

Abstract
Pandemics pose a more significant threat to pregnant women than to the nonpregnant population and may have a detrimental effect on the well being of the fetus. We have developed an animal model to evaluate the consequences of a viral infection characterized by lack of fetal transmission. The experiments described in this work show that viral infection of the placenta can elicit a fetal inflammatory response that, in turn, can cause organ damage and potentially downstream developmental deficiencies. Furthermore, we demonstrate that viral infection of the placenta may sensitize the pregnant mother to bacterial products and promote preterm labor. It is critical to take into consideration the fact that during pregnancy it is not only the maternal immune system responding, but also the fetal/placental unit. Our results further support the immunological role of the placenta and the fetus affecting the global response of the mother to microbial infections. This is relevant for making decisions associated with treatment and prevention during pandemics.
Pregnant women are more susceptible to the effects of microbial products (i.e., endotoxins) and were the most vulnerable subjects during the 1918 pandemic (influenza A subtype H1N1), with a mortality rate that ranged between 50 and 75% (1). Exposure to the virus during pregnancy may also have overt or subclinical effects that become apparent only over time.
Although substantial progress has been made in the understanding of the immunology of pregnancy, many unanswered questions remain, especially those associated with the susceptibility and severity of infectious agents of mothers and unborn children (2), (3).
Epidemiological studies have demonstrated an association between viral infections and preterm labor (4, 5) and fetal congenital anomalies of the CNS and the cardiovascular system (6–8). Although some viral infections during pregnancy may be asymptomatic (9), approximately one-half of all preterm deliveries are associated with histological evidence of inflammation of the placenta, termed acute chorioamnionitis (10), or chronic chorioamnionitis (10). Despite the high incidence of acute chorioamnionitis, only a fraction of fetuses have demonstrable infection (11). Most viral infections affecting the mother do not cause congenital fetal infection, and only in a small number of cases is the virus found in the fetuses (12–17), attesting to the unique ability of the placenta to act as a potent barrier with an immune-regulatory function that protects the fetus from systemic infection (10, 12, 18, 19).
Recent observations indicate that rather than acting as a mechanical barrier, the placenta functions as a regulator of the trafficking between the fetus and the mother (20–22). Fetal and maternal cells move in two directions (23, 24); similarly, some viruses and bacteria can reach the fetus by transplacental passage with adverse consequences (25). Although viral infections are common during pregnancy (26), transplacental passage and fetal infection appear to be the exception rather than the rule (27, 28; reviewed in Ref. 29 and subsequent references).
There is a paucity of evidence that viral infections lead to preterm labor (10, 19, 22–24); however, there are several areas of controversy and open questions. For example, what effects do subclinical viral infections of the decidua and/or placenta during early pregnancy have in response to other microorganisms, such as bacteria; and what is the effect of a subclinical viral infection of the placenta on the fetus?
The trophoblast is an important component of the placenta, and it is able to recognize and respond to microorganisms and their products through the expression of TLRs (30–32). TLRs are a family of innate immune receptors that have an essential role in the recognition of pathogen-associated molecular patterns (33–35). Trophoblasts are able to produce cytokines/chemokines and antiviral factors following TLR-3 ligation in vitro, suggesting the potentially active role of these cells in the control of viral infections (20, 36). Some of these receptors (chemokine and TLRs) may also function as viral receptors mediating viral recognition and entry into the trophoblast. Signaling through TLRs has been shown to induce murine γ-herpesvirus 68 (MHV-68) reactivation in vivo (37).
Herpesviruses are the most common cause of viral-related perinatal neurologic injury in the United States (38). However, among the eight known human herpesviruses, most reported adverse pregnancy and neonatal outcomes are the result of the HSVs (HSV-1 and HSV-2) and CMV (39) and usually occur due to a primary infection of the mother during the first trimester or infection of the infant during delivery. MHV-68 (murid herpesvirus 4 [NC_001826.2]) is a γ-herpesvirus of rodents that shares significant genomic colinearity with two human pathogens, EBV and Kaposi's sarcoma-associated herpesvirus (40). As in these two viruses, the effect of MHV-68 in pregnancy is unknown.
We developed a novel murine model to evaluate the role of viral infection in pregnancy and fetal development. Our data suggest that even in the absence of placental passage of the virus, the fetus could be adversely affected by an inflammatory response mounted in response to viral invasion of the placenta. Furthermore, we demonstrate that a viral infection in early pregnancy sensitizes the pregnant mother to the effects of bacterial products later on in gestation, and specifically, to premature labor. These data suggest that exposure to early viral infections may program the immune response of mother and fetus. Such observations have important consequences for understanding the potential risk of viral infections during pregnancy and the importance of adequate surveillance to prevent maternal mortality and subclinical fetal injury, leading to long-term consequences.
Materials and Methods
Virus culture
MHV-68 expressing GFP (provided by R. Sun, University of California, Los Angeles, CA) was passaged in NIH 3T3 cells with DMEM plus 10% FCS. After lysis, supernatant was harvested, filtered (0.45-μm pore), and titered by 2-fold serial dilutions. To determine virus load in infected mice, frozen homogenized tissues were minced and subjected to 10-fold serial dilutions, and endpoint titers were determined in NIH 3T3 cells by GFP (41, 42). A single virion or DNA copy was sufficient to show a positive result by plaque assay or PCR.
Animal procedures
C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME), and TLR-3 knockout (KO) was provided by R. A. Flavell (Yale University, New Haven, CT). Adult mice (8–12 wk of age) with vaginal plugs were infected i.p. at embryonic day (E) 8.5 postconception with either 1 × 106 PFU MHV-68 expressing GFP (in 200 μl vol) or DMEM (vehicle). Three or 9 d postinfection (dpi), animals were sacrificed, and organs were removed, fixed in 4% paraformaldehyde, and/or stored at −80°C. All animals were maintained in the Yale University School of Medicine Animal Facility under specific pathogen-free conditions. All experiments were approved by the Yale Animal Resource Committee.
Reagents and Abs
LPS (Escherichia coli O111:B4) was purchased from Sigma-Aldrich (St. Louis, MO). Lymphocyte separation media was purchased from MP Biomedicals (Solon, OH).
For NK and macrophage detection, biotinylated lectin Dolichos biflorus agglutinin from Sigma-Aldrich and rat anti-mouse F4/80 Ab (eBioscience, San Diego, CA) were used, respectively. Anti–NF-κB p65 mAb was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Dominant-negative (DN) TLR1 Toll/IL-1 receptor (TIR) and TLR2TIR, incapable of tranducing a signal after ligand binding, were purchased from InvivoGen (San Diego, CA). Bio-plex Pro custom 18-plex panel (catalogue M500FHB86U;171B6007M) for cytokine detection was purchased from Bio-Rad (Hercules, CA).
Cell lines
Human first trimester trophoblast HTR-8 cells were gifted from C. Graham (Queen's University, Kingston, Ontario, Canada). Human first trimester trophoblast 3A cells were stably transfected with DN TLR2 and TLR1 genes, as previously described (22). Following transfection, cells expressing the TLR1TIR (TLR1-DN) or the TLR2TIR (TLR2-DN) were selected with puromycin. Vector alone-transfected cells served as negative control.
Human trophoblast isolation
First trimester trophoblast cells were isolated and cultured, as described (43). Briefly, after washing, tissues were minced and incubated in PBS, 0.125% trypsin, and 30 U/ml DNase I for 1 h at 37°C. A 70-μm filtered suspension was layered on lymphocyte separation media (MP Biomedicals) and centrifuged. The interface layer was collected, washed, and resuspended with MEM. Cells were cultured in MEM with d-valine (Caisson Laboratories, North Logan, UT), 10% human serum, placed in a type IV collagen-coated plate (BD Biosciences, Franklin Lakes, NJ), and kept at 37°C/5% CO2.
Mouse embryonic fibroblast cell preparation
Embryos were harvested on days 11.5–13.5, and fibroblast cells were prepared according to the method described by Bowtell's laboratory (44–46). This is a standard method used for the preparation of supporting fibroblast cells for stem cell growth. Mice embryonic fibroblasts were propagated and maintained according to the 3T3 protocol (47).
Cytokine analysis
Cytokine concentration was determined, as previously described (48–50), using the cytokine multiplex assays from Bio-Rad. Briefly, wells of a 96-well filter plate were loaded with either 50 μl prepared standard solution or 50 μl cell-free supernatant and incubated on an orbital shaker at ±500 rpm for 2 h in the dark at room temperature. Wells were then vacuum washed three times with 100 μl wash buffer. Samples were then incubated with 25 μl biotinylated detection Ab at ±500 rpm for 30 min in the dark at room temperature. After three washes, 50 μl streptavidin-PE was added to each well and incubated for 10 min at ±500 rpm in the dark at room temperature. After a final wash, the beads were resuspended in 125 μl assay buffer for measurement with the LUMINEX 200 (LUMINEX, Austin, TX). The cytokines included in the Multiplex assay were as follows: IL-1β, IL-10, GM-CSF, IFN-γ, TNF-α, IL-1α, IL-6, IL-12p40, IL-12p70, G-CSF, KC, MIP-1α, RANTES, MCP-1, and MIP-1β.
Immunohistochemistry
After Ag retrieval with Retrievagen A (pH 6.0; BD Biosciences), macrophages were detected in paraffin-embedded murine uteri with rat anti-mouse F4/80 Ab at 1:20. NK cell detection was performed, as previously reported (31, 51).
For the localization of the p65 subunit of NF-κB, MHV-68–infected human trophoblast cells were fixed and incubated with the mouse anti–NF-κB p65 Ab. Slides were then incubated with Alexa Fluor546 anti-mouse IgG and counterstained with Hoechst 33342 dye (Molecular Probes, Eugene, OR).
Total RNA isolation
Total RNA was extracted using TRIzol, and cDNA was prepared using a Verso cDNA kit per manufacturer's protocol (Thermo Scientific, Wal-tham, MA).
Real-time PCR
Real-time PCR was performed in duplicate using SYBR Green (Invitrogen, Carlsbad, CA) in an ABI Prism 7500 (Applied Biosystems, Foster City, CA). cDNA sample (1 μl) was amplified with gene-specific primers using optimized PCR cycles. GAPDH was used as an endogenous control for relative comparison of human TLR-2, TLR-3, and TLR-4. GAPDH expression did not vary with treatments. TLR-2 forward, 5′-ATGCCTACT-GGGTGGAGAAC-3′; TLR-2 reverse, 5′-TGCACCACTCACTCACA-3′; TLR-3 forward, 5′-GTGCCGTCTATTTGCCA-3′; TLR-3 reverse, 5′-AG-TCTGTCTCATGATTCTGTTG-3′; TLR-4 forward, 5′-CAGCTCTTGGT-GGAAGTTGA-3′; TLR-4 reverse, 5′-GCAAGAAGCATCAGGTGAAA-3′; GAPDH forward, 5′-GAGTCAACGGATTTGGTCGT-3′; GAPDH reverse, 5′-GACAAGCTTCCCGTTCTCAGCC-3′.
The TLR-2, TLR-3, and TLR-4 cycle threshold value was analyzed using the ΔΔ cycle threshold Livak method (52).
Elisa
Serum from wild-type (wt) and TLR3 KO mice was analyzed for the presence of anti-viral IgM or IgG Abs. For coating of ELISA plates, MHV-68 GFP stocks were filtered through a 0.45-μm-pore membrane, and virions were centrifuged through a 5% sucrose cushion (SW28, 20,000 rpm for 75 min at 4°C). Virion pellets were resuspended in PBS containing 0.5% Triton X-100 and 0.5% FBS to achieve 5000× concentration. Maxisorb plates (Nunc-immuno plates) (Corning Life Sciences, Corning, NY) were coated with 91 μg virus protein/well, washed, blocked with 0.3 mg BSA, and incubated with serum from either wt or TLR3 KO with or without MHV-68 infection. After washing, goat anti-mouse IgG or IgM, HRP-conjugated Ab (Southern Biotechnology Associates, Birmingham, AL) was added at 1/4000 dilution in PBS/1% BSA for 2 h at room temperature, washed, and detected with tetramethylbenzidine substrate at 405 nm.
Statistical analysis
Data are expressed as mean ± SE for in vitro study and median ± first or third quartiles for in vivo study. Statistical significance (p < 0.05) was determined using either two-tailed unpaired Student t tests or Mann-Whitney U test for nonparametric data. Unless stated otherwise, all experiments were performed in duplicate.
Results
Maternal infection with MHV-68 does not induce preterm labor
Systemic administration of polyinosinic:polycytidylic acid [poly (I:C)] to pregnant mice induced preterm labor and delivery, and production of proinflammatory cytokines (53, 54). The inflammatory response to the TLR-3 ligand was found in the placenta at E17.5 (local response) as well as in the spleen (systemic response) (30, 55) characterized by upregulation of IL-6, IL-12p40, MCP-1, MIP-1β, growth-related oncogene-α, and RANTES. These observations indicated that the placenta was able to recognize and respond to viral products. To understand the effects of viral infection in pregnancy, we used MHV-68. C57BL/6 pregnant mice received 1 × 106 PFU MHV-68 i.p. on E8.5 of pregnancy, and were followed up to E17.5. Control animals received media as a placebo. This viral dose has previously been shown to infect mice organs and produce a systemic viral response (37).
Maternal infection with MHV-68 had no effect on pregnancy outcome, including litter size, weight, or gestational age at delivery (Fig. 1). We then evaluated whether the absence of viral infection to the placenta and decidua was responsible for the lack of effect in pregnancy outcome. To test this hypothesis, replicative viral loads (PFU/ml) were determined using a limiting dilution plaque assay on frozen tissue taken from the placenta and decidua (local), and spleen and lymph node (systemic and classical target organ for MHV-68) (56) from pregnant mice who had received MHV-68 on E8.5 of pregnancy and sacrificed 3 or 9 d after viral administration (dpi).
FIGURE 1.
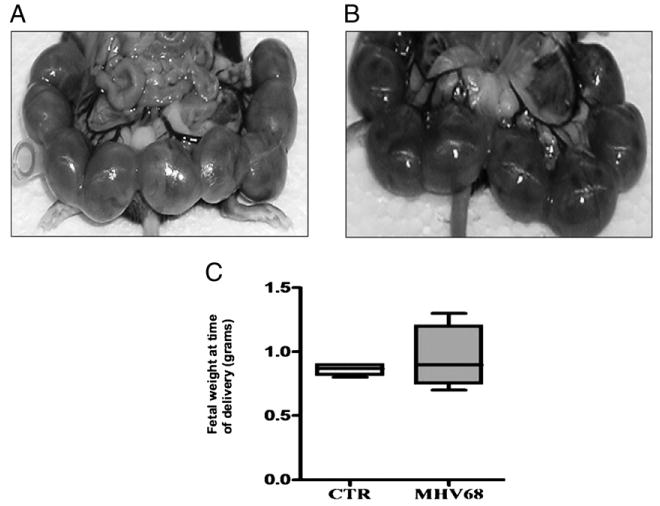
Pregnancy outcome in wt mice infected with MHV-68. Wt pregnant mice were infected i.p. with 1 × 106 PFU MHV-68 or vehicle at E8.5 and sacrificed at E17.5. A, Pups, uterus, and gestational sacs from wt treated with vehicle, and B, pups, uterus, and gestational sac from wt infected with MHV-68, showing no differences in gross anatomy. C, Fetal weight at the time of delivery. Note the lack of difference between the two groups. n = 6 mice per group.
Three days after viral administration, we observed a high viral load in the spleen and lymph nodes. Interestingly, viral titers in the decidua were significantly higher than those in the spleen (Fig. 2A). The placenta was also infected, but overall viral titers were lower than those in the spleen.
FIGURE 2.
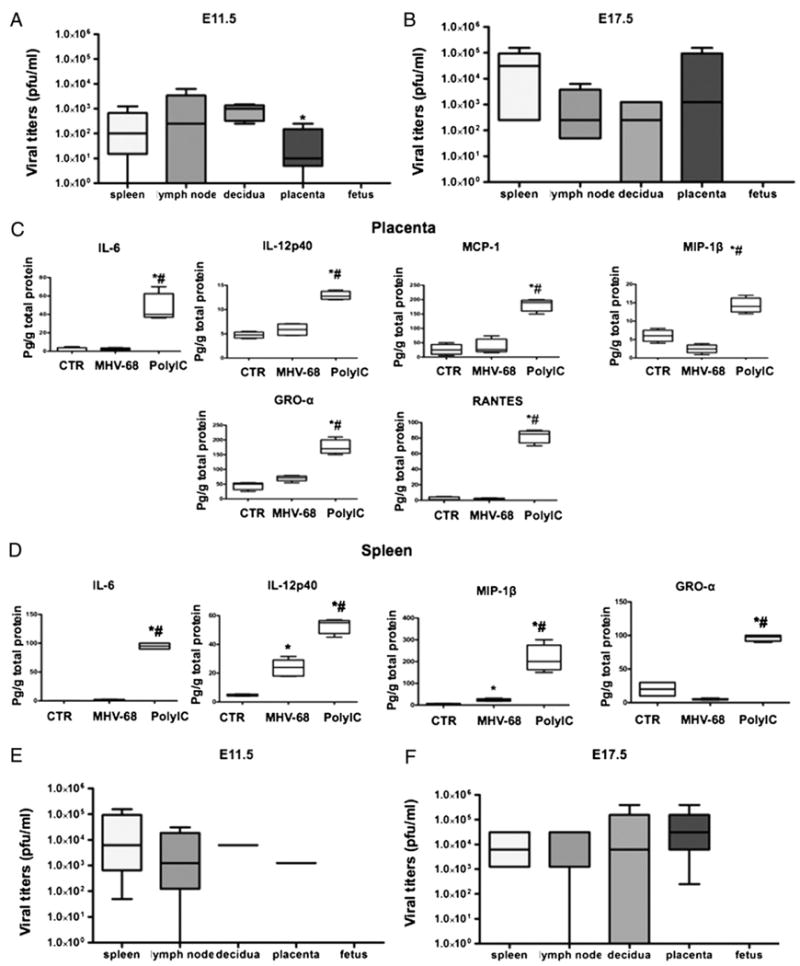
Effect of MHV-68 viral infection in pregnant mice. Viral titers as PFU/ml were determined in wt pregnant mice infected with MHV-68 (1 × 106 PFU) 3 d (E11.5; A) and 9 d (E17.5; B) postinfection. Viral titers were observed in lymph nodes, placenta, decidua, and spleen, but were absent in the fetuses. *p < 0.05, decidua versus spleen. Placenta (C) and spleen (D) cytokine profile was determined in wt pregnant mice treated with poly(I:C) or MHV-68 4 and 9 d postinfection, respectively. *p < 0.05, MHV-68 versus control; #p < 0.05, poly(I:C) versus MHV-68. E and F, Viral titers as PFU/ml were determined in TLR-3 KO pregnant mice infected with MHV-68: E, 3 d (E11.5), and F, 9 d (E17.5) postinfection. Note the high levels of viral titers in lymph nodes, placenta, decidua, and spleen, but absent in the fetuses. Bars show median ± SEM. n = 6 mice per group.
Nine days after viral administration, we observed a substantial increase in splenic viral titers (higher than 3 logs), a slight decrease in the decidua, and increasing viral titers in the placenta (Fig. 2B). Most notably, no viruses were detected in the fetus of infected mothers using the plaque assay or by PCR, even 9 d after viral administration (Fig. 2A and data not shown).
These results suggest that the viral load administered to the mice is able to infect all the organs, including placenta and decidua; however, in contrast to the effect observed with poly(I:C), the viral infection in the placenta and decidua did not seem to have an effect on pregnancy outcome. To better understand the differences between the two responses [virus versus poly(I:C)], we evaluated the cytokine/chemokine profile in the placenta and spleen of mice infected with MHV-68. MHV-68 infection did not induce the production of chemokines and inflammatory cytokines seen with poly (I:C) administration (Fig. 2C, 2D). Moreover, we observed inhibition on the production levels of IL-6, MIP-1β, and RANTES in the placenta of MHV-68–infected mice. These data suggest that the change in the cytokine profile may play an important role in the induction of preterm labor/delivery.
TLR-3 is necessary for the control of early viral infection
The effects of poly(I:C) in pregnancy outcome (and, in particular, preterm labor) are mediated through TLR-3 (30, 35), and this pattern recognition receptor plays an important role in the immune response to herpesviruses (57). Moreover, some viruses such as influenza A and Kaposi's sarcoma-associated herpesvirus have been associated with activation of the TLR-3 pathway in humans (58, 59). Thus, we evaluated the role of TLR-3 on MHV-68 infection during pregnancy by using TLR-3 KO mice. Similarly, as in the wt mice, administration of MHV-68 had no effect on the duration of pregnancy (i.e., there was no premature labor). However, we observed higher viral titers in all tissues, including the decidua and placenta of TLR-3 KO mice, both at 3 and 9 dpi (Fig. 2E, 2F). The fetuses of either wt or TLR-3 KO-infected mothers were not infected, suggesting that TLR-3 is not required for the protection of fetuses against viral invasion.
We then evaluated whether the viral dose used in this study is able to mount an adaptive immune response by assessing seroconversion. IgG anti–MHV-68 were significantly higher in both wt and TLR-3 KO-infected pregnant mice than in noninfected animals. However, when we compared the response between wt and TLR-3 KO, we observed that IgG anti MHV-68 levels were significantly lower in the TLR-3 KO (Fig. 3). These results confirm that MHV-68 infections during pregnancy are able to mount a specific adaptive immune response characterized by the presence of anti–MHV-68 IgG. The presence of higher viral titers and low levels of anti–MHV-68 IgG in the TLR-3 KO mice indicated that TLR-3 expression is required to elicit a potent antiviral Ab response against this dsDNA virus.
FIGURE 3.
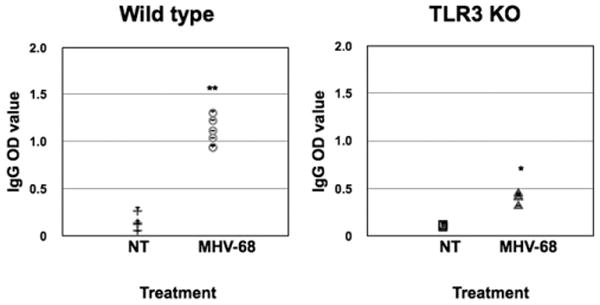
Seroconversion in wt and TLR-3 KO pregnant mice infected with MHV-68. Wt and TLR-3 KO mice were infected i.p. with MHV-68 (1 × 106 PFU) or vehicle at E8.5. Serum samples were collected 9 dpi, and levels of IgG Abs were determined by ELISA. Note the high levels of IgG anti–MHV-68 Abs in the wt treated group compared with controls. A significantly lower response was observed in TLR-3 KO-treated mice. n = 6 mice per group. *p < 0.05.
Effect of MHV-68 infection on the placenta
Because we observed viral infection of the placenta and decidua in mothers, the next objective was to determine the effect of MHV-68 infection on the placental and decidual pathology. Thus, utero-placental units were collected at 9 dpi, and H&E staining was performed. All histological samples were analyzed in a blinded manner by an independent animal pathologist (C.B.). Sites of edema were observed only in the decidua of infected mice; necrosis and inflammation foci were observed in the labyrinth of infected mice (Fig. 4A). Significant pathologic changes present within the labyrinth of TLR3 KO-treated mice included an overall tissue hyperesinophilia, nuclear pyknosis, cellular fragmentation, and multifocal loss of tissue of architecture (necrosis) in the labyrinth (Fig. 4B).
FIGURE 4.
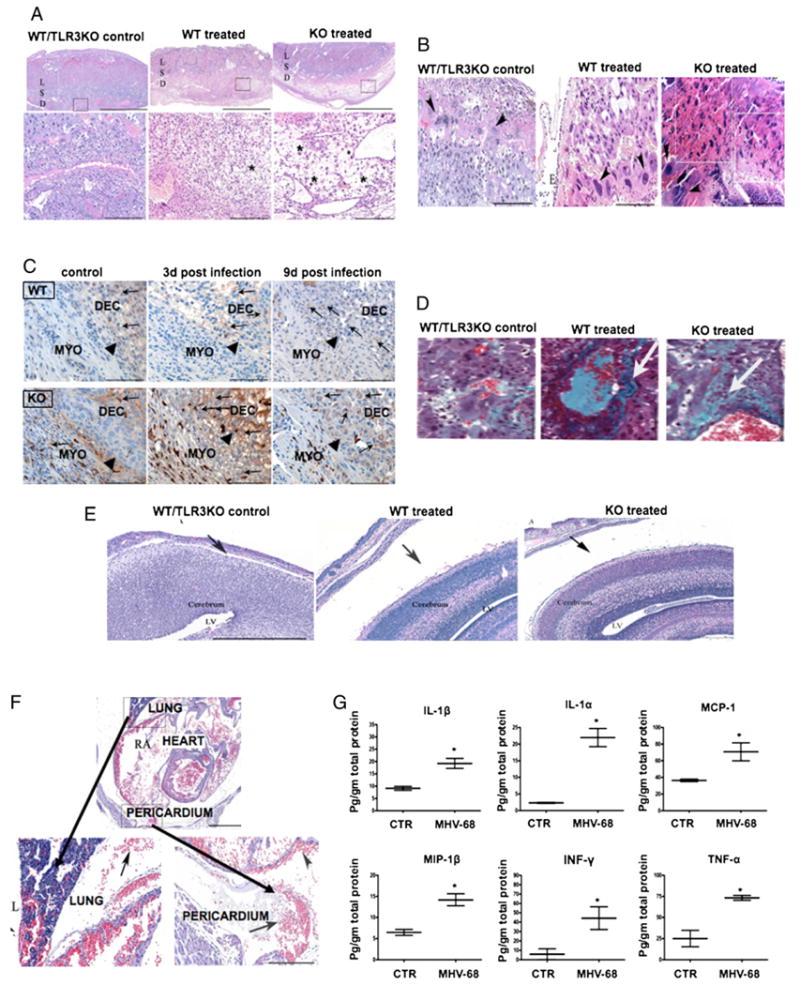
Effect of MHV-68 viral infection on the maternal/fetal interface. Morphological changes were observed in the placenta and decidua of MHV-68–infected pregnant mice associated with the following: A, edema (*) in the D, but absent in the L and S. Upper and lower panel, scale bar, 200 and 300 μm, respectively. B, Necrosis in placenta, marked loss of cellular detail, fragmentation, hypereosinophilia (boxes) in the labyrinth subjacent to the epithelium (E), and necrosis of scattered giant cells (arrowheads). These changes were more accentuated in the TLR-3 KO compared with wt mice. Scale bars, 200 μm. C, Immunohistochemistry for F4/80-positive macrophages (brown) localized in the myometrium (MYO) and decidua (DEC) at E11.8 and E17.5. Black arrows show the edge between myometrium and decidua. Original magnification ×20. D, Increase in collagen deposition (arrows) in the labyrinth layer of MHV-68–infected mice using Trichromic Mason staining (original magnification ×20. E, Presence of fetal brain hydrocephalus (black arrows) in wt and TLR-3 KO MHV-68–infected mice (middle and right panel) compared with normal controls (left panel). Note the width of LVs. Original magnification ×10. F, Fetal thoracic cavity from WT and TLR-3 KO pregnant mice infected with MHV-68. Note the areas of hemorrhage in lung right middle lobe and pericardium. G, Fetal cytokine profile from pregnant mice infected with MHV-68. Fetal lysates were obtained, and cytokines/chemokines were measured by Luminex. Bars show median ± SEM. n = 6 mice per group. *p < 0.05. Figures are representative of six animals per group and three independent experiments. D, decidua; L, labyrinth; LV, lateral ventricle; S, sponginous layer.
We then evaluated changes of the number and distribution of NK (lectin-positive) cells and macrophages (F4/80 positive). NK cells were observed in decidua of control as well as infected mice. No change in the location of these cells was observed as a result of the infection (data not shown). Macrophages are mainly localized in the myometrium and decidua of the pregnant mice (Fig. 4C, arrows). Few macrophages are also found in the placenta. No differences in the distribution and number of macrophages were found in the decidua and placenta from infected and control groups.
In addition, we observed an increase in collagen deposition in the perivascular spaces of infected animals, predominantly in the labyrinth layer, as compared with control mice (Fig. 4D). The presence of collagen in the perivascular areas suggests that an active repair process was taking place in the placenta of infected mice. Similar changes were observed in TLR-3 KO mice (Fig. 4D).
Fetal response to placental infection
Although we observed high viral titers in the placenta, no virus was detected in any of the fetuses, as determined by the limiting dilution plaque assay and confirmed by PCR. To determine whether the lack of fetal infection was due to inability of the virus to infect fetal cells, mouse embryonic fibroblast cells were isolated and infected with MHV-68 in vitro with a similar dose as that used for trophoblast cells (see below). Eighty percent of embryonic fibroblasts were infected by MHV-68 in less than 12 h, as shown by GFP-positive signal; however, the viral infection induced a lytic effect (data not shown). These results suggest that the placenta is functioning as an immunological barrier, capturing the virus and preventing it from reaching the fetus. To determine whether the infection of the placenta could have an effect on the developing fetus, we next assessed fetal morphology from mice infected with MHV-68 during pregnancy versus those receiving a placebo.
Analysis of the fetuses revealed that viral infection of the mother has a transient effect on development. Three dpi, fetuses of infected mothers were smaller and had a lower weight (in both wt and TLR-3 KO mice), although this effect was more evident in the TLR-3 KO group (Supplemental Fig. 1A). Furthermore, we observed a delay in the process of differentiation of the eye, tails, and limbs (Supplemental Fig. 1B). However, after 9 d, the differences between the fetuses from infected and noninfected mice from both wt and TLR-3 KO were no longer detectable (Supplemental Fig. 1C). These important observations are evidence of the remarkable plasticity of the developing fetus.
Because we observed an early effect on fetal development, we then evaluated the integrity of fetal organs and tissues using microscopic sections. Despite the absence of viruses in the fetuses, we noted severe pathological changes in the fetal tissues of infected mothers from both wt and TLR-3 KO. We observed hydrocephalus, defined as an increase in the subarachnoid space, in the brains of all fetuses from infected mothers (Fig. 4E). We did not see any changes in the lateral ventricles, nor did we detect abnormal immune infiltration or white matter damage.
In the thoracic cavity, the pathological changes were characterized by the presence of hemorrhage inside the lungs and pericardium in all treated animals compared with the controls (Fig. 4F). However, there was no damage in the abdominal cavity or the limbs.
We then evaluated the cytokine profile in fetuses from infected and control mothers. Interestingly, 9 dpi, we observed a significant increase in the levels of fetal proinflammatory cytokines (Fig. 4G), including high levels of IFN-γ and TNF-α. The presence of these two cytokines may explain some of the morphological changes observed in these fetuses.
Collectively, these data suggest that although there is no demonstrable fetal viral infection, the presence of an active inflammatory response in the placenta and decidua can have a direct effect on fetal development.
Trophoblast-viral interaction
To understand the implications of these observations in humans, trophoblast cells were isolated from first trimester human placentas and infected with GFP-MHV-68 for either 24 or 48 h. Infection was monitored by the presence of GFP. Positive GFP-MHV-68–infected trophoblast cells were observed ≈12 h postinfection and remained viable up to 6 dpi (Fig. 5A).
FIGURE 5.
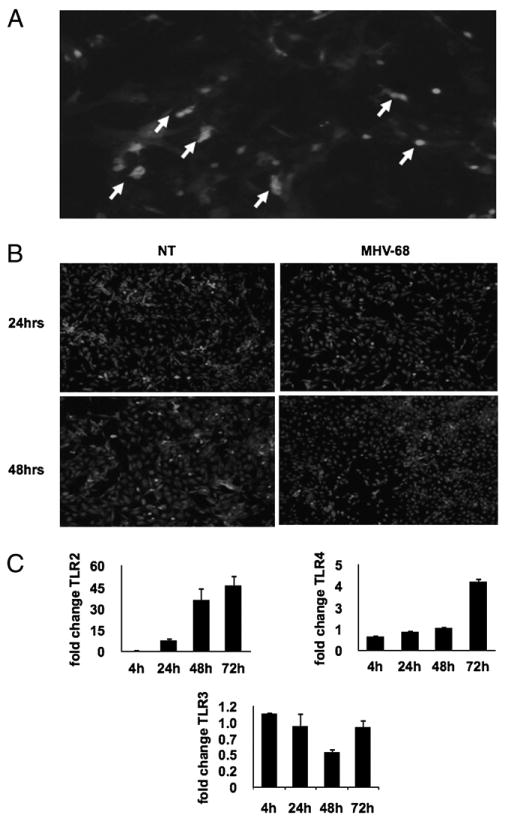
Primary cultures of human first trimester trophoblast cells infected with GFP-MHV-68. A, Infection was monitored by the presence of GFP-labeled MHV-68. Positive GFP-MHV-68–infected trophoblast cells (white arrows) were observed ≈12 h postinfection. B, Inhibition of NF-κB activity in MHV-68–infected trophoblast cells. Expression of p65 was determined by immunofluorescence. Note the decrease in the number of trophoblast cells with nuclear p65 (white dots) following MHV-68 infection. C, Expression of TLR-2, -3, and -4 by human first trimester trophoblast cells following MHV-68 infection. TLR-2, -3, and -4 expressions were determined by real-time quantitative RT-PCR. Note the significant increase on TLR-2 and -4 expression and decrease in TLR-3 in MHV-68–infected cells compared with the control. n = 3 samples per group. *p < 0.05.
Next, we determined the cytokine response induced by MHV-68 in trophoblast cells in vitro. Contrary to what we observed with poly (I:C) treatment (which induces robust proinflammatory cytokine/ chemokine production [30]), MHV-68 has a unique profile characterized by inhibition of chemokines and lack of production of proinflammatory cytokines (Table I). However, we observed a mild increase in modulatory cytokines, such as IL-6, IL-1β, and the immune suppressor vascular endothelial growth factor (VEGF) (60) (Table I). Evaluation of the cytokine response by trophoblast isolated from wt mice or TLR-3 KO mice to MHV-68 infection revealed a similar profile in terms of the inhibition of chemokines. However, contrary to the wt mice, we did not observe an increase in IL-1β and IL-6, suggesting that TLR-3 expression might be necessary for the production of these two cytokines (data not shown).
Table I. Cytokine/chemokine profile of poly(I:C)- or MHV-68–infected human trophoblast.
| Factor | Poly(I:C) | MHV-68 |
|---|---|---|
| IL-6 | ↑6.3 | ↑3.3 |
| IL-1B | — | ↑4.5 |
| VEGF | — | ↑1.2 |
| FGF-2 | ↑2.0 | ↑5.7 |
| IL-8 | ↑7.2 | ↓11.5 |
| MCP-1 | ↑1.7 | ↓162.5 |
| RANTES | ↑2.3 | ↓2.4 |
| GRO-α | ↑6.5 | ↓8.7 |
| IL-1α | ↑115.8 | — |
| GM-CSF | ↑261.1 | — |
| IL-12p70 | ↑2.2 | ↓2.0 |
| IFN-γ | ↑6.9 | ↓1.5 |
| IP-10 | ↑225.2 | ↓3.3 |
| IFN-α | ↑2.6 | ↓1.5 |
| IFN-β | ↑60.0 | ↑3.0 |
Cytokine/chemokine profile of poly(I:C)-treated and MHV-68–infected human primary trophoblast. Isolated human first trimester trophoblast cells were treated with either 25 μg/ml poly(I:C) or MHV-68 at a multiplicity of infection of 1.4 for 72 h. Supernatants were collected, and cytokines and chemokines were measured by Multiplex. Fold changes (mean ± SEM) of cytokine/chemokine secretions with poly (I:C) and MHV-68. n = 3 samples per group. *p < 0.05.
↓ Decrease; ↑, increase; FGF-2, fibroblast growth factor-2; GRO-α, growth-related oncogene-α; IP-10, IFN-γ–inducible protein-10.
Differential regulation and role of the TLR/NF-κB pathway during MHV-68 infection in trophoblast cells
The generalized inhibition of chemokine expression and the increased secreted levels of the immune suppressor VEGF represent an important immune-regulatory mechanism by which MHV-68 can escape immune surveillance and successfully infect trophoblast cells.
Our next objective was to determine the potential mechanism by which MHV-68 inhibits chemokine production. Because the NF-κB pathway is a major regulator of cytokine and chemokine production, we evaluated the status of p65 (a regulatory subunit of NF-κB) in trophoblast cells following MHV-68 infection. Trophoblast cells are characterized by constitutive NF-κB activity and cytokine production (20, 30). Therefore, p65 is localized in the nuclei of the cells (Fig. 5B). Following MHV-68 infection, no nuclear staining was observed along with a decrease in cytoplasmic expression (Fig. 5B), suggesting that the NF-κB pathway is inhibited in trophoblast cells infected by MHV-68. The viral-induced inhibition of NF-κB correlates with the inhibition on chemokine production observed in the infected trophoblast and placenta.
TLR/MyD88 signal has been shown to be important in the regulation of MHV-68 replication (61). Trophoblast cells express TLRs and are able to respond to TLR ligands (20, 30); therefore, we evaluated whether viral infection could affect the expression or function of TLRs. MHV-68 infection induces TLR-2 and TLR-4 mRNA expression in human first trimester trophoblast cells in a time-dependent manner. In contrast, TLR-3 mRNA levels were not affected or were decreased postinfection (Fig. 5C).
The significant increase in TLR-2 expression following MHV-68 infection indicates a potential association between TLR-2 and the viral adaptation to the host. Thus, wt trophoblasts, trophoblasts stably transfected with a TLR-2 DN or TLR-1 DN, were infected with MHV-68, and 48 h postinfection, supernatants from these cultures were collected and transferred to new cultures of wt first trimester trophoblast cells. No de novo infection was observed following transfer of supernatants obtained from TLR-2 DN trophoblasts, and similarly, TLR1 DN reinfectivity was greatly reduced, suggesting that TLR-2 is necessary for viral reactivation, replication, or egress (Supplemental Fig. 2).
MHV-68 infection sensitizes to bacterial LPS
Because we observed that MHV-68 infection leads to an increase in the expression levels of TLR-4 and TLR-2, we next tested the hypothesis that viral infection in early pregnancy could affect the response to microbial products. We injected MHV-68 into pregnant wt mice at E8.5 (early pregnancy), followed by LPS injection at E15.5 (late pregnancy). We selected a dose of LPS that has a modest effect on pregnancy outcome (20 μg/kg) (30). Control mice received only vehicle or only LPS at E15.5. LPS treatment of MHV-68–infected pregnant mice induced preterm labor/delivery in less than 24 h in all of the treated mice (Fig. 6). Anatomical examination of the mothers showed vaginal bleeding and 100% fetal death in the MHV-68 plus LPS-treated group (Supplemental Fig. 3). LPS administration, without previous viral infection, induced preterm labor/delivery in 29% of cases, and we did not observe major anatomical changes in the mother or the fetus (Supplemental Fig. 4).
FIGURE 6.
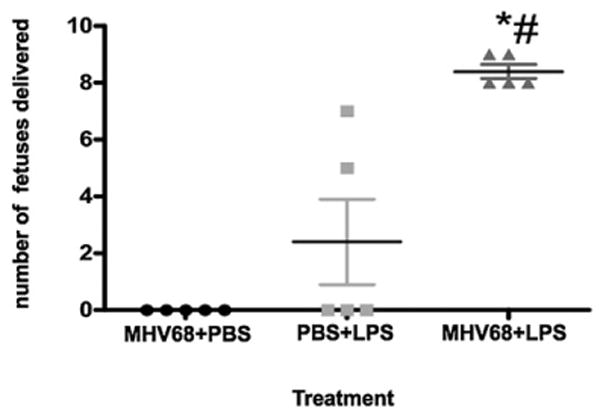
MHV-68 infection sensitizes to bacterial LPS. Wt mice were infected i.p. with MHV-68 at E8.5, followed by a single dose of LPS (20 μg/kg) at E15.5. LPS induced preterm labor in all of the animals that received prior MHV-68 infections (triangles), compared with animals receiving only LPS (squares) or MHV-68 infection (circles). Bars show median ± SEM. n = 6 mice per group. *p < 0.05. MHV-68 plus LPS versus PBS plus LPS. #p < 0.05. MHV-68 plus LPS versus PBS plus MHV-68.
Discussion
We demonstrated that maternal viral infection can lead to productive replication in the placenta and a fetal inflammatory response, even though the virus is not detected in the fetus. The experiments described in this work are intended to show that viral infection of the placenta can elicit a fetal inflammatory response, which in turn can cause organ damage and, potentially, downstream developmental deficiencies. Furthermore, we demonstrated that a viral infection of the placenta may sensitize to bacterial infection and promote preterm labor.
Pregnant women are exposed to many infectious agents that are potentially harmful to the fetus. The risk evaluation has been focused on whether there is a maternal viremia or fetal transmission (62). Viral infections that are able to reach the fetus by crossing the placenta might have a detrimental effect on the pregnancy (63, 64). It is well accepted that in those cases infection can lead to embryonic and fetal death, induce miscarriage, or induce major congenital anomalies (62, 65). However, even in the absence of fetal viral infection, the fetus could be adversely affected by the maternal response to the infection. Examples are infections with HIV, hepatitis B, varizella zoster virus, and parvovirus B19, among others (5, 28, 66, 67). Indeed, viral crossing of the placenta may be the exception rather than the rule.
One of the main questions of this study was how a microorganism, in this case a virus, might initiate a response that may not lead to preterm labor, but would alter the immunologic balance at the maternal fetal interface. Poly(I:C) has been used in several studies as a model for TLR3 activation and shown to be a potent inducer of preterm labor (68). However, the use of MHV-68, which is able to activate TLR-3 (61), did not show the same outcome. Our results indicate that only a condition characterized by the expression of inflammatory cytokines at the maternal-fetal interface will trigger a cascade of events leading to the termination of the pregnancy. In contrast, a viral infection in the placenta that triggers a mild inflammatory response will not terminate the pregnancy, but is able to activate the immune system not only of the mother, but of the fetus as well.
It is critical to take into consideration the fact that during pregnancy it is not only the maternal immune system responding, but also the fetal/placental unit. Our results further support the immunological role of the placenta and the fetus affecting the global response of the mother to microbial infections. This is relevant for making decisions associated with treatment and prevention during pandemics.
An important observation in this study is the fact that even though there is a high viral titer in the placenta and decidua, no virus was detected in the fetus. This result further confirms our and others' studies suggesting that the placenta is an active barrier, able to control an infection and protect the fetus (49, 69–72). However, the inflammatory response originated on the maternal side has a negative impact on the fetus and triggers a fetal inflammatory condition.
Fetal inflammatory response syndrome (FIRS) is a condition in which, despite an absence of cultivable microorganisms, neonates with placental infections have very high circulating levels of inflammatory cytokines, such as IL-1, IL-6, IL-8, and TNF-α (73–75). We observed a similar outcome in our animal model in which MHV-68 infection of the placenta triggers a fetal inflammatory response similar to the one observed in FIRS, even though the virus was not able to reach the fetus. In the case of human FIRS, these cytokines have been shown to affect the CNS and the circulatory system (76). In this study, we showed that fetal morphologic abnormalities may be caused by fetal proinflammatory cytokines, such as IL-1, TNF-α, MCP-1, MIP1-β, and IFN-γ. Beyond morphological effects on the fetal brain, the presence of FIRS increases the future risk for schizophrenia, neurosensorial deficits, and psychosis induced in the neonatal period (77–79).
Therefore, we propose that an inflammatory response of the placenta, which alters the cytokine balance in the fetus, may affect the normal development of the fetal immune system, leading to anomalous responses during childhood or later in life (77–79). One example of this is the differential responses in children to vaccination or the development of allergies (11, 80). Antenatal infections can have a significant impact on later vaccine responses. We can observe this type of outcome in other conditions associated with placental infection, such as malaria. A few studies suggest that surviving infants with placental malaria may suffer adverse neurodevelopmental sequelae and may have an abnormal response to a later infection with the parasite (81). In the majority of the cases, the parasite did not reach the fetus, but the inflammatory process in the placenta affected the normal fetal development (82).
The differential cytokine response observed between poly(I:C) and MHV-68 infection questioned the role of TLRs during a viral infection. However, our finding demonstrates a unique interaction between the virus and TLR expression and function at the placenta and decidua. We confirmed that TLR-3 is necessary for the control of viral replication, as demonstrated by the presence of higher titers of MHV-68 found in the TLR-3 KO mice. According to this, clinical data proved that TLR-3 controls herpesvirus infection, because children with a TLR-3 deficiency are very susceptible to HSV-1–induced encephalitis (83). In contrast, the virus requires TLR-2 and TLR-1 expression for its own replication. These findings open the possibility of using TLR-2 or TLR-1 antagonists as potential agents for preventing herpes viral replication.
Viral infection may influence the outcome of a concurrent bacterial infection (84); however, to date there is no evidence indicating whether a viral infection sensitizes to bacterial infection during pregnancy. We showed that MHV-68–infected pregnant mice undergo preterm labor following injection of a low dose of LPS, which has almost no effect on noninfected mice. These results suggest that a viral infection during pregnancy increases the risk of preterm labor or maternal death in response to other microorganisms, such as bacterial infection. In the pandemic of 1918, high rates of pregnancy loss and preterm delivery were reported (1), and during the pandemic of 1957–1958, an increase in CNS defects and other adverse outcomes were reported. In the more recent H1N1 influenza virus infection, 13% of the deaths were pregnant women (3). In all these cases, a bacterial-associated complication was reported.
In conclusion, we demonstrate that even in the absence of fetal viral infection, the inflammatory response originating in the placenta and decidua induces an inflammatory process with potential damage in fetal organs. It is therefore essential to evaluate the presence of maternal viral infections prenatally to prevent long-term adverse outcomes for the child and the mother. Future studies are needed to develop useful biomarkers for viral infections during pregnancy even in a subclinical state as a strategy of early detection and prevention of fetal damage and maternal mortality. Furthermore, it is extremely important to take into consideration the possibility of placental infection when determining a response to emerging infectious disease threats.
Supplementary Material
Acknowledgments
This work was in part supported by grants from the National Institutes of Health (NICDH P01HD054713 and 3N01 HD23342) and the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services.
Abbreviations used in this paper
- DN
dominant negative
- dpi
days postinfection
- E
embryonic day
- FGF-2
fibroblast growth factor-2
- FIRS
fetal inflammatory response syndrome
- GRO-α
growth-related oncogene-α
- IP-10
IFN-γ–inducible protein-10
- KO
knockout
- MHV-68
murine γ-herpesvirus 68
- poly(I:C)
polyinosinic:polycytidylic acid
- TIR
Toll/IL-1R
- VEGF
vascular endothelial growth factor
- wt
wild type
Footnotes
The online version of this article contains supplemental material.
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Nuzum JW, Pilot I, Stangl FH, Bonar BE. 1918 pandemic influenza and pneumonia in a large civil hospital. IMJ Ill Med J. 1976;150:612–616. [PubMed] [Google Scholar]
- 2.Romero R, Espinoza J, Chaiworapongsa T, Kalache K. Infection and prematurity and the role of preventive strategies. Semin Neonatol. 2002;7:259–274. doi: 10.1016/s1084-2756(02)90121-1. [DOI] [PubMed] [Google Scholar]
- 3.Jamieson DJ, Honein MA, Rasmussen SA, Williams JL, Swerdlow DL, Biggerstaff MS, Lindstrom S, Louie JK, Christ CM, Bohm SR, et al. Novel Influenza A (H1N1) Pregnancy Working Group H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet. 2009;374:451–458. doi: 10.1016/S0140-6736(09)61304-0. [DOI] [PubMed] [Google Scholar]
- 4.Burguete T, Rabreau M, Fontanges-Darriet M, Roset E, Hager HD, Köppel A, Bischof P, Schlehofer JR. Evidence for infection of the human embryo with adeno-associated virus in pregnancy. Hum Reprod. 1999;14:2396–2401. doi: 10.1093/humrep/14.9.2396. [DOI] [PubMed] [Google Scholar]
- 5.Basurko C, Carles G, Youssef M, Guindi WE. Maternal and fetal consequences of dengue fever during pregnancy. Eur J Obstet Gynecol Reprod Biol. 2009;147:29–32. doi: 10.1016/j.ejogrb.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 6.Han YW, Ikegami A, Bissada NF, Herbst M, Redline RW, Ashmead GG. Transmission of an uncultivated Bergeyella strain from the oral cavity to amniotic fluid in a case of preterm birth. J Clin Microbiol. 2006;44:1475–1483. doi: 10.1128/JCM.44.4.1475-1483.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seubert DE, Maymon E, Pacora P, Gervasi MT, Berry SM, Torry DS, Romero R. A study of the relationship between placenta growth factor and gestational age, parturition, rupture of membranes, and intrauterine infection. Am J Obstet Gynecol. 2000;182:1633–1637. doi: 10.1067/mob.2000.107437. [DOI] [PubMed] [Google Scholar]
- 8.Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342:1500–1507. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- 9.Sekirime WK, Lule JC. Outcome of cesarean section in asymptomatic HIV-1 infection in Kampala, Uganda. J Obstet Gynaecol Res. 2009;35:679–688. doi: 10.1111/j.1447-0756.2008.01002.x. [DOI] [PubMed] [Google Scholar]
- 10.Mel'nikova VF, Aksenov OA. Infectious placentitis and characterization of the placenta as an immune barrier. Arkh Patol. 1993;55:78–81. [PubMed] [Google Scholar]
- 11.Chatterjee A, Chartrand SA, Harrison CJ, Felty-Duckworth A, Bewtra C. Severe intrauterine herpes simplex disease with placentitis in a newborn of a mother with recurrent genital infection at delivery. J Perinatol. 2001;21:559–564. doi: 10.1038/sj.jp.7210573. [DOI] [PubMed] [Google Scholar]
- 12.Van den Veyver IB, Ni J, Bowles N, Carpenter RJ, Jr, Weiner CP, Yankowitz J, Moise KJ, Jr, Henderson J, Towbin JA. Detection of intrauterine viral infection using the polymerase chain reaction. Mol Genet Metab. 1998;63:85–95. doi: 10.1006/mgme.1997.2651. [DOI] [PubMed] [Google Scholar]
- 13.Euscher E, Davis J, Holzman I, Nuovo GJ. Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet Gynecol. 2001;98:1019–1026. doi: 10.1016/s0029-7844(01)01625-8. [DOI] [PubMed] [Google Scholar]
- 14.Satosar A, Ramirez NC, Bartholomew D, Davis J, Nuovo GJ. Histologic correlates of viral and bacterial infection of the placenta associated with severe morbidity and mortality in the newborn. Hum Pathol. 2004;35:536–545. doi: 10.1016/j.humpath.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Redline RW. Placental inflammation. Semin Neonatol. 2004;9:265–274. doi: 10.1016/j.siny.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Redline RW. Infections and other inflammatory conditions. Semin Diagn Pathol. 2007;24:5–13. doi: 10.1053/j.semdp.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Redline RW, Faye-Petersen O, Heller D, Qureshi F, Savell V, Vogler C, Society for Pediatric Pathology, Perinatal Section, Amniotic Fluid Infection Nosology Committee Amniotic infection syndrome: nosology and reproducibility of placental reaction patterns. Pediatr Dev Pathol. 2003;6:435–448. doi: 10.1007/s10024-003-7070-y. [DOI] [PubMed] [Google Scholar]
- 18.Kiehl K, Schlehofer JR, Schultz R, Zugaib M, Armbruster-Moraes E. Adeno-associated virus DNA in human gestational trophoblastic disease. Placenta. 2002;23:410–415. doi: 10.1053/plac.2002.0827. [DOI] [PubMed] [Google Scholar]
- 19.Mor G, Romero R, Aldo PB, Abrahams VM. Is the trophoblast an immune regulator? The role of Toll-like receptors during pregnancy. Crit Rev Immunol. 2005;25:375–388. doi: 10.1615/critrevimmunol.v25.i5.30. [DOI] [PubMed] [Google Scholar]
- 20.Abrahams VM, Bole-Aldo P, Kim YM, Straszewski-Chavez SL, Chaiworapongsa T, Romero R, Mor G. Divergent trophoblast responses to bacterial products mediated by TLRs. J Immunol. 2004;173:4286–4296. doi: 10.4049/jimmunol.173.7.4286. [DOI] [PubMed] [Google Scholar]
- 21.Abrahams VM, Schaefer TM, Fahey JV, Visintin I, Wright JA, Aldo PB, Romero R, Wira CR, Mor G. Expression and secretion of antiviral factors by trophoblast cells following stimulation by the TLR-3 agonist, poly(I:C) Hum Reprod. 2006;21:2432–2439. doi: 10.1093/humrep/del178. [DOI] [PubMed] [Google Scholar]
- 22.Abrahams VM, Aldo PB, Murphy SP, Visintin I, Koga K, Wilson G, Romero R, Sharma S, Mor G. TLR6 modulates first trimester trophoblast responses to peptidoglycan. J Immunol. 2008;180:6035–6043. doi: 10.4049/jimmunol.180.9.6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevens AM, McDonnell WM, Mullarkey ME, Pang JM, Leisenring W, Nelson JL. Liver biopsies from human females contain male hep-atocytes in the absence of transplantation. Lab Invest. 2004;84:1603–1609. doi: 10.1038/labinvest.3700193. [DOI] [PubMed] [Google Scholar]
- 24.Mold JE, Michaëlsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, Lee TH, Nixon DF, McCune JM. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. 2008;322:1562–1565. doi: 10.1126/science.1164511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kudesia G, Ball G, Irving WL. Vertical transmission of hepatitis C. Lancet. 1995;345:1122–1123. [PubMed] [Google Scholar]
- 26.Nigro G, Torre RL, Pentimalli H, Taverna P, Lituania M, de Tejada BM, Adler SP. Regression of fetal cerebral abnormalities by primary cytomegalovirus infection following hyperimmunoglobulin therapy. Prenat Diagn. 2008;28:512–517. doi: 10.1002/pd.2013. [DOI] [PubMed] [Google Scholar]
- 27.Arrivé E, Dabis F. Prophylactic antiretroviral regimens for prevention of mother-to-child transmission of HIV in resource-limited settings. Curr Opin HIV AIDS. 2008;3:161–165. doi: 10.1097/COH.0b013e3282f51b89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connor EM, Sperling RS, Gelber R, Kiselev P, Scott G, O'Sullivan MJ, VanDyke R, Bey M, Shearer W, Jacobson RL, et al. Reduction of maternal-infant transmission of human immunodeficiency virus type 1 with zidovudine treatment: Pediatric AIDS Clinical Trials Group Protocol 076 Study Group. N Engl J Med. 1994;331:1173–1180. doi: 10.1056/NEJM199411033311801. [DOI] [PubMed] [Google Scholar]
- 29.Faye-Petersen OM. The placenta in preterm birth. J Clin Pathol. 2008;61:1261–1275. doi: 10.1136/jcp.2008.055244. [DOI] [PubMed] [Google Scholar]
- 30.Koga K, Cardenas I, Aldo P, Abrahams VM, Peng B, Fill S, Romero R, Mor G. Activation of TLR3 in the trophoblast is associated with preterm delivery. Am J Reprod Immunol. 2009;61:196–212. doi: 10.1111/j.1600-0897.2008.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakada E, Walley KR, Nakada T, Hu Y, von Dadelszen P, Boyd JH. Toll-like receptor-3 stimulation upregulates sFLT-1 production by trophoblast cells. Placenta. 2009;30:774–779. doi: 10.1016/j.placenta.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez JM, Xu H, Ofori E, Elovitz MA. Toll-like receptors in the uterus, cervix, and placenta: is pregnancy an immunosuppressed state? Am J Obstet Gynecol. 2007;197:296.e1–6. doi: 10.1016/j.ajog.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 33.Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105–38108. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 34.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 35.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 36.Trinh QD, Izumi Y, Komine-Aizawa S, Shibata T, Shimotai Y, Kuroda K, Mizuguchi M, Ushijima H, Mor G, Hayakawa S. H3N2 influenza A virus replicates in immortalized human first trimester trophoblast cell lines and induces their rapid apoptosis. Am J Reprod Immunol. 2009;62:139–146. doi: 10.1111/j.1600-0897.2009.00723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gargano LM, Forrest JC, Speck SH. Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J Virol. 2009;83:1474–1482. doi: 10.1128/JVI.01717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, Berman SM, Markowitz LE. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. J Am Med Assoc. 2006;296:964–973. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- 39.Haun L, Kwan N, Hollier LM. Viral infections in pregnancy. Minerva Ginecol. 2007;59:159–174. [PubMed] [Google Scholar]
- 40.Olivadoti M, Toth LA, Weinberg J, Opp MR. Murine gammaherpesvirus 68: a model for the study of Epstein-Barr virus infections and related diseases. Comp Med. 2007;57:44–50. [PubMed] [Google Scholar]
- 41.Krug LT, Moser JM, Dickerson SM, Speck SH. Inhibition of NF-kappaB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 2007;3:e11. doi: 10.1371/journal.ppat.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW., IV Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol. 1996;70:6775–6780. doi: 10.1128/jvi.70.10.6775-6780.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Straszewski-Chavez SL, Abrahams VM, Alvero AB, Aldo PB, Ma Y, Guller S, Romero R, Mor G. The isolation and characterization of a novel telomerase immortalized first trimester trophoblast cell line, Swan 71. Placenta. 2009;30:939–948. doi: 10.1016/j.placenta.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frew IJ, Dickins RA, Cuddihy AR, Del Rosario M, Reinhard C, O'Connell MJ, Bowtell DD. Normal p53 function in primary cells deficient for Siah genes. Mol Cell Biol. 2002;22:8155–8164. doi: 10.1128/MCB.22.23.8155-8164.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dickins RA, Frew IJ, House CM, O'Bryan MK, Holloway AJ, Haviv I, Traficante N, de Kretser DM, Bowtell DD. The ubiquitin ligase component Siah1a is required for completion of meiosis I in male mice. Mol Cell Biol. 2002;22:2294–2303. doi: 10.1128/MCB.22.7.2294-2303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blesofsky WA, Mowen K, Arduini RM, Baker DP, Murphy MA, Bowtell DD, David M. Regulation of STAT protein synthesis by c-Cbl. Oncogene. 2001;20:7326–7333. doi: 10.1038/sj.onc.1204919. [DOI] [PubMed] [Google Scholar]
- 47.Nilausen K, Green H. Reversible arrest of growth in G1 of an established fibroblast line (3T3) Exp Cell Res. 1965;40:166–168. doi: 10.1016/0014-4827(65)90306-x. [DOI] [PubMed] [Google Scholar]
- 48.Aldo PB, Mulla MJ, Romero R, Mor G, Abrahams VM. Viral ssRNA induces first trimester trophoblast apoptosis through an inflammatory mechanism. Am J Reprod Immunol. 2010 doi: 10.1111/j.1600-0897.2010.00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Straszewski-Chavez SL, Abrahams VM, Aldo PB, Romero R, Mor G. AKT controls human first trimester trophoblast cell sensitivity to FAS-mediated apoptosis by regulating XIAP expression. Biol Reprod. 2010;82:146–152. doi: 10.1095/biolreprod.109.078972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvero AB, Chen R, Fu HH, Montagna M, Schwartz PE, Rutherford T, Silasi DA, Steffensen KD, Waldstrom M, Visintin I, Mor G. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle. 2009;8:158–166. doi: 10.4161/cc.8.1.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abrahams VM, Kim YM, Straszewski SL, Romero R, Mor G. Macrophages and apoptotic cell clearance during pregnancy. Am J Reprod Immunol. 2004;51:275–282. doi: 10.1111/j.1600-0897.2004.00156.x. [DOI] [PubMed] [Google Scholar]
- 52.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 53.Abrahams VM, Romero R, Mor G. TLR-3 and TLR-4 mediate differential chemokine production and immune cell recruitment by first trimester trophoblast cells. Am J Reprod Immunol. 2005;53:279. ASRI205-202. [Google Scholar]
- 54.Ilievski V, Lu SJ, Hirsch E. Activation of Toll-like receptors 2 or 3 and preterm delivery in the mouse. Reprod Sci. 2007;14:315–320. doi: 10.1177/1933719107302959. [DOI] [PubMed] [Google Scholar]
- 55.Abrahams VM, Fahey JV, Schaefer TM, Wright JA, Wira CR, Mor G. Stimulation of first trimester trophoblast cells with poly(I:C) induces SLPI secretion. Am J Reprod Immunol. 2005;53:280. ASRI205-204. [Google Scholar]
- 56.Alvarez F, Flaño E, Castillo A, López-Fierro P, Razquin B, Villena A. Tissue distribution and structure of barrier cells in the hematopoietic and lymphoid organs of salmonids. Anat Rec. 1996;245:17–24. doi: 10.1002/(SICI)1097-0185(199605)245:1<17::AID-AR4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 57.Gregory SM, Damania B. KSHV and the toll of innate immune activation. Cell Cycle △. 2009:3246–3247. doi: 10.4161/cc.8.20.9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 59.West J, Damania B. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J Virol. 2008;82:5440–5449. doi: 10.1128/JVI.02590-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, Benencia F, Stan RV, Keler T, Sarobe P, et al. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res. 2008;68:7684–7691. doi: 10.1158/0008-5472.CAN-08-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gargano LM, Moser JM, Speck SH. Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J Virol. 2008;82:3853–3863. doi: 10.1128/JVI.02577-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibson CS, Goldwater PN, MacLennan AH, Haan EA, Priest K, Dekker GA, South Australian Cerebral Palsy Research Group Fetal exposure to herpesviruses may be associated with pregnancy-induced hypertensive disorders and preterm birth in a Caucasian population. BJOG. 2008;115:492–500. doi: 10.1111/j.1471-0528.2007.01653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gomez LM, Ma Y, Ho C, McGrath CM, Nelson DB, Parry S. Placental infection with human papillomavirus is associated with spontaneous preterm delivery. Hum Reprod. 2008;23:709–715. doi: 10.1093/humrep/dem404. [DOI] [PubMed] [Google Scholar]
- 64.Johansson S, Buchmayer S, Harlid S, Iliadou A, Sjöholm M, Grillner L, Norman M, Sparén P, Dillner J, Cnattingius S. Infection with parvovirus B19 and herpes viruses in early pregnancy and risk of second trimester miscarriage or very preterm birth. Reprod Toxicol. 2008;26:298–302. doi: 10.1016/j.reprotox.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 65.Srinivas SK, Ma Y, Sammel MD, Chou D, McGrath C, Parry S, Elovitz MA. Placental inflammation and viral infection are implicated in second trimester pregnancy loss. Am J Obstet Gynecol. 2006;195:797–802. doi: 10.1016/j.ajog.2006.05.049. [DOI] [PubMed] [Google Scholar]
- 66.Vernochet C, Azoulay S, Duval D, Guedj R, Cottrez F, Vidal H, Ailhaud G, Dani C. Human immunodeficiency virus protease inhibitors accumulate into cultured human adipocytes and alter expression of adipocytokines. J Biol Chem. 2005;280:2238–2243. doi: 10.1074/jbc.M408687200. [DOI] [PubMed] [Google Scholar]
- 67.Vernochet C, Caucheteux SM, Gendron MC, Wantyghem J, Kanellopoulos-Langevin C. Affinity-dependent alterations of mouse B cell development by noninherited maternal antigen. Biol Reprod. 2005;72:460–469. doi: 10.1095/biolreprod.104.035048. [DOI] [PubMed] [Google Scholar]
- 68.Koga K, Mor G. Expression and function of Toll-like receptors at the maternal-fetal interface. Reprod Sci. 2008;15:231–242. doi: 10.1177/1933719108316391. [DOI] [PubMed] [Google Scholar]
- 69.Abzug MJ, Rotbart HA, Magliato SA, Levin MJ. Evolution of the placental barrier to fetal infection by murine enteroviruses. J Infect Dis. 1991;163:1336–1341. doi: 10.1093/infdis/163.6.1336. [DOI] [PubMed] [Google Scholar]
- 70.Mor G. Inflammation and pregnancy: the role of Toll-like receptors in trophoblast-immune interaction. Ann NY Acad Sci. 2008;1127:121–128. doi: 10.1196/annals.1434.006. [DOI] [PubMed] [Google Scholar]
- 71.Bai H, Zhang L, Ma L, Dou XG, Feng GH, Zhao GZ. Relationship of hepatitis B virus infection of placental barrier and hepatitis B virus intra-uterine transmission mechanism. World J Gastroenterol. 2007;13:3625–3630. doi: 10.3748/wjg.v13.i26.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koi H, Zhang J, Makrigiannakis A, Getsios S, MacCalman CD, Strauss JF, III, Parry S. Syncytiotrophoblast is a barrier to maternal-fetal transmission of herpes simplex virus. Biol Reprod. 2002;67:1572–1579. doi: 10.1095/biolreprod.102.004325. [DOI] [PubMed] [Google Scholar]
- 73.Davies JK, Shikes RH, Sze CI, Leslie KK, McDuffie RS, Jr, Romero R, Gibbs RS. Histologic inflammation in the maternal and fetal compartments in a rabbit model of acute intra-amniotic infection. Am J Obstet Gynecol. 2000;183:1088–1093. doi: 10.1067/mob.2000.108888. [DOI] [PubMed] [Google Scholar]
- 74.Romero R, Gotsch F, Pineles B, Kusanovic JP. Inflammation in pregnancy: its roles in reproductive physiology, obstetrical complications, and fetal injury. Nutr Rev. 2007;65:S194–S202. doi: 10.1111/j.1753-4887.2007.tb00362.x. [DOI] [PubMed] [Google Scholar]
- 75.Madsen-Bouterse SA, Romero R, Tarca AL, Kusanovic JP, Espinoza J, Kim CJ, Kim JS, Edwin SS, Gomez R, Draghici S. The transcriptome of the fetal inflammatory response syndrome. Am J Reprod Immunol. 2010;63:73–92. doi: 10.1111/j.1600-0897.2009.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 77.Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23:116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meyer U, Feldon J, Yee BK. A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull. 2009;35:959–972. doi: 10.1093/schbul/sbn022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology. 2005;48:903–917. doi: 10.1016/j.neuropharm.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 80.Qureshi F, Jacques SM. Maternal varicella during pregnancy: correlation of maternal history and fetal outcome with placental histopathology. Hum Pathol. 1996;27:191–195. doi: 10.1016/s0046-8177(96)90374-3. [DOI] [PubMed] [Google Scholar]
- 81.Labeaud AD, Malhotra I, King MJ, King CL, King CH. Do antenatal parasite infections devalue childhood vaccination? PLoS Negl Trop Dis. 2009;3:e442. doi: 10.1371/journal.pntd.0000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Desai M, ter Kuile FO, Nosten F, McGready R, Asamoa K, Brabin B, Newman RD. Epidemiology and burden of malaria in pregnancy. Lancet Infect Dis. 2007;7:93–104. doi: 10.1016/S1473-3099(07)70021-X. [DOI] [PubMed] [Google Scholar]
- 83.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 84.Nansen A, Randrup Thomsen A. Viral infection causes rapid sensitization to lipopolysaccharide: central role of IFN-alpha beta. J Immunol. 2001;166:982–988. doi: 10.4049/jimmunol.166.2.982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.