

Preface
Cytochromes P450 constitute a broad class of heme monooxygenase enzymes with more than 11,500 isozymes which have been identified in organisms from all biological kingdoms [1]. These enzymes are responsible for catalyzing dozens chemical oxidative transformations such as hydroxylation, epoxidation, N-demethylation, etc., with very broad range of substrates [2-3]. Historically these enzymes received their name from ‘pigment 450’ due to the unusual position of the Soret band in UV-Vis absorption spectra of the reduced CO-saturated state [4-5]. Despite detailed biochemical characterization of many isozymes, as well as later discoveries of other ‘P450-like heme enzymes’ such as nitric oxide synthase and chloroperoxidase, the phenomenological term ‘cytochrome P450’ is still commonly used as indicating an essential spectroscopic feature of the functionally active protein which is now known to be due to the presence of a thiolate ligand to the heme iron [6]. Heme proteins with an imidazole ligand such as myoglobin and hemoglobin as well as an inactive form of P450 are characterized by Soret maxima at 420 nm [7]. This historical perspective highlights the importance of spectroscopic methods for biochemical studies in general, and especially for heme enzymes, where the presence of the heme iron and porphyrin macrocycle provides rich variety of specific spectroscopic markers available for monitoring chemical transformations and transitions between active intermediates of catalytic cycle.
Introduction
All cytochromes P450 have a similar overall protein fold and the same heme coordination complex with cysteinate as the fifth ligand. This provides the basis for the very similar spectroscopic properties of all cytochromes P450, despite a broad variability of their biochemical properties and functional roles. The heme-oxygen intermediates shown in the Scheme 1 ([4] – [6]), as well as prosthetic group complexes with other small ligands and/or inhibitors, have the same or similar spectral properties in the various P450 isoforms. This can be observed from the heme and residues in proximity, such as the axial ligands (probed by EPR, Mössbauer, EXAFS and Raman spectroscopy), or the porphyrin components (revealed by optical absorption, Raman and MCD). Spectroscopic studies can also utilize the signal from individual amino acids, such as high-resolution NMR or fluorescence spectroscopy. Hence, a general survey of the spectroscopic probes common for all P450 enzymes that are sensitive to the changes observed at different steps in the P450 catalytic cycle (Scheme 1) [8], provides a useful review for understanding the functionally important mechanistic similarities of the P450 superfamily and also markers for reflecting the distinct features specific for different isozymes.
Scheme 1.
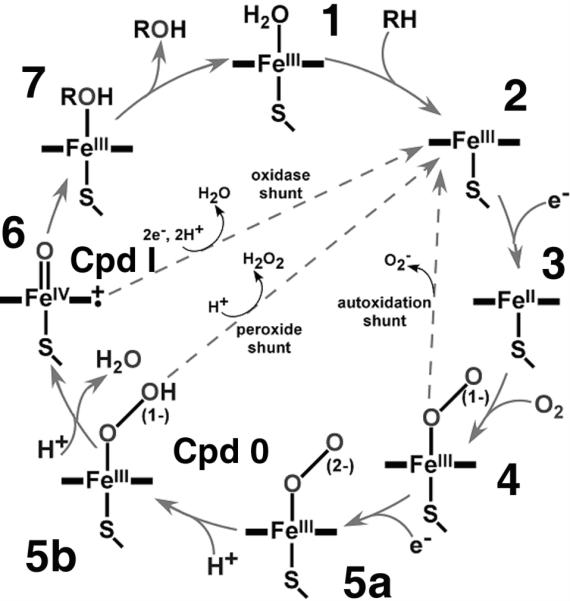
Catalytic cycle of cytochrome P450 showing the intermediates involved in the oxygenase mechanism.
As is depicted in Scheme 1, the catalytic cycle of the cytochromes P450 includes seven or eight distinct states, with three different formal oxidation states of iron. These include Fe2+ [3, 4] (although [4] has partial ferric character), Fe3+ [1, 2, 4, 5, 7] and Fe4+ [6]. The axial sixth ligand provides another distinguishing descriptor; in the substrate free form it is an aquo/hydroxo entity, dioxygen (superoxide) in the oxygenated complex, peroxo-dianion or protonated hydroperoxide (Cpd 0) in [5a, 5b] and product alcohol in [7]. Following cleavage of the dioxygen bond and release of water, the higher valent “oxo” state (Cpd I) contains a single oxygen atom [6]. Following oxygen atom transfer to substrate there are potential transient complexes with the product molecule prior to dissociation. Detailed knowledge about the most important steps of the reaction, including the structure and main features of the intermediate species on the path from reactants to products, is necessary for a deep understanding of the mechanism of P450 catalysis. This review focuses on the application of various types of spectroscopic tools to characterize the reaction intermediates of cytochromes P450 and the advancements that they have provided in understanding this enzyme system which plays a central role in metabolism. Although the focus is on spectroscopy, we make connection with structural data where available.
1. The ferric resting state of cytochrome P450
EPR Spectroscopy
Electron paramagnetic resonance (EPR, also known as ESR for electron spin resonance) spectroscopy allows one to measure the energy separation between different electronic-spin states and the nuclear hyperfine splitting constants [9]. In an ideal case, the information available from an EPR spectra are: the spin-state of the paramagnetic center, magnitude of hyperfine interactions, the zero-field splitting of half-integer S > 1/2 states, and the identity of the paramagnetic center (free radical or metals in metal cluster).
Due to their low redox potential, all cytochromes P450 tend to exist in the ferric state under aerobic conditions, where the heme iron is formally described as Fe3+ with five electrons in the 3d orbital. In the absence of substrates and inhibitors, the active sites of structurally characterized P450s show a hydrogen bonded cluster of several water molecules [10]. This network of hydrogen bonds stabilizes an iron-aquo or iron-hydroxo coordination at the sixth ligand position to the heme iron, trans to a sulfur atom provided by a conserved cysteine residue that serves as the axial ligand to the iron. In this case, iron is in the low-spin configuration, S = 1/2, having an EPR spectrum with g-values ~ [2.45, 2.26, 1.91] [11]. Binding of hydrophobic substrates results in a displacement of multiple water molecules, usually destabilizing water ligation to the iron, and allowing a 5-coordinated high-spin (HS) configuration, S = 5/2. The ionic radius of the iron in the S = 5/2 state is larger than the space provided by the four planar pyrolle ligands and the iron with its cysteine ligand moves out of plane. EPR spectra of HS cytochromes P450 have rhombically split g-values at ~ [8, 4, 1.7] [11]. There is a thermodynamic equilibrium between the high-spin and low-spin (LS) states of cytochrome P450, with the HS state favored at high temperatures, as confirmed using optical absorption spectroscopy [12-13], EPR [11], Mössbauer [14], and NMR [15-16]. This equilibrium has been documented in solution by a variety of optical spectroscopies and can be quantitated at lower temperatures by EPR and Mössbauer. Where the measurements are made below the solvent glass temperature, the spin state equilibrium is frozen in place, and even at helium temperatures (~ 4 K) there is substantial population of the high spin form [11].
Optical absorption spectroscopy
Optical absorption spectroscopy is one of the most sensitive of all spectroscopic methods, providing measurable data even with only a single monolayer of the heme protein. Optical methods also benefit from fast signal detection, where continuous data can be measured routinely on the nanosecond and faster scales, and pulse – probe methods extend measurements to the sub-picosecond range [17-19]. The ease and readily availability of optical absorption instrumentation leads it to dominate the physicochemical characterization of cytochromes P450, starting from concentration measurements during purification, monitoring substrate and inhibitor binding, kinetic studies in steady-state turnover experiments, as well as in different kinetic methods, such as stopped flow, laser photolysis, temperature jump etc..
Optical absorption spectra of the heme are sensitive to the spin state of the heme iron reflected in the characteristic positions of Soret band in UV absorption spectra; 415 – 417 nm for low-spin (LS) P450s and 390 – 394 nm for the high-spin (HS) state. Substrate binding to cytochromes P450 is often monitored by the concomitant transition from LS to HS using optical absorption difference spectra. The classic “Type I” difference spectra corresponding to the displacement of the active site water molecules by substrate binding generates a minimum at 420 and maximum at 390 nm. The absolute amount of cytochrome P450 converted from LS to HS state can be estimated with good precision from the amplitude of the observed difference spectrum. As all cytochromes P450 have the same heme prosthetic group, their optical absorption spectra vary only by a minor extent in the different isozymes. As a result, the difference between the spectra of pure LS and pure HS states is approximately the same for all cytochromes P450, with the peak-to-trough extinction coefficient (ε390 – ε420) ≈ 100 mM-1cm-1. Using this value one can estimate the fraction of the enzyme converted to the HS state by given substrate if the total enzyme concentration is known. Importantly, the presence of Type I spectral changes after addition of putative substrates to a given cytochrome P450 provides a crude means to estimate the ability of the enzyme to metabolize this compound.
Kinetic studies of substrate binding often indicate the presence of two or three distinct phases [20-22] in the optical changes. This can be interpreted as indication of multiple fractions of the protein with different accessibility or as complex multi-step sequential processes involving conformational changes of the hemoprotein. In addition, fast optical methods make possible the expansion of kinetic studies into the nanosecond and picosecond range and allow one to resolve relaxation processes in the binding and dissociation of substrates as well as diatomic ligands, using such methods as temperature jump [12, 23], rapid laser photoreduction [24-26], photolysis of diatomic ligands and nanosecond kinetics of geminate rebinding [17, 27].
In some cases, ‘Reverse Type I’ spectra are observed during titration with a given small molecule compound. This indicates the presence of a significant population of the HS state in the initial preparation of the enzyme, which is decreased or eliminated as a result of binding of the target compound. Such a situation suggests either the presence of a weakly bound endogenous substrate or substrate analog, which is replaced by the target compound thereby shifting the spin state equilibrium to the LS state. The latter case is has been documented in several isozymes such as CYP1A2 [28] and P450BioI [29] .
Difference spectra of opposite sign, historically termed “Type II”, with a positive maximum at 425 – 440 nm and negative minimum at 415 nm or lower are observed as a result of formation of 6-coordinated complexes with strong ligands, including many clinically important drugs and other inhibitors [30]. The coordination of strong ligands to the heme iron of cytochromes P450 shifts the Soret band to the longer wavelengths, from 422 – 435 nm for nitrogen containing compounds (imidazoles, pyridines, amines), 440 – 450 nm for cyanide and phosphine and 450 – 460 nm for thiolates. The optical changes on heme complex formation have been elegantly reviewed by Dawson [31-32]. The position of the Soret band and the presence of a “hyperporphyrin split” for the 6-coordinated heme complexes depends on the relative ability of the axial ligands to serve as π-acceptors, or as σ-donors, where the former type causes the simple red shift of the main electronic π–π* transition of the porphyrin and the latter results in the split Soret band [32-33]. A study published recently [34] described the binding of Type II nitrogen coordinating inhibitors to CYP2C9 and CYP3A4. As expected, imidazole, aniline, fluconazole, itraconazole and several other inhibitors, all produce Type II spectra with the Soret band red shifted by 2 – 8 nm and the amplitude at maximum decreased to different extents. These studies indicate a notable difference between the spectral properties of the different Type II ligands. The amplitude and shape of Type II spectra depends on the position of Soret bands and corresponding molar absorption coefficients, which may vary significantly for different ligands. Hence, the general quantitative approach to the spectral analysis of Type II binding similar to described above for Type I spectra, is impractical.
MCD Spectroscopy
Magnetic circular dichroism (MCD) involves measurement of CD spectra in a longitudinal magnetic field, the direction of which is parallel to the wave vector of circularly polarized light. This method directly probes the Zeeman splitting between degenerate energy levels of the paramagnetic iron atom, giving rise to a derivative-shaped and temperature independent contribution to the MCD spectra (A-term) [35-37]. At low temperatures, T<50K, a significant decrease of the Boltzmann population of the higher energy levels gives rise to the temperature dependent C-term with an absorption band shape [35, 38]. Another temperature independent MCD signal is observed from the transition between non-degenerate energy levels, if they are mixed in the magnetic field (B-term). In addition to strong porphyrin bands at 350 – 450 nm (Soret) and 500 – 600 nm (Q-band), several weaker transitions can be resolved at 600 – 1300 nm in MCD spectra of cytochromes P450 [37, 39]. They have been assigned to porphyrin – iron and iron-sulphur charge-transfer bands. For non-Kramers metal ions with integer spins the low-temperature MCD spectroscopy provides indispensable information about the details of electronic structure of metal centers, as reviewed by Solomon and coauthors [38]. The works of Dawson and collaborators documented the effects of the electronic properties of various ligands on the features of MCD spectra of P450 CYP101 in the ferric and ferrous state [32, 40-41]. The sensitivity of MCD spectra to the details of the active site hydration and to the presence and absence of substrate was further demonstrated by comparing the MCD spectra of different cytochromes P450 [42] and the spectra of CYP101 in the presence of different substrates [43]. In general, MCD spectra offer an excellent “fingerprint” for defining the oxidation, spin and ligation states of hemoproteins [6, 39, 44].
Resonance Raman Spectroscopy
Raman spectroscopy is a direct probe of the vibrational manifold of a protein. When the incident wavelength is near the absorption band of the heme prosthetic group, resonance enhancement occurs, with a dramatic increase in the intensity of the scattering. The main advantage of Resonance Raman (RR) spectroscopy is that when the wavelength of the exciting laser line is adjusted to coincide with that of an allowed electronic transition in a molecule, only the vibrational modes associated with that transition are enhanced, and thus one can selectively study a single chromophoric group in complex biomolecules. In P540s, RR spectra characterizes those modes directly linked to the porphyrin and iron – axial ligand if they are of the appropriate selection rules [45]. RR spectroscopy provides detailed and site specific information, monitoring vibrational modes of the porphyrin ring, or involving the vinyl, propionic, or methyl side chains. Additional opportunities for Raman spectroscopy are provided by reconstitution of cytochrome P450 with isotopically substituted protoporphyrin IX, or other porphyrins, which allow one to identify specific modes and detect site-specific protein-heme interactions [46-47].
In many cases, resonance Raman spectroscopy has been used to monitor the spectral changes caused by ligand binding, by following the prosthetic group core size and spin state sensitive modes ν2 (1584 and 1570 cm-1) and ν3 (1503 and 1488 cm-1) for 6-coordinated LS and 5-coordinated HS states respectively [46, 48-51]. Ligand vibrational modes have been used as a probe for interaction of the heme with substrates in resonance Raman studies of ferric CYP101 complexes with NO [52] and CN [53], and in isocyanide complexes of CYP2B4 [54].
Changes in the porphyrin vinyl modes upon the binding of imidazole inhibitors to CYP102 indicated a decrease of out-of-plane distortion of the porphyrin ring and in-plane alignment of vinyl side chains [55]. The same in-plane movement of the vinyl groups upon reduction have been observed in CYP102 [49], and the similar spectral changes in Phe393 mutants correlate with the changes in the heme redox potential caused by mutations in this position. These results provide a good example of the potential of resonance Raman spectroscopy to identify the local effects caused by point mutations or by the binding of small molecules.
NMR Spectroscopy
NMR methods used in the studies of cytochromes P450 have been recently reviewed by Pochapsky and coauthors [56]. The application of powerful NMR methods to large heme proteins is limited by the requirement of highly concentrated isotopically substituted samples and by the complicated assignment of the high resolution spectra, but can provide unprecedented selectivity and detailed insight into the equilibrium structural properties and dynamic behavior of these enzymes, even in complex with their redox partners [57-59]. Recently, a specific cis-trans isomerization of the Ile88-Pro89 amide bond induced by putidaredoxin (Pdx) binding has been identified in CYP101 in both oxidized and CO-bound reduced states [60]. This conformational switch changes orientation of the bound substrate camphor [61] and stabilizes Pdx-CYP101 complex.
Using perdeuterated substrate analog, adamantane, McDermott and coauthors studied substrate binding to CYP101 by monitoring paramagnetic shifts in solid-state deuterium magic angle spinning NMR [62]. Signals from different deuterium atoms were averaged, due to the fast rotation of the bound adamantane with respect to the heme iron. Thus, it was evident that this substrate could bind in different orientations and undergo fast rotational relaxation on a sub-millisecond time scale. A similar approach was used to reveal the correlation of the spin state change with the shift of the bound substrate analog N-palmitoylglycine in the heme iron substrate binding pocket of CYP102 [15, 63]. Another example of detecting the repositioning and reorientation of the substrate by monitoring NMR paramagnetic relaxation was documented for acetaminophen bound to CYP3A4 in the presence of caffeine [64]. One can also monitor the NMR signal from a diatomic ligand coordinated to the heme iron, such as 15N enriched cyanide. Using this method the presence of substrate camphor in the binding pocket and the subsequent conformational rearrangement caused by putidaredoxin binding has been detected by shift of 15N NMR signal. [65].
In addition, NMR spectroscopy can be used to study protein – protein interaction by monitoring distance – dependent perturbations of the signal caused by specific paramagnetic labels. An example of such an approach is described in the recent study of the complex between adrenodoxin and adrenodoxin reductase that was covalently labeled with caged lanthanide tags [66]. Pseudo-contact shifts induced by paramagnetic Tm3+ and paramagnetic relaxation enhanced by Gd3+ have been probed at the binding interface, and the distance restraints obtained from these experiments allowed for the refinement of the docking solutions to the 3.2 Ǻ resolution.
Mössbauer Spectroscopy
Mössbauer spectroscopy is a precise tool for characterizing the electronic structure of the heme iron atom in all possible oxidation states. It measures the electric and magnetic interactions of the iron nucleus with its environment and provides information about the spin state as well as the oxidation state of the Mössbauer atom as well as important structural parameters such as the zero-field splitting (D) and symmetry such as the rhombicity (E/D) and the electron spin density at the iron [67]. However, this method requires metals with an odd nuclear spin. Placing an 57Fe isotope in the heme prosthetic group, requiring either reconstitution with a labeled heme or heterologous expression to yield 57Fe isotope enrichment. In addition, low temperatures and relatively high quantities of the material, i.e. ~0.5 ml of > 0.2 mM 57Fe atoms [68] are needed for a manageable signal-to-noise ratio; hence, this method has mostly been limited to those microbial P450s where large quantities of material from bacterial expression are available.
In the first application of Mössbauer spectroscopy to P450 studies, Sharrock et al. [14] reported the parameters of the substrate free and substrate bound CYP101, and identified the mixture of HS and LS states in the substrate bound enzyme. Subsequent investigations at high magnetic fields by Champion and colleagues [69] were instrumental in defining the electronic structure of this cytochrome P450.
X-ray Spectroscopy
X-ray spectroscopic methods are useful tools for investigating the structure and redox properties of metal coordination compounds. In particular, extended X-ray absorption fine structure (EXAFS) Spectroscopy can provide highly accurate metal-ligand bond distances, and the number and identity of coordinating atoms in metalloenzymes [70]. Here the intensity of the iron absorption beyond the edge is modulated by the virtual backscatter from nearby atoms. Due to the large phase shift provided by sulfur backscattering, EXAFS has proved to be very valuable in investigations of the thiolate ligated heme of the cytochromes P450. By focusing on the edge region of the x-ray absorption envelope, X-ray absorption near edge spectroscopy (XANES) has been used to probe the efficient oxidation state of redox active metals and the ligand coordination geometry/symmetry for selected metals. In contrast with EXAFS, XANES spectroscopy focuses on the region about 100 eV around the Fe-K edge absorption. The XANES spectrum carries information about the geometry of heme iron, such as Fe-O-O bond angle, the distortion in coordination geometry around the iron and the electronic structure of the iron coordination sphere [71]. In many cases X-ray absorption spectroscopy can provide the most accurate metal-ligand bond distances, and structural information can be obtained from samples in either frozen solution or solid state. In addition, this spectroscopy is very element selective with little interference from other centers. However, the method requires high intensity X-rays from a synchrotron light source and often does not give a unique determination of ligand environment due to similarity of oxygen and nitrogen in scattering. Moreover, the conclusions often depend strongly on theoretical simulation and curve fitting.
Strong EXAFS experimental proof for the presence of thiolate as an axial ligand in both cytochrome P450cam and choloroperoxidase has been provided for ferric enzymes [72] and for ferrous complexes [73]. For CYP101, the presence of sulfur in the first coordinating sphere was confirmed, and the short Fe-S bond has been suggested as a strong indication of the unprotonated anionic cysteinate.
2. Ferrous cytochrome P450
Optical Absorption Spectroscopy
The absorption spectra of all heme proteins are sensitive to the redox state of the heme iron, and oxidation/reduction processes can be monitored by the changes in Soret region. For cytochromes P450, the position of Soret band maximum for the ferrous protein with no ligands to the heme iron (5-coordinated high-spin Fe2+) is 408 – 410 nm , while binding of ligands results in a splitting and red-shifting of the longer wavelength peak to 420 – 425 nm (dioxygen), 430 – 435 nm (NO), or 445 – 450 nm (CO). Optical absorption can be used to monitor the reduction of cytochromes P450 in spectroelectrochemical titration studies, as it was done with CYP101 [13], CYP102 [74], P450BioI [50], and CYP3A4 [75]. In addition, optical absorption is the most convenient method for kinetic methods, such as stopped flow studies of cytochrome P450 reduction with the native redox partners [76-77], or using chemical reductants [78]. Substrate binding to the ferrous state can be studied optically by monitoring the competitive binding of the relatively weak sixth ligand to the heme iron. For instance, camphor binding to CYP101 displaces pyridine from the substrate binding pocket with concomitant spectral changes [79]. The same approach was used with 4-cyanopyridine to study substrate binding to the ferrous CYP102 [80]. Reduced enzymes that retain coordination with inhibitors exhibit red-shifted Soret bands, for instance at 427 nm in CYP121 complexed with the antifungal drug ketoconazole [81].
The kinetics of diatomic ligand binding to ferrous cytochromes P450 was studied using stopped flow UV-Vis spectroscopy on bacterial soluble cytochromes CYP101, CYP102, CYP108 [82-83] and microsomal human CYP3A4 in Nanodiscs [84]. It was shown that binding of dioxygen and CO is relatively fast with the second order rates 105 – 106 M-1s-1, and is not rate-limiting in the steady-state kinetics of substrate turnover. Optical monitoring of nanosecond kinetics of photolysis of ferrous - CO complexes with the short laser pulses and subsequent recombination of diatomic ligand provides useful information about the structure and relaxation of the substrate binding pocket in several cytochromes P450, such as CYP101 [17], CYP102 [27], and CYP3A4 [84]. Changes of the geminate rebinding amplitude in the presence of substrate can indicate the extent of steric restriction caused by substrate binding, even in the absence of other spectroscopic signals [17, 27, 84]. The dynamics and relaxation of CYP101 have also been studied by cryogenic photolysis of the Fe-CO complex followed by thermal annealing, which allowed the estimation of kinetic barriers for the diatomic ligand escape form the protein matrix [19].
MCD Spectroscopy
Low-temperature MCD has been used to evaluate the differences between P450 and P420 [85]. Analysis of the low-temperature MCD together with available Mössbauer data [86] proved that the different spectral properties of histidine ligated Mb and HRP compared to the cysteine ligated cytochrome P450, indicate the distinct electron distributions in the ground state.
Mössbauer and EXAFS
Sharrock et al. [14, 87] documented Mössbauer spectra of reduced CYP101 without ligands, and complexes with dioxygen and CO. For the 5-coordinated ferrous CYP101 the minimal temperature dependence of the spectra indicate higher energy of the excited electronic levels than observed in deoxyhemoglobin, which shows pronounced temperature dependence. Spectra of the ferrous-CO complexes of CYP101 and hemoglobin are very similar, as are the spectra of oxy-ferrous complexes of these two proteins. Analysis of EXAFS data on the ferrous and ferrous-CO complexes of CYP101 provided the first estimate of the length of Fe-S bond formed by the heme iron and axial sulphur from Cys357. The distances were measured to be 2.20 Ǻ for the ferric and 2.34 Ǻ for the ferrous state, both shorter than in model porphyrin complexes. Binding of CO does not change the Fe-S bond length significantly, similar to that revealed in model complexes [70].
3. The oxygenated complex of P450
Oxygen binding to reduced P450 gives species 4 (Scheme I), the Fe2+-OO (ferrous dioxygen), or Fe3+- OO- (ferric superoxide) complex [88]. This is a quasi-stable intermediate and is the origin of an uncoupling pathway, termed the autoxidation shunt, wherein oxidation results in the concomitant production of a superoxide anion regenerating the ferric resting state. The rates of autoxidation vary for different cytochromes P450 with or without substrate from 0.005 s−1 for CYP101 with camphor to 20 s−1 for CYP3A4 without substrates. Sometimes autoxidation represents the main uncoupling pathway [89-90], especially if it is significantly faster than the rate of the second electron transfer, as shown for CYP3A4 [91]. The stability of the oxy-ferrous P450s is intimately related to the redox potential of the complex and as described recently in detail [92-93].
Splitting of the O-O bond requires the transfer of a second electron and concomitant delivery of two protons to species [4]. To elucidate the mechanism of dioxygen activation, extensive studies have been carried out to characterize each step of this reaction by biochemical, site-directed mutagenic, and biophysical methods. CYP101 has served as a paradigm for the mechanistic study of these P450 mechanisms due to the fact that it is a soluble protein, its reaction is highly coupled and its crystal structure was the first to be solved.
UV-Vis and MCD Characterization
The dioxygen-adduct of ferrous cytochrome P450 was first reported by Peterson and co-workers in 1971 for CYP101 [94]. Since then the UV-Vis absorption spectra of the dioxygen adduct of various cytochromes P450 have been published [95-100]. The Soret maximum for the oxy-ferrous complex of P450 usually exists at 417 – 418 nm, but may also be found at 424 nm as in CYP3A4 [84, 91] or even at 430 nm in CPO [41]. The Q-band has a maximum at 552 – 557 nm and a shoulder at ~580 nm [95]. Some P450s, such as the thermophilic CYP119 [101] and CYP102 [83, 99], display slightly unusual spectral properties, possibly due to changes in the structural parameters of the proximal thiolate ligand or by different types of interactions of the bound dioxygen molecule in the distal pocket of these two proteins.
The first cytochrome P450 whose oxy-ferrous form was characterized by MCD was the camphor-bound form of CYP101 [102]. Owing to similarities in the Mössbauer and optical absorption properties of oxy-CYP101 and oxy-Mb it was speculated that neutral histidine was the axial ligand in CYP101 as well [103]. Later, Dawson and Cramer reported the MCD spectra of oxy-CYP101 in its camphor-bound form and conclusively showed the presence of cysteinate sulphur as the axial heme ligand [102]. Sono et al. reported the MCD spectra of oxy-CYP101 in the substrate free and camphor bound forms and found them to be very similar [43]. These authors also showed that the electronic structure of the complex is sensitively influenced by the size and electronic properties of the bound substrate. They correlated this to the differences in the spin-state change of oxidized enzyme upon binding of different substrates and substrate-analogs and remarked that the ability of the substrate to modulate the reactivity of P450 intermediates could be a relevant factor in explaining the remarkable diversity of reactions catalyzed by the enzyme [43].
Raman Spectroscopy
For some time, Raman studies of oxy-complexes of P450s were limited by the instability of this intermediate at ambient temperature (t1/2 ~ 90 s at 298 K for CYP101) and the fact that this complex is photosensitive to laser illumination. However, studies with thiolate model compounds had identified the ν(O-O) stretching vibration at 1140 cm-1. Bangcharoenpaurpong et al. [104], using a special spinning optical cell cooled to cryogenic temperatures for their Raman experiments, reported the dioxygen stretching frequency for a complex with cytochrome P450. They observed a strong peak at 1140 cm-1 in the 16O2 spectrum for CYP101. 18O2 substitution resulted in a downshift of 66 cm-1. In studies with model compounds this isotopic shift was found to be 65 cm-1. These observations lead to the assignment of the stretching frequency of bound dioxygen, ν (O-O) to the 1140 cm -1 mode, establishing the nature of this intermediate as a ferric superoxide complex The ν(Fe-O2) mode could not be detected in this study. An important observation was the distinctive difference in the pattern of resonance Raman enhancement of ν(Fe-O2) and ν(O-O) in thiolate complexes and in associated nitrogen base complexes as in dioxygen bound to Mb or Hb [104].
The key mode ν(Fe-O2), which carries much information about the bond strength and geometry of the Fe-O2 oxygenated state, had eluded detection until Hu and Kincaid reported resonance Raman spectroscopic characterization of oxygenated cytochrome CYP101 in the presence of camphor and adamantanone, two of its substrates [105]. They observed, and identified by using 16O2/ 18O2 isotopic substitution, a ν(Fe-O2) mode at 541 cm-1 for camphor-bound oxygenated CYP101. The low frequency resonance Raman spectra of the Fe-O2 state indicated that this bond is substantially weakened in cytochrome P450. This lowering of frequencies was observed previously for ferrous-carbonyl [106] and ferric-nitrosyl [52] P450 adducts, owing to the electron donating effect of the axial thiolate ligand. Also in this study [105], the oxidation marker band, ν4, of cytochrome CYP101 was observed at 1374 cm-1, lower that that of oxyhemoproteins possessing histidyl as a proximal ligand. This showed the presence of a strong electron-releasing thiolate axial ligand. The significantly lowered frequencies of ν(O-O) and ν(Fe-O2) and their sensitivities to different substrates provides a structural basis for cleavage of the bound dioxygen to generate a regio- and stereospecific hydroxylation agent. Comparing resonance Raman spectra of oxy-CYP101 saturated with camphor and adamantanone, Hu et al. demonstrated that the vibrational frequencies associated with the bound dioxygen are sensitive to steric bulk of the substrate and provide direct evidence for its contact with bound dioxygen [105].
The first report of the bending mode of the Fe-O-O moiety came from Paul Champion at Northeastern University [107]. A new-oxygen sensitive mode was observed at 401 cm-1 in oxygenated wild type oxy-CYP10, and was assigned to the deformation motion of the iron-dioxygen ligand, δ (Fe-O-O) based on comparison with Co- and Fe-oxyporphyrin complexes .
Vibrational spectroscopy revealed that the O–O and Fe–O bonds are slightly weaker in oxy-P450 (1128 – 1140 cm−1 and 537 – 541 cm−1) [104-105] as compared to those seen in oxygenated myoglobin and hemoglobin (1148 cm−1 and 572 cm−1) [45]. This difference is explained by the more electron-rich thiolate ligand to the heme and an increased donation of electron density to the antibonding orbitals of Fe–O–O moiety in cytochrome P450. Sometimes, more than one band can be resolved at 1136 – 1138 cm−1 and 1126 – 1128 cm−1 for (O–O) mode [108-109] where the downshifted mode is attributed to the different conformer and/or stronger hydrogen bonding of the distal oxygen atom.
Mössbauer Spectroscopy
Sharrock et al. reported Mössbauer spectra of oxy-CYP101 in zero magnetic field as a doublet with a large quadrupole splitting and only a moderate isomer shift [87]. The Mössbauer spectrum of oxy-CYP101 was found to be very similar to that of oxyhemoglobin despite the other spectral differences in these two proteins. The values of quadrupole splitting and isomer shifts obtained are not commonly found in ferric and ferrous heme proteins and therefore, were thought to be characteristic of the heme-O2 linkage. Mössbauer data indicated that the complex corresponds more closely to a ferric superoxide complex than a ferrous dioxygen complex having a thiolate ligand [87].
EXAFS/XANES Spectroscopy
Long before the X-ray structure of the oxy-complex was known, Dawson et al. structurally characterized oxygenated CYP101 using EXAFS spectroscopy [110]. By comparing EXAFS spectra of oxy-CYP101 to that of oxy-chloroperoxidase they were able to confirm that the two complexes had identical heme iron coordination spheres as it was proposed earlier [41] and that the discrepancies in their UV-Vis absorption and MCD spectra were due to differences in the heme environments. Furthermore, the Fe-S bond distance of 2.37 Å was calculated for oxy-CYP101. Mechanistic implications of having thiolate ligand in the P450 system were subject of many discussions and have been summarized in [6]. Shiro et al. compared the XANES spectra of oxy-forms of CYP101, HRP and Mb [71]. They could show that the site-symmetry around the iron and the Fe-O-O bond angle is similar among these hemoproteins. However, the π-electron donation from the thiolate S to the Fe-O-O moiety is substantiated in the XANES spectrum of CYP101. This observation was consistent with its biological function where the weakening of the oxygen-oxygen bond favors its heterolytic scission and formation of the main ferryl-oxo catalytic intermediate.
X-ray Structure
The determination of the oxy-ferrous complex of CYP101 structure by Schlichting et al. in 2000 was a major breakthrough in the structural characterization of cytochromes P450 [111]. From the crystal structure, it was found that oxygen is coordinated in the bent ‘end-on’ mode with the angle Fe-O-O of 142°, indicating no steric conflict with the bound substrate molecule. This Fe-O-O angle is similar to that observed in myoglobin of 110 – 123° [112-116], 135 – 160° in hemoglobin [116-120], 131° in cytochrome c peroxidase [121], 126° in HRP [122], 114 – 134° in guanylate cyclase [123] and 101 – 114° in heme oxygenase [124]. Subsequently, the X-ray structures of oxy–ferrous complexes in mutant CYP101 [125] CYP107 [126], and CYP158 [127] were solved. Comparison of the structures of oxy-ferrous complexes in these three P450s revealed a common structural arrangement of the heme iron and axial ligand. The major differences concerned the detailed stereochemistry of the amino acid side chains and water molecules in the immediate vicinity of the bound dioxygen [111, 128].
4. The Peroxo States of P450
The heme-oxygen intermediates following the oxy-ferrous complex in the catalytic cycle can be formed by radiolytic reduction of the oxy-complex in frozen solution and subsequent thermal annealing. Symons, Davydov, Hutterman and others pioneered this approach by the introduction of low-temperature radiolytic reduction of frozen heme proteins [129-133]
Electron Paramagnetic Resonance Spectroscopy
Electron paramagnetic resonance (EPR) spectroscopy has been invaluable in defining the peroxo states of cytochrome P450. Davydov et al. reported the first EPR and ENDOR spectra of P450 peroxo-intermediate states prepared by radiolysis using 60Co γ-irradiation at low temperatures [130, 134-135]. This resulted in cryoreduction of 4 (Scheme 1) at 77K for CYP101 and its mutants, Thr252Ala and Asp251Asn. The primary reduced oxy-P450 species at 77 K was identified as a hydroperoxo-Fe3+-heme complex with g = 2.29, 2.166, 1.96 for CYP101 (WT) [135]. These EPR experiments also contributed to the understanding of the roles of the famous ‘acid-alcohol pair’ (Asp251-Thr252 in CYP101). It was observed that cryoreduced oxy-Thr252Ala does not yield product where the wild-type enzyme does. Therefore, 5B is a key intermediate at or near the branch point that leads either to product formation or to “uncoupling” in which O-O bond cleavage is replaced by H2O2 production. Moreover, since Thr252Ala mutation did not interfere with the formation of 5B, it can be concluded that this mutation perturbs the delivery of the second proton of catalysis. The EPR spectra of the (hydro)peroxo complexes of CYP101 documented in this study were similar to those reported for the same complexes in Hb, Mb and HRP, with a signature narrow span of g values (2.3-2.25, 2.2-2.14, 1.94-1.97) [129-133].
ENDOR Spectroscopy
The application of 1H ENDOR to cryogenically irradiated wild-type and mutant oxy-complexes enabled the visualization of proton(s) coordinated the ferric-peroxo species and the delineation of a covalently bound and hydrogen bonded proton. In CYP101, a detailed ENDOR analysis of exchangeable protons throughout the annealing profile of reduced oxy-ferrous CYP101 strongly indicated the involvement of a high-valency intermediate, although neither optical nor EPR studies could directly detect the Compound I state [135]. As shown for CYP101 and later, CYP2B4, the g-values of cryoreduced hydroperoxo-ferric in substrate free and substrate bound forms are different, which indicates that the active site structure in this state can be modulated by the substrate [136-137].
UV-Vis Absorption Spectroscopy
Electronic absorption spectroscopy is an essential tool to characterize cytochrome P450 intermediates and complements paramagnetic methods that are blind to important diamagnetic or antiferromagnetically coupled states. UV-Vis spectroscopy is a particularly important tool for documenting peroxo states since it connects low temperature radiolysis to ambient temperature rapid-mix experiments.
The first UV-visible spectra of the reduced oxygenated state (hydroperoxo-ferric complex) of P450 with CYP101 used cryogenic radiolytic reduction with phosphorus-32 [101]. Following the increase in the absorbed dose of 32P radioactivity (β-radiation from 32P decay), the absorbance at 417 nm (maximum of the oxy-P450 spectrum at low temperatures) decreased, and a new peak at ~440 nm emerged with time. These changes reflected the progress of one-electron reduction of oxy-P450 and the formation of reduced oxy-P450 complex. The irreversible evolution of the reduced oxy-P450 reaction intermediate upon its careful annealing from 77 to 240 K was also monitored by UV-Vis spectroscopy. Formation of the low-spin ferric cytochrome P450 (having an absorbance maxima at 417 nm for CYP101) as an intermediate step before the product release that was previously determined by EPR spectroscopy [135] was also confirmed in this study. The results of these and subsequent investigations are summarized in [138], and show UV-Vis characterization of the hydroperoxo states generated by low-temperature radiolysis of the ferrous-oxy complex in CYP101, CYP119 and chloroperoxidase among other systems.
X-Ray Absorption Spectroscopy (XAS)
All of the spectroscopic techniques discussed so far have clearly been important in providing a fingerprint of the various iron-ligand states of cytochrome P450. However, they do not provide detailed information about the geometry of the ligand. Precise structural determination of the heme iron coordination environment and associated changes is important in elucidating the catalytic mechanism in these systems.
Sligar and co-workers used XAS to characterize the ligand environment of the radiolytically produced states in CYP101 [138]. Comparison of the edge positions of oxyferrous and low-temperature radiolytically reduced form of CYP101 showed a shift to higher edge energy, indicating a metal centered oxidation. This observation indicated a major structural change concomitant with a redistribution of electron density upon the input of the second electron required for catalysis. Fitting the EXAFS region showed a decrease in symmetry attributable to the elongation of the proximal thiolate bond.
Raman Spectroscopy
Resonance Raman spectroscopy is the most powerful method for providing direct information about Fe−X−Y bonds and, through the observation of hydrogen−deuterium isotope shifts of corresponding vibrational modes, can reveal important details concerning the protonation state of exchangeable groups, such as the peroxo-ferric fragments of interest. In contrast, neither EPR/ ENDOR nor XAS can define the electrostatics of the bound peroxo or delineate subtle differences between a short, strong hydrogen bond from a full covalent protonation event. The first systematic RR characterization of the peroxo and hydroperoxo intermediates in WT and Asp251Asn mutant of CYP101 resulted from a collaboration of our group with that of Prof. Kincaid at Marquette University [139-140]. It was noted that cryoradiolysis of oxygenated WT CYP101 at 77K directly yields the hydroperoxo derivative with ν(O-O) and ν(Fe-O) at 799 cm-1 and 559 cm-1 respectively. The unprotonated precursor was not observed for WT CYP101. Resonance Raman investigations of the Asp251Asn mutant of CYP101, in which the proton delivery is hampered, enabled the observation of the peroxo intermediate with ν(O-O) and ν(Fe-O) at 792 cm-1 and 553 cm-1 respectively. The most interesting observation in experiments with mutant CYP101 was the direct observation of a 18 cm-1 downshift in the ν(O-O) mode, indicating a weakening of the O-O bond due to the protonation of the peroxo-anion coordinated to iron. This was accompanied with a concurrent increase in the ν(Fe-O) mode by 11 cm-1 to 564 cm-1. Furthermore, the ν(O-O) in peroxo-ferric intermediates in cytochrome P450 was found to be lower than that in myoglobin substituted with cobalt-substituted heme. This showed that the trans effect of thiolate weakens the O-O bond and promotes heterolytic cleavage with concomitant formation of the high-valent catalytically active ferryl-oxo intermediate [6].
5. The Compound I and Compound II Ferryl-oxo Species
Heterolytic scission of O-O bond in the hydroperoxo-ferric intermediate yields the main catalytically active intermediate of cytochrome P450, the ferryl-oxo complex with a pi-cation radical on the porphyrin ring. This intermediate is called Compound I (Cpd I) following traditional phenomenological classification in peroxidase literature [141-142]. Properties of high valent ferryl oxo complexes in metalloporphyrins and heme proteins have been reviewed recently [143-145]. In addition, a detailed comprehensive review on Cpd I was recently published by Jung [146]. Cpd I in heme proteins can be detected by the broad band at 650 – 690 nm in optical absorption spectra, short Fe-O distance determined by EXAFS (1.65 – 1.68 Ǻ) and high Fe-O stretch frequency (790 – 810 cm-1) [147] accompanied by ~20 cm-1 downshift of ν2 in resonance Raman spectra [145-146, 148]. EPR and Mössbauer spectroscopy also allow for distinguishing between Cpd I and Cpd II, although they yield spectroscopic parameters with considerable variations (see Table 1 in the reference [146]), possibly due to perturbations caused by other factors, such as pH variations at cryogenic temperatures, difference in coupling energy between iron spin and porphyrin radical, and easy radiolytic reduction of Cpd I.
The porphyrin-π-cation radical in model complexes has a characteristic UV-visible spectrum with the Soret band between 390 – 406 nm, and a weak band between 670 – 690 nm [149] which appear in CPO at about 370 nm and 690 nm, respectively [150]. In general, the most characteristic feature of Cpd I is the broad weak band at 650 – 690 nm in absorption spectra (ε = 4 – 12 mM-1cm-1), which is indicative of the porphyrin radical [151]. If this band is not seen in the optical spectra, the assignment of the observed intermediate as Cpd I can be questioned, at least for the porphyrin radical part. In addition, this band can be observed at the ambient conditions and does not require high concentrations of the protein. Because of the unstable transient nature of this intermediate in cytochromes P450, most data have been obtained using stopped flow and freeze-quench techniques [152-161]. Numerous attempts to observe Cpd I by EPR [135, 137] or optical absorption spectroscopy [101, 138] by careful annealing of hydroperoxo-ferric CYP101 have been unsuccessful. However, analysis of the product formed in H2O and D2O [135] unambiguously demonstrated the rebound hydroxylation [143] catalyzed by Cpd I.
These studies show (see Table 1 of reference [146]) that the iron-oxo character is proved by the crystal structure showing electron density corresponding to a single oxygen atom bound to the iron in a distance of ~1.65 Ǻ [122]. The longer Fe-O bond distance corresponds to the protonated state of the oxygen, as indicate EXAFS studies on Compound I and Compound II species [162]. Resonance Raman studies show that the Fe=O stretch vibration frequency for six-coordinated porphyrin model complexes with non-radical character of the porphyrin (Compound II) appears between 818 cm-1 and 841 cm-1 [163]. If the porphyrin is a pi-cation radical (Compound I) this frequency shifts to lower values at about 760–780 cm-1 [149].
The high-valent Fe4+ state has a characteristic doublet signal in the 57Fe Mössbauer spectrum with a low isomer shift ~ 0.03 – 0.14 mm/s [149]. The iron–porphyrin radical system reveals an unusual axial EPR spectrum, which depends on the ratio of the spin exchange interaction energy J and the zero-field splitting parameter D for the iron spin / porphyrin-spin coupling [164-165]. Recent ENDOR studies on the Compound I of CPO showed that the radical is mainly located in the porphyrin π-system with some admixture of ~0.2 spin density at the sulfur ligand [161] which is suggested to be also the case in P450.
When this review was in revision, the first direct spectroscopic characterization of Cpd I in cytochrome P450 was published by Rittle and Green [166]. Using highly purified substrate free cytochrome P450 CYP119 from the thermophilic organism Sulfolobus acidocaldarius, they were able to freeze-quench the product of the reaction of this heme enzyme with m-chloroperbenzoic acid which was identified as Cpd I based on the optical absorption (signature porphyrin pi-cation band at 690 nm and broad Soret band at ~370 nm with low intensity) and Mössbauer parameters (δ = 0.11 mm/s, ΔE = 0.90 mm/s) similar to the spectral characteristics of the Cpd I in chloroperoxidase. However, EPR spectra of this intermediate in CYP119 are different from those in CPO due to the higher exchange coupling parameter J = 1.3 (J = 1.02 in CPO). The origin of this difference is not clear and may be attributed to the higher spin density or shorter Fe-S distance in CYP119 [166].
Conclusion
In the field of bioinorganic chemistry there is a constant quest by the x-ray crystallographer and the spectroscopist to define the functionally important aspects of an enzyme. Obviously, both approaches are needed for a deepest understanding of biological systems. In the case of heme proteins in general, and the cytochromes P450 in particular, spectroscopy has provided indispensable guidance to the quest for a precise mechanistic description of these systems.
Acknowledgments
This work was supported by NIH grants GM31756 and GM33775 to S.G. Sligar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nelson DR. Human Genomics. 2009;4:59–65. doi: 10.1186/1479-7364-4-1-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guengerich FP. Chem. Res. Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 3.Ortiz de Montellano PR, De Voss JJ. Substrate Oxidation by Cytochrome P450 Enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Function, Genetics. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 183–245. [Google Scholar]
- 4.Klingenberg M. Arch. Biophys. Biochem. 1958;75:376–386. doi: 10.1016/0003-9861(58)90436-3. [DOI] [PubMed] [Google Scholar]
- 5.Omura T, Sato R. J. Biol. Chem. 1962;237:1375–1376. [PubMed] [Google Scholar]
- 6.Dawson JH, Sono M. Chem. Rev. 1987;87:1255–1276. [Google Scholar]
- 7.Wells AV, Li P, Champion PM, Martinis SA, Sligar SG. Biochemistry. 1992;31:4384–4393. doi: 10.1021/bi00133a002. [DOI] [PubMed] [Google Scholar]
- 8.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 9.Hales BJ. Electron Paramagnetic Resonance (EPR) Spectroscopy. In: Scott RA, Lukehart CM, editors. Applications of Physical Methods to Inorganic and Bioinorganic Chemistry. John Wiley & Sons, Ltd.; 2007. [Google Scholar]
- 10.Poulos TL, Meharenna YT, Sigel A, Sigel H, Sigel RKO, editors. Metal Ions in Life Sciences. Vol. 3. 2007. Structures of P450 proteins and their molecular phylogeny. pp. 57–96. [Google Scholar]
- 11.Lipscomb JD. Biochemistry. 1980;19:3590–3599. doi: 10.1021/bi00556a027. [DOI] [PubMed] [Google Scholar]
- 12.Fisher MT, Sligar SG. Biochemistry. 1987;26:4797–4803. doi: 10.1021/bi00389a029. [DOI] [PubMed] [Google Scholar]
- 13.Sligar SG. Biochemistry. 1976;15:5399–5406. doi: 10.1021/bi00669a029. [DOI] [PubMed] [Google Scholar]
- 14.Sharrock M, Münck E, Debrunner PG, Marshall V, Lipscomb JD, Gunsalus IC. Biochemistry. 1973;12:258–265. doi: 10.1021/bi00726a013. [DOI] [PubMed] [Google Scholar]
- 15.Jovanovic T, Farid R, Friesner RA, McDermott AE. J. Am. Chem. Soc. 2005;127:13548–13552. doi: 10.1021/ja0524604. [DOI] [PubMed] [Google Scholar]
- 16.Ravindranathan KP, Gallicchio E, Friesner RA, McDermott AE, Levy RM. J. Am. Chem. Soc. 2006;128:5786–5791. doi: 10.1021/ja058465i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian WD, Wells AV, Champion PM, Di Primo C, Gerber N, Sligar SG. J. Biol. Chem. 1995;270:8673–8679. doi: 10.1074/jbc.270.15.8673. [DOI] [PubMed] [Google Scholar]
- 18.Gruia F, Ionascu D, Kubo M, Ye X, Dawson J, Osborne RL, Sligar SG, Denisov I, Das A, Poulos TL, Terner J, Champion PM. Biochemistry. 2008;47:5156–5167. doi: 10.1021/bi7025485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tetreau C, Mouawad L, Murail S, Duchambon P, Blouquit Y, Lavalette D. Biophys. J. 2005;88:1250–1263. doi: 10.1529/biophysj.104.050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isin EM, Guengerich FP. J. Biol. Chem. 2006;281:9127–9136. doi: 10.1074/jbc.M511375200. [DOI] [PubMed] [Google Scholar]
- 21.Isin EM, Guengerich FP. Anal. Bioanal. Chem. 2008;392:1019–1030. doi: 10.1007/s00216-008-2244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davydov DR, Halpert JR. Expert Opin. Drug Metab. Toxicol. 2008;4:1523–1535. doi: 10.1517/17425250802500028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brenner S, Hay S, Girvan HM, Munro AW, Scrutton NS. J. Phys. Chem. B. 2007;111:7879–7886. doi: 10.1021/jp073036n. [DOI] [PubMed] [Google Scholar]
- 24.Dunford AJ, McLean KJ, Sabri M, Seward HE, Heyes DJ, Scrutton NS, Munro AW. J. Biol. Chem. 2007;282:24816–24824. doi: 10.1074/jbc.M702958200. [DOI] [PubMed] [Google Scholar]
- 25.Sevrioukova IF, Hazzard JT, Tollin G, Poulos TL. Biochemistry. 2001;40:10592–10600. doi: 10.1021/bi010874d. [DOI] [PubMed] [Google Scholar]
- 26.Dunn AR, Dmochowski IJ, Winkler JR, Gray HB. J. Am. Chem. Soc. 2003;125:12450–12456. doi: 10.1021/ja0294111. [DOI] [PubMed] [Google Scholar]
- 27.McLean MA, Yeom H, Sligar SG. Biochimie. 1996;78:700–705. doi: 10.1016/s0300-9084(97)82527-8. [DOI] [PubMed] [Google Scholar]
- 28.Sandhu P, Guo Z, Baba T, Martin MV, Tukey RH, Guengerich FP. Arch. Biochem. Biophys. 1994;309:168–177. doi: 10.1006/abbi.1994.1099. [DOI] [PubMed] [Google Scholar]
- 29.Green AJ, Rivers SL, Cheeseman M, Reid GA, Quaroni LG, Macdonald ID, Chapman SK, Munro AW. J. Biol. Inorg. Chem. 2001;6:523–533. doi: 10.1007/s007750100229. [DOI] [PubMed] [Google Scholar]
- 30.McLean KJ, Dunford AJ, Neeli R, Driscoll MD, Munro AW. Arch. Biochem. Biophys. 2007;464:228–240. doi: 10.1016/j.abb.2007.03.026. [DOI] [PubMed] [Google Scholar]
- 31.Andersson LA, Sono M, Dawson JH. Biochim. Biophys. Acta. 1983;748:341–352. doi: 10.1016/0167-4838(83)90178-4. [DOI] [PubMed] [Google Scholar]
- 32.Dawson JH, Andersson LA, Sono M, Hager LP. New J. Chemistry. 1992;16:577–582. [Google Scholar]
- 33.Hanson LK, Eaton WA, Sligar SG, Gunsalus IC, Gouterman M, Connell CR. J. Am. Chem. Soc. 1976;98:2672–2674. doi: 10.1021/ja00425a050. [DOI] [PubMed] [Google Scholar]
- 34.Locuson CW, Hutzler JM, Tracy TS. Drug Metab. Dispos. 2007;35:614–622. doi: 10.1124/dmd.106.012609. [DOI] [PubMed] [Google Scholar]
- 35.McMaster J, Oganesyan VS. Curr Opin Struct Biol. 2010;20:615–22. doi: 10.1016/j.sbi.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Oganesyan VS, Sharonov YA. Biochim Biophys Acta. 1998;1429:163–75. doi: 10.1016/s0167-4838(98)00228-3. [DOI] [PubMed] [Google Scholar]
- 37.Thomson AJ, Cheesman MR, George SJ. Methods Enzymol. 1993;226:199–232. doi: 10.1016/0076-6879(93)26011-w. [DOI] [PubMed] [Google Scholar]
- 38.Solomon EI, Bell CB. Inorganic and Bioinorganic Spectroscopy. In: Bakac A, editor. Physical Inorganic Chemistry: Principles, Methods, and Models. John Wiley & Sons, Inc.; New York: 2010. pp. 1–37. [Google Scholar]
- 39.Cheesman MR, Greenwood C, Thomson AJ. Magnetic circular dichroism of hemoproteins. In: Sykes AG, editor. Adv. Inorg. Chem. Academic Press; San Diego: 1991. pp. 201–255. [Google Scholar]
- 40.Dawson JH, Andersson LA, Sono M. J. Biol. Chem. 1982;257:3606–3617. [PubMed] [Google Scholar]
- 41.Sono M, Eble KS, Dawson JH, Hager LP. J. Biol. Chem. 1985;260:15530–15535. [PubMed] [Google Scholar]
- 42.Andersson LA, Johnson AK, Peterson JA. Arch. Biochem. Biophys. 1997;345:79–87. doi: 10.1006/abbi.1997.0248. [DOI] [PubMed] [Google Scholar]
- 43.Sono M, Perera R, Jin S, Makris TM, Sligar SG, Bryson TA, Dawson JH. Arch. Biochem. Biophys. 2005;436:40–49. doi: 10.1016/j.abb.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 44.Cheek J, Dawson JH. Magnetic circular dichroism spectroscopy of heme proteins and model systems. In: Kadish KM, Smith KM, Guilard R, editors. Porphyrin Handbook. Academic Press; 2000. pp. 339–369. [Google Scholar]
- 45.Kincaid JR. Resonance Raman spectra of heme proteins and model compounds. In: Kadish KM, Smith KM, Guilard R, editors. Porphyrin Handbook. Academic Press; N.Y.: 2000. pp. 225–291. [Google Scholar]
- 46.Mak PJ, Kaluka D, Manyumwa ME, Zhang H, Deng T, Kincaid JR. Biopolymers. 2008;89:1045–1053. doi: 10.1002/bip.21058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh UP, Obayashi E, Takahashi S, Iizuka T, Shoun H, Shiro Y. Biochim. Biophys. Acta. 1998;1384:103–111. doi: 10.1016/s0167-4838(98)00006-5. [DOI] [PubMed] [Google Scholar]
- 48.Niaura G, Reipa V, Mayhew MP, Holden M, Vilker VL. Arch. Biochem. Biophys. 2003;409:102–112. doi: 10.1016/s0003-9861(02)00581-7. [DOI] [PubMed] [Google Scholar]
- 49.Chen Z, Ost TWB, Schelvis JPM. Biochemistry. 2004;43:1798–1808. doi: 10.1021/bi034920g. [DOI] [PubMed] [Google Scholar]
- 50.Lawson RJ, Leys D, Sutcliffe MJ, Kemp CA, Cheesman MR, Smith SJ, Clarkson J, Smith WE, Haq I, Perkins JB, Munro AW. Biochemistry. 2004;43:12410–12426. doi: 10.1021/bi049132l. [DOI] [PubMed] [Google Scholar]
- 51.Tosha T, Kagawa N, Ohta T, Yoshioka S, Waterman Michael R, Kitagawa T. Biochemistry. 2006;45:5631–5640. doi: 10.1021/bi060094a. [DOI] [PubMed] [Google Scholar]
- 52.Hu S, Kincaid JR. J. Am. Chem. Soc. 1991;113:2843–2850. [Google Scholar]
- 53.Deng TJ, Macdonald IDG, Simianu MC, Sykora M, Kincaid JR, Sligar SG. J. Am. Chem. Soc. 2001;123:269–278. doi: 10.1021/ja001517d. [DOI] [PubMed] [Google Scholar]
- 54.Tomita T, Ogo S, Egawa T, Shimada H, Okamoto N, Imai Y, Watanabe Y, Ishimura Y, Kitagawa T. J. Biol. Chem. 2001;276:36261–36267. doi: 10.1074/jbc.M104932200. [DOI] [PubMed] [Google Scholar]
- 55.Smith SJ, Munro AW, Smith WE. Biopolymers. 2003;70:620–627. doi: 10.1002/bip.10502. [DOI] [PubMed] [Google Scholar]
- 56.Pochapsky TC, Kazanis S, Dang M. Antioxid. Redox Signal. 2010;13:1273–1296. doi: 10.1089/ars.2010.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pochapsky SS, Dang M, OuYang B, Simorellis AK, Pochapsky TC. Biochemistry. 2009;48:4254–4261. doi: 10.1021/bi900002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pochapsky SS, Pochapsky TC, Wei JW. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- 59.Asciutto EK, Madura JD, Pochapsky SS, OuYang B, Pochapsky TC. J. Mol. Biol. 2009;388:801–814. doi: 10.1016/j.jmb.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.OuYang B, Pochapsky SS, Dang M, Pochapsky TC. Structure. 2008;16:916–923. doi: 10.1016/j.str.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei JY, Pochapsky TC, Pochapsky SS. J. Am. Chem. Soc. 2005;127:6974–6976. doi: 10.1021/ja051195j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee H, Ortiz de Montellano PR, McDermott AE. Biochemistry. 1999;38:10808–10813. doi: 10.1021/bi990463l. [DOI] [PubMed] [Google Scholar]
- 63.Jovanovic T, McDermott AE. J. Am. Chem. Soc. 2005;127:13816–13821. doi: 10.1021/ja0438314. [DOI] [PubMed] [Google Scholar]
- 64.Cameron MD, Wen B, Roberts AG, Atkins WM, Campbell AP, Nelson SD. Chem. Res. Toxicol. 2007;20:1434–1441. doi: 10.1021/tx7000702. [DOI] [PubMed] [Google Scholar]
- 65.Shiro Y, Iizuka T, Makino R, Ishimura Y, Morishima I. J. Am. Chem. Soc. 1989;111:7707–7711. [Google Scholar]
- 66.Keizers PHJ, Mersinli B, Reinle W, Donauer J, Hiruma Y, Hannemann F, Overhand M, Bernhardt R, Ubbink M. Biochemistry. 2010;49:6846–6855. doi: 10.1021/bi100598f. [DOI] [PubMed] [Google Scholar]
- 67.Schünemann V, Paulsen H. Mössbauer Spectroscopy. In: Scott RA, Lukehart CM, editors. Applications of Physical Methods to Inorganic and Bioinorganic Chemistry. John Wiley & Sons, Ltd.; 2007. pp. 243–269. [Google Scholar]
- 68.Martinho M, Münck E. 57Fe Mossbauer Spectroscopy in Chemistry and Biology. In: Bakac A, editor. Physical Inorganic Chemistry: Principles, Methods, and Models. John Wiley & Sons, Inc; New York: 2010. pp. 39–67. [Google Scholar]
- 69.Champion PM, Lipscomb JD, Münck E, Debrunner P, Gunsalus IC. Biochemistry. 1975;14:4151–4158. doi: 10.1021/bi00690a001. [DOI] [PubMed] [Google Scholar]
- 70.Andersson LA, Dawson JH. Struct. Bonding (Berlin) 1991;74:1–40. [Google Scholar]
- 71.Shiro Y, Makino R, Sato F, Oyanagi H, Matsushita T, Ishimura Y, Iizuka T. Biochim. Biophys. Acta. 1991;1115:101–107. doi: 10.1016/0304-4165(91)90018-c. [DOI] [PubMed] [Google Scholar]
- 72.Cramer SP, Dawson JH, Hodgson KO, Hager LP. J. Am. Chem. Soc. 1978;100:7282–7290. [Google Scholar]
- 73.Hahn JE, Hodgson KO, Andersson LA, Dawson JH. J. Biol. Chem. 1982;257:10934–10941. [PubMed] [Google Scholar]
- 74.Ost TWB, Miles CS, Munro AW, Murdoch J, Reid GA, Chapman SK. Biochemistry. 2001;40:13421–13429. doi: 10.1021/bi010716m. [DOI] [PubMed] [Google Scholar]
- 75.Das A, Grinkova YV, Sligar SG. J. Am. Chem. Soc. 2007;129:13778–13779. doi: 10.1021/ja074864x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hintz MJ, Peterson JA. J. Biol. Chem. 1981;256:6721–6728. [PubMed] [Google Scholar]
- 77.Hintz MJ, Mock DM, Peterson LL, Tuttle K, Peterson JA. J. Biol. Chem. 1982;257:14324–14332. [PubMed] [Google Scholar]
- 78.Hintz MJ, Peterson JA. J. Biol. Chem. 1980;255:7317–7325. [PubMed] [Google Scholar]
- 79.Griffin BW, Peterson JA. Biochemistry. 1972;11:4740–4746. doi: 10.1021/bi00775a017. [DOI] [PubMed] [Google Scholar]
- 80.Ost TW, Clark JP, Anderson JL, Yellowlees LJ, Daff S, Chapman SK. J. Biol. Chem. 2004;279:48876–48882. doi: 10.1074/jbc.M408601200. [DOI] [PubMed] [Google Scholar]
- 81.McLean KJ, Cheesman MR, Rivers SL, Richmond A, Leys D, Chapman SK, Reid GA, Price NC, Kelly SM, Clarkson J, Smith WE, Munro AW. J. Inorg. Biochem. 2002;91:527–541. doi: 10.1016/s0162-0134(02)00479-8. [DOI] [PubMed] [Google Scholar]
- 82.Peterson JA, Griffin BW. Arch. Biochem. Biophys. 1972;151:427–433. doi: 10.1016/0003-9861(72)90518-8. [DOI] [PubMed] [Google Scholar]
- 83.Sevrioukova IF, Peterson JA. Arch. Biochem. Biophys. 1995;317:397–404. doi: 10.1006/abbi.1995.1180. [DOI] [PubMed] [Google Scholar]
- 84.Denisov IG, Grinkova YV, McLean MA, Sligar SG. J. Biol. Chem. 2007;282:26865–26873. doi: 10.1074/jbc.M704747200. [DOI] [PubMed] [Google Scholar]
- 85.Sharonov YA, Pismensky VF, Greschner S, Ruckpaul K. Biochem. Biophys. Res. Commun. 1987;146:165–172. doi: 10.1016/0006-291x(87)90706-6. [DOI] [PubMed] [Google Scholar]
- 86.Oganesyan VS, Sharonov YA. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1997;53A:433–449. doi: 10.1016/s1386-1425(96)01802-1. [DOI] [PubMed] [Google Scholar]
- 87.Sharrock M, Debrunner PG, Schulz C, Lipscomb JD, Marshall V, Gunsalus IC. Biochim. Biophys. Acta. 1976;420:8–26. doi: 10.1016/0005-2795(76)90340-8. [DOI] [PubMed] [Google Scholar]
- 88.Momenteau M, Reed C. Chem. Rev. 1994;94:659–698. [Google Scholar]
- 89.Hanukoglu I. Drug Metab. Rev. 2006;38:171–196. doi: 10.1080/03602530600570040. [DOI] [PubMed] [Google Scholar]
- 90.Kuthan H, Ullrich V. Europ. J. Biochem. 1982;126:583–588. doi: 10.1111/j.1432-1033.1982.tb06820.x. [DOI] [PubMed] [Google Scholar]
- 91.Denisov IG, Grinkova YV, Baas BJ, Sligar SG. J. Biol. Chem. 2006;281:23313–23318. doi: 10.1074/jbc.M605511200. [DOI] [PubMed] [Google Scholar]
- 92.Denisov IG, Sligar SG. Cytochrome P450 enzymes. In: Kadish KM, Smith KM, Guilard R, editors. Handbook of Porphyrin Chemistry. World Scientific; London: 2010. pp. 165–210. [Google Scholar]
- 93.Ost TWB, Clark J, Mowat CG, Miles CS, Walkinshaw MD, Reid GA, Chapman SK, Daff S. J. Am. Chem. Soc. 2003;125:15010–15020. doi: 10.1021/ja035731o. [DOI] [PubMed] [Google Scholar]
- 94.Ishimura Y, Ullrich V, Peterson JA. Biochem. Biophys. Res. Comm. 1971;42:140–146. doi: 10.1016/0006-291x(71)90373-1. [DOI] [PubMed] [Google Scholar]
- 95.Eisenstein L, Debey P, Douzou P. Biochem. Biophys. Res. Comm. 1977;77:1377–1383. doi: 10.1016/s0006-291x(77)80131-9. [DOI] [PubMed] [Google Scholar]
- 96.Denisov IG, Hung S-C, Weiss KE, McLean MA, Shiro Y, Park S-Y, Champion PM, Sligar SG. J. Inorg. Biochem. 2001;87:215–226. doi: 10.1016/s0162-0134(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 97.Bonfils C, Debey P, Maurel P. Biochem. Biophys. Res. Comm. 1979;88:1301–1307. doi: 10.1016/0006-291x(79)91122-7. [DOI] [PubMed] [Google Scholar]
- 98.Larroque C, Lange R, Maurin L, Bienvenue A, van Lier JE. Arch. Biochem. Biophys. 1990;282:198–201. doi: 10.1016/0003-9861(90)90104-7. [DOI] [PubMed] [Google Scholar]
- 99.Bec N, Anzenbacher P, Anzenbacherova E, Gorren ACF, Munro AW, Lange R. Biochem. Biophys. Res. Comm. 1999;266:187–189. doi: 10.1006/bbrc.1999.1794. [DOI] [PubMed] [Google Scholar]
- 100.Tuckey RC, Kamin H. J. Biol. Chem. 1982;257:9309–9314. [PubMed] [Google Scholar]
- 101.Denisov IG, Makris TM, Sligar SG. J. Biol. Chem. 2001;276:11648–11652. doi: 10.1074/jbc.M010219200. [DOI] [PubMed] [Google Scholar]
- 102.Dawson JH, Cramer SP. FEBS Lett. 1978;88:127–130. doi: 10.1016/0014-5793(78)80623-1. [DOI] [PubMed] [Google Scholar]
- 103.Chevion M, Peisach J, Blumberg W. J. Biol. Chem. 1977;252:3637–3645. [PubMed] [Google Scholar]
- 104.Bangcharoenpaurpong O, Rizos AK, Champion PM, Jollie D, Sligar SG. J. Biol. Chem. 1986;261:8089–8092. [PubMed] [Google Scholar]
- 105.Hu S, Schneider AJ, Kincaid JR. J. Am. Chem. Soc. 1991;113:4815–4822. [Google Scholar]
- 106.Uno T, Nishimura Y, Makino R, Iizuka T, Ishimura Y, Tsuboi M. J. Biol. Chem. 1985;260:2023–2026. [PubMed] [Google Scholar]
- 107.Macdonald IDG, Sligar SG, Christian JF, Unno M, Champion PM. J. Am. Chem. Soc. 1999;121:376–380. [Google Scholar]
- 108.Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SG, Hoffman BM, Champion PM. Biochemistry. 2001;40:6852–6859. doi: 10.1021/bi002510b. [DOI] [PubMed] [Google Scholar]
- 109.Tosha T, Kagawa N, Arase M, Waterman MR, Kitagawa T. J. Biol. Chem. 2008;283:3708–3717. doi: 10.1074/jbc.M707338200. [DOI] [PubMed] [Google Scholar]
- 110.Dawson JH, Kau LS, Penner-Hahn JE, Sono M, Eble KS, Bruce GS, Hager LP, Hodgson KO. J. Am. Chem. Soc. 1986;108:8114–8116. [Google Scholar]
- 111.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 112.Carver TE, Brantley RE, Jr., Singleton EW, Arduini RM, Quillin ML, Phillips GN, Jr., Olson JS. J. Biol. Chem. 1992;267:14443–14450. [PubMed] [Google Scholar]
- 113.Brucker E, Olson J, Phillips G, Dou Y, Ikeda-Saito M. J. Biol. Chem. 1996;271:25419–25422. doi: 10.1074/jbc.271.41.25419. [DOI] [PubMed] [Google Scholar]
- 114.Hirota S, Li T, Phillips GN, Olson JS, Mukai M, Kitagawa T. J. Am. Chem. Soc. 1996;118:7845–7846. [Google Scholar]
- 115.Vojtechovsky J, Chu K, Berendzen J, Sweet R, Schlichting I. Biophys. J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shaanan B. J. Mol. Biol. 1983;171:31–59. doi: 10.1016/s0022-2836(83)80313-1. [DOI] [PubMed] [Google Scholar]
- 117.Liddington R, Derewenda Z, Dodson E, Hubbard R, Dodson G. J. Mol. Biol. 1992;228:551–579. doi: 10.1016/0022-2836(92)90842-8. [DOI] [PubMed] [Google Scholar]
- 118.Harutyunyan E, Safonova T, Kuranova I, Popov A, Teplyakov A, Obmolova G, Rusakov A, Vainshtein B, Dodson G, Wilson J. J. Mol. Biol. 1995;251:104–115. doi: 10.1006/jmbi.1995.0419. [DOI] [PubMed] [Google Scholar]
- 119.Condon P, Royer W. J. Biol. Chem. 1994;269:25259–25267. doi: 10.2210/pdb1hbi/pdb. [DOI] [PubMed] [Google Scholar]
- 120.Paoli M, Liddington R, Tame J, Wilkinson A, Dodson G. J. Mol. Biol. 1996;256:775–792. doi: 10.1006/jmbi.1996.0124. [DOI] [PubMed] [Google Scholar]
- 121.Miller M, Shaw A, Kraut J. Nature Struct. Mol. Biol. 1994;1:524–531. doi: 10.1038/nsb0894-524. [DOI] [PubMed] [Google Scholar]
- 122.Berglund G, Carlsson G, Smith A, Szoke H, Henriksen A, Hajdu J. Nature. 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 123.Pellicena P, Karow D, Boon E, Marletta M, Kuriyan J. Proc. Natl. Acad. Sci. USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Unno M, Matsui T, Chu G, Couture M, Yoshida T, Rousseau D, Olson J. J. Biol. Chem. 2004;279:21055–21061. doi: 10.1074/jbc.M400491200. [DOI] [PubMed] [Google Scholar]
- 125.Nagano S, Poulos TL. J. Biol. Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 126.Nagano S, Cupp-Vickery JR, Poulos TL. J. Biol. Chem. 2005;280:22102–22107. doi: 10.1074/jbc.M501732200. [DOI] [PubMed] [Google Scholar]
- 127.Zhao B, Guengerich FP, Voehler M, Waterman MR. J. Biol. Chem. 2005;280:42188–42197. doi: 10.1074/jbc.M509220200. [DOI] [PubMed] [Google Scholar]
- 128.Poulos TL. Drug Metab. Rev. 2007;39:557–566. doi: 10.1080/03602530701498240. [DOI] [PubMed] [Google Scholar]
- 129.Davydov R. Biofizika. 1980;25:203. [PubMed] [Google Scholar]
- 130.Davydov R, Kappl R, Huettermann J, Peterson J. FEBS Lett. 1991;295:113–115. doi: 10.1016/0014-5793(91)81398-r. [DOI] [PubMed] [Google Scholar]
- 131.Kappl R, Höhn-Berlage M, Hüttermann J, Bartlett N, Symons M. Biochim. Biophys. Acta. 1985;827:327–343. [Google Scholar]
- 132.Leibl W, Nitschke W, Hüttermann J. Biochim. Biophys. Acta. 1986;870:20–30. doi: 10.1016/0167-4838(86)90004-x. [DOI] [PubMed] [Google Scholar]
- 133.Symons M, Petersen R. Proc. Roy. Soc. London, Ser. B, Biol. Sci. 1978;201:285–300. doi: 10.1098/rspb.1978.0046. [DOI] [PubMed] [Google Scholar]
- 134.Davydov R, Macdonald IDG, Makris TM, Sligar SG, Hoffman BM. J. Amer. Chem. Soc. 1999;121:10654–10655. [Google Scholar]
- 135.Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM. J. Am. Chem. Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 136.Davydov R, Razeghifard R, Im SC, Waskell L, Hoffman BM. Biochemistry. 2008;47:9661–9666. doi: 10.1021/bi800926x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Davydov R, Perera R, Jin S, Yang T-C, Bryson TA, Sono M, Dawson JH, Hoffman BM. J. Am. Chem. Soc. 2005;127:1403–1413. doi: 10.1021/ja045351i. [DOI] [PubMed] [Google Scholar]
- 138.Sligar SG, Makris TM, Denisov IG. Biochem. Biophys. Res. Commun. 2005;338:346–354. doi: 10.1016/j.bbrc.2005.08.094. [DOI] [PubMed] [Google Scholar]
- 139.Denisov IG, Mak PJ, Makris TM, Sligar SG, Kincaid JR. J. Phys. Chem. A. 2008;112:13172–13179. doi: 10.1021/jp8017875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mak PJ, Denisov IG, Victoria D, Makris TM, Deng T, Sligar SG, Kincaid JR. J. Am. Chem. Soc. 2007;129:6382–6383. doi: 10.1021/ja071426h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dunford HB, Stillman JS. Coord. Chem. Rev. 1976;19:187–251. [Google Scholar]
- 142.Dunford HB. Heme Peroxidases. Wiley; New York: 1999. p. 507. [Google Scholar]
- 143.Groves JT. J. Inorg. Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 144.Makris TM, von Koenig K, Schlichting I, Sligar SG. J. Inorg. Biochem. 2006;100:507–518. doi: 10.1016/j.jinorgbio.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 145.Terner J, Palaniappan V, Gold A, Weiss R, Fitzgerald MM, Sullivan AM, Hosten CM. J. Inorg. Biochem. 2006;100:480–501. doi: 10.1016/j.jinorgbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 146.Jung C. Biochim. Biophys. Acta. 2011;1814:46–57. doi: 10.1016/j.bbapap.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 147.Kincaid JR, Zheng Y, Al-Mustafa J, Czarnecki K. J. Biol. Chem. 1996;281:28805–28811. doi: 10.1074/jbc.271.46.28805. [DOI] [PubMed] [Google Scholar]
- 148.Terner J, Gold A, Weiss R, Mandon D, Trautwein AX. J. Porphyrins Phtalocyanines. 2001;5:357–364. [Google Scholar]
- 149.Weiss R, Gold A, Trautwein AX, Terner J. High-valent iron and manganese complexes of porphyrins and related macrocycles. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Academic Press, Elsevier Science (USA); New York: 2000. pp. 65–96. [Google Scholar]
- 150.Palcic MM, Rutter R, Araiso T, Hager LP, Dunford HB. Biochem. Biophys. Res. Commun. 1980;94:1123–1127. doi: 10.1016/0006-291x(80)90535-5. [DOI] [PubMed] [Google Scholar]
- 151.Dolphin D, Forman A, Borg DC, Fajer J, Felton RH. Proc. Natl. Acad. Sci. U. S. A. 1971;68:614–618. doi: 10.1073/pnas.68.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Behan RK, Hoffart LM, Stone KL, Krebs C, Green MT. J. Am. Chem. Soc. 2006;128:11471–11474. doi: 10.1021/ja062428p. [DOI] [PubMed] [Google Scholar]
- 153.Egawa T, Proshlyakov DA, Miki H, Makino R, Ogura T, Kitagawa T, Ishimura Y. J. Biol. Inorg. Chem. 2001;6:46–54. doi: 10.1007/s007750000181. [DOI] [PubMed] [Google Scholar]
- 154.Egawa T, Shimada H, Ishimura Y. Biochem. Biophys. Res. Comm. 1994;201:1464–1469. doi: 10.1006/bbrc.1994.1868. [DOI] [PubMed] [Google Scholar]
- 155.Green MT, Dawson JH, Gray HB. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 156.Kellner DG, Hung S-C, Weiss KE, Sligar SG. J. Biol. Chem. 2002;277:9641–9644. doi: 10.1074/jbc.C100745200. [DOI] [PubMed] [Google Scholar]
- 157.Schuenemann V, Lendzian F, Jung C, Contzen J, Barra A-L, Sligar SG, Trautwein AX. J. Biol. Chem. 2004;279:10919–10930. doi: 10.1074/jbc.M307884200. [DOI] [PubMed] [Google Scholar]
- 158.Schuenemann V, Trautwein AX, Jung C, Terner J. Hyperfine Interactions. 2002;141/142:279–284. [Google Scholar]
- 159.Schunemann V, Jung C, Terner J, Trautwein AX, Weiss R. J. Inorg. Biochem. 2002;91:586–596. doi: 10.1016/s0162-0134(02)00476-2. [DOI] [PubMed] [Google Scholar]
- 160.Schunemann V, Jung C, Trautwein AX, Mandon D, Weiss R. FEBS Lett. 2000;479:149–154. doi: 10.1016/s0014-5793(00)01886-x. [DOI] [PubMed] [Google Scholar]
- 161.Kim SH, Perera R, Hager LP, Dawson JH, Hoffman BM. J. Am. Chem. Soc. 2006;128:5598–5599. doi: 10.1021/ja060776l. [DOI] [PubMed] [Google Scholar]
- 162.Behan RK, Green MT. J. Inorg. Biochem. 2006;100:448–59. doi: 10.1016/j.jinorgbio.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 163.Nakamoto K. Coord. Chem. Rev. 2002;226:163–165. [Google Scholar]
- 164.Rutter R, Hager LP, Dhonau H, Hendrich M, Valentine M, Debrunner P. Biochemistry. 1984;23:6809–6816. doi: 10.1021/bi00321a082. [DOI] [PubMed] [Google Scholar]
- 165.Rutter R, Valentine M, Hendrich MP, Hager LP, Debrunner PG. Biochemistry. 1983;22:4769–4774. doi: 10.1021/bi00289a024. [DOI] [PubMed] [Google Scholar]
- 166.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]