay System (Promega, Madison, WI) according to the manufacturer's protocol. Briefly, 10 ug of each protein sample in a total volume of 100 ul was mixed with 20 ul of the Luciferase Assay Reagent, and luminescence measured using TD 20/20 Luminometer (Turner Designs, Sunnyvale, CA). Relative activity was calculated based on readings measured from untreated cells after subtracting blank values. Each sample was done in triplicates.
Cytokine profiling
Cytokine profiling was performed from protein extracts or culture supernatants using the Bio-Rad 100 IS System (Bio-Rad, Hercules, CA) Assays were performed using Beadlyte® Human Multi-Cytokine Beadmaster™ Kit according to the manufacturer's instructions. In summary, 50 ul of standard or sample plus 25 ul of microparticle mixture was added to 96-well plates, and incubated at room temperature on an orbital shaker (600 rpm) for 2 hours. The plates were then washed twice with Beadlyte Cell Signaling Assay Buffer (Upstate, Charlottesville, VA) and the microparticles resuspended in 75 ul of the Assay Buffer. 25 ul of biotinylated detection Ab was added to each well. After 1.5 hours of incubation at RT a 1:20 Streptavadin-PE Assay buffer mixture was prepared and 25 ul added to each well and incubated for an additional 0.5 hour. The plates were washed twice with Assay Buffer and resuspended in 125 ul of Sheath buffer. The plates were then read on the Bio-Plex Reader.
Supplementary Material
Acknowledgments
This study was supported in part by grants from NCI/NIH 1R01CA118678-01A2 and 1R01CA127913-01A2 and the Janet Burros Memorial Foundation.
Thanks to Lisa Baker and Martha Luther for consenting and enrolling patients for this study. Special thanks to Dr. Fortune Kohen for reviewing the manuscript.
Footnotes
Note: Supplementary materials can be found at: www.landesbioscience.com/supplement/AlveroCC8-1-Sup.pdf
References
- 1.Schwartz PE. Current diagnosis and treatment modalities for ovarian cancer. Cancer Treat Res. 2002;107:99–118. doi: 10.1007/978-1-4757-3587-1_4. [DOI] [PubMed] [Google Scholar]
- 2.Onnis A. The management of ovarian cancer: an update. Eur J Gynaecol Oncol. 1997;18:157–60. [PubMed] [Google Scholar]
- 3.Torri V, Harper PG, Colombo N, Sandercock J, Parmar MK. Paclitaxel and cisplatin in ovarian cancer. J Clin Oncol. 2000;18:2349–51. [PubMed] [Google Scholar]
- 4.Markman M, Kennedy A, Webster K, Peterson G, Kulp B, Belinson J. Combination chemotherapy with carboplatin and docetaxel in the treatment of cancers of the ovary and fallopian tube and primary carcinoma of the peritoneum. J Clin Oncol. 2001;19:1901–5. doi: 10.1200/JCO.2001.19.7.1901. [DOI] [PubMed] [Google Scholar]
- 5.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–5. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Hermann PC, Huber SL, Heeschen C. Metastatic cancer stem cells: a new target for anti-cancer therapy? Cell Cycle. 2008;7:188–93. doi: 10.4161/cc.7.2.5326. [DOI] [PubMed] [Google Scholar]
- 7.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 8.Hambardzumyan D, Becher OJ, Holland EC. Cancer stem cells and survival pathways. Cell Cycle. 2008;7:1371–8. doi: 10.4161/cc.7.10.5954. [DOI] [PubMed] [Google Scholar]
- 9.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 10.Friel AM, Sergent PA, Patnaude C, Szotek PP, Oliva E, Scadden DT, Seiden MV, Foster R, Rueda BR. Functional analyses of the cancer stem cell-like properties of human endometrial tumor initiating cells. Cell Cycle. 2008;7:242–9. doi: 10.4161/cc.7.2.5207. [DOI] [PubMed] [Google Scholar]
- 11.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 12.Akashi K, Reya T, Dalma-Weiszhausz D, Weissman IL. Lymphoid precursors. Curr Opin Immunol. 2000;12:144–50. doi: 10.1016/s0952-7915(99)00064-3. [DOI] [PubMed] [Google Scholar]
- 13.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 14.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 15.Collins AT, Maitland NJ. Prostate cancer stem cells. Eur J Cancer. 2006;42:1213–8. doi: 10.1016/j.ejca.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 16.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 17.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–73. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025–9. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- 20.Zhang K, Wang Q, Xie Y, Mor G, Sega E, Low PS, Huang Y. Receptor-mediated delivery of siRNAs by tethered nucleic acid base-paired interactions. Rna. 2008;14:577–83. doi: 10.1261/rna.739308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong DJ, Segal E, Chang HY. Stemness, cancer and cancer stem cells. Cell Cycle. 2008;7 doi: 10.4161/cc.7.23.7104. [DOI] [PubMed] [Google Scholar]
- 22.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their Toll—the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27:225–33. doi: 10.1038/sj.onc.1210907. [DOI] [PubMed] [Google Scholar]
- 25.Akira S, Hoshino K. Myeloid differentiation factor 88-dependent and -independent pathways in toll-like receptor signaling. J Infect Dis. 2003;187:356–63. doi: 10.1086/374749. [DOI] [PubMed] [Google Scholar]
- 26.Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, Visintin I, Rutherford T, Mor G. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66:3859–68. doi: 10.1158/0008-5472.CAN-05-3948. [DOI] [PubMed] [Google Scholar]
- 27.Karin M, Cao Y, Greten FR, Li ZW. NFkappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 28.Leung CH, Grill SP, Lam W, Gao W, Sun HD, Cheng YC. Eriocalyxin B inhibits nuclear factor-kappaB activation by interfering with the binding of both p65 and p50 to the response element in a noncompetitive manner. Mol Pharmacol. 2006;70:1946–55. doi: 10.1124/mol.106.028480. [DOI] [PubMed] [Google Scholar]
- 29.Dingli D, Traulsen A, Pacheco JM. Stochastic dynamics of hematopoietic tumor stem cells. Cell Cycle. 2007;6:461–6. doi: 10.4161/cc.6.4.3853. [DOI] [PubMed] [Google Scholar]
- 30.Zhou J, Zhang Y. Cancer stem cells: Models, mechanisms and implications for improved treatment. Cell Cycle. 2008;7:1360–70. doi: 10.4161/cc.7.10.5953. [DOI] [PubMed] [Google Scholar]
- 31.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–37. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 32.Strizzi L, Abbott DE, Salomon DS, Hendrix MJ. Potential for cripto-1 in defining stem cell-like characteristics in human malignant melanoma. Cell Cycle. 2008;7:1931–5. doi: 10.4161/cc.7.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 34.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NFkappaB in cancer cells converts inflammation-induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 36.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu MT. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:1055–65. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 37.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 38.Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, Leiser A, Schwartz PE, Rutherford T, Mor G. Regulation of IKKbeta by miR-199a affects NFkappaB activity in ovarian cancer cells. Oncogene. 2008;27:4712–23. doi: 10.1038/onc.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.