

Abstract
The TMPRSS2/ERG (T/E) fusion gene is present and thought to be an oncogenic driver of approximately half of all prostate cancers. Fusion of the androgen regulated TMPRSS2 promoter to the ERG oncogene results in constitutive high level expression of ERG which promotes prostate cancer invasion and proliferation. Here we report the characterization of multiple alternatively spliced T/E fusion gene isoforms which have differential affects on invasion and proliferation. We found that T/E fusion gene isoforms differentially increase NF-κB mediated transcription, which may explain in part the differences in biological activities of the T/E fusion isoforms. This increased activity is due to phosphorylation of NF-κB p65 on Ser536. Tissue microarray immunochemistry revealed that p65 phospho-Ser536 is present in the majority of prostate cancers where it is associated with ERG protein expression. The T/E fusion gene isoforms differentially increase expression of a number of NF-κB associated genes including PAR1, CCL2, FOS, TLR3 and TLR4 (Toll-like receptor 4). TLR4 activation is known to promote p65 Ser536 phosphorylation and knockdown of TLR4 with ShRNA decreases Ser536 phosphorylation in T/E fusion gene expressing cells. TLR4 can be activated by proteins in the tumor microenvironment and lipopolysacharide from Gram (−) bacteria. Our findings suggest that bacterial infection of the prostate and/or endogenous microenvironment proteins may promote progression of high-grade prostatic intraepithelial neoplasia and/or prostate cancers that express the T/E fusion gene, where the NF-κB pathway might be targeted as a rational therapeutic approach.
Keywords: prostate cancer, NF-κB, ERG, fusion gene
INTRODUCTION
The discovery of recurrent fusion of the androgen-regulated TMPRSS2 gene to the ETS transcription factors (1), particularly the ERG gene, in the majority of prostate cancer (PCa) lesions, has led to intensive study of the biological and clinical consequences of these rearrangements in PCa. The TMPRSS2/ERG (T/E) fusion gene is by far the most common fusion gene and is present in 40–60% of PCas (2–3). In all cases the TMPRSS2 promoter and one or more TMPRSS2 5' exons are juxtaposed to the ERG exons, with deletion of the ERG promoter. Therefore, the fusion transcripts are under the control of the androgen regulated TMPRSS2 promoter, resulting in the constitutive high level expression of ERG fusion transcripts in the neoplastic prostatic epithelium bearing this fusion gene. ERG shares a conserved 85 amino acid DNA-binding domain with other ETS factors, which are generally oncogenic and promote tumor progression in a number of malignancies.
There is significant heterogeneity in the structures of the T/E fusion transcripts, both at the 5' end of the mRNA (2–3) and downstream ERG coding exons (4). We previously characterized 8 fusion isoforms based on the fusion junction structure (3). The most common transcript contains the TMPRSS2 exon 1 fused to ERG exon 4, which we have designated as the Type III isoform (3). This variant is expressed in the vast majority (80–90%) of fusion gene expressing PCa, either alone or in combination with other isoforms. Translation would have to arise from an internal ATG codon since the native ERG ATG is absent and would thus give rise to a slightly truncated ERG protein. Of particular interest is an isoform in which TMPRSS2 exon 2 is fused with ERG exon 4 (designated Type VI). This variant was present in 26% of the cases with T/E fusion gene expression (3). For this isoform, the in-frame fusion results in a true fusion protein containing the first five amino acids of the TMPRSS2 gene fused to a slightly truncated ERG protein. We found that expression of this isoform is associated with aggressive disease. Subsequent studies have shown that the Type VI isoform enhances proliferation and invasion of immortalized prostatic epithelial cells to a greater extent than the Type III isoform, consistent with the clinical associations (4). In addition, we have identified multiple alternative splicing exons of ERG by cloning and sequencing of the fusion gene transcripts (4). In particular, alternative splicing leading to inclusion or exclusion of a 72-bp exon (genomic exon 11) is common. We have shown that the presence of this 72-bp exon significantly enhances proliferation and invasion of immortalized prostate epithelial cells when compared to mRNAs with the same 5' structure that lack this exon. Thus alternative splicing of the 5' region and the coding exons of the T/E fusion gene, as well as the ratio of the various isoforms, can impact the biological activities of the fusion gene.
The T/E fusion gene can promote PCa invasion and to a lesser extent proliferation and decrease differentiation via several known pathways such as uPA, MMPs, and C-myc that have previously been implicated in PCa initiation and progression (5–8). Here we report that NF-kB transcriptional activity is increased by T/E fusions. The NF-kB pathway activation is associated with aggressive clinical behavior in PCa (9). Phosphorylation of p65 has been shown by many groups to enhance p65 transcriptional activity (10). We have found that T/E fusion type isoforms enhance NF-κB transcriptional activity 3-fold in prostatic epithelial cells. Significantly increased phosphorylation of the NF-κB p65 subunit on Serine 536 was observed in stably selected PNT1a cells expressing T/E fusion isoforms while VCaP cells with stable knockdown of the T/E fusion gene by shRNA show a marked decrease in p65 Ser536 phosphorylation. Using immunohistochemistry we have found that p65 phospho-Ser536 is present in the majority of PCa tissues and its expression is associated with expression of the ERG protein. Further investigation revealed upregulation of several genes related to NF-kB pathway by the fusion gene, including Toll-like receptor-4 (TLR4). The TLR4 protein can activate the NF-kB pathway and increase p65 Ser536 phosphorylation when activated (11–12). We verified upregulation of TLR-4 mRNA by T/E fusions and decreased p65 phospho-Ser536 in PNT1a cells expressing the T/E fusion was seen when TLR4 was knocked down. Thus the NF-kB pathway is activated by phosphorylation via TLR4 activation in fusion gene expressing cells, linking the expression of this common fusion gene to a key pathway in PCa progression which can potentially be activated by bacterial lipopolysacharide and/or tumor microenvironment proteins.
MATERIALS and METHODS
Cell culture
PNT1a cells were maintained in the RPMI with 10% fetal bovine serum ((FBS). HEK293T cells and Cos7 cells were maintained in DMEM medium with 10% FBS.
Transfection and NF-kB reporter assays
Transient transfection was conducted in triplicate in 24-well plates as described previously (13). The NF-kB luciferase reporter vector was obtained from Stratagene (Cat# 219077, PathDetect, Stratagene, La Jolla, CA). Luciferase activity was determined on triplicate samples and each experiment was repeated at least three times.
Proliferation Assay
VCaP Cells (1.0 × 105) of each cell line were trypsinized and plated in 6-well dishes in complete medium. The next day, medium containing 20uM PS1145 (Sigma Aldrich, P6624) were added to the wells in treatment group. Cells were trypsinized and counted using a Coulter counter at different time points in triplicate. The experiment was repeated twice.
Western Blotting
Anti-p65 and anti-phos-p65 Ser536 (both from Cell Signaling Technology) were used at 1:1000 dilution for Western blotting using procedures described previously (13). Anti-β-actin control was performed as described previously (13).
Immunohistochemistry
Immunohistochemistry of VCaP orthotopic tumors was performed using a rabbit polyclonal p65 anti-phospho-Ser536 from Abgent, Inc (AP3178a) as described previously (14). Slides were scanned and photographed using a Nikon Eclipse E400 microscope connected with Nuance Multispectral Imaging System at 200× magnification with 3.3 megapixel resolution. Images were saved as JPEG files with 6–8 images were taken for each slide, covering the entire tumor area. All staining signals are localized to cell nuclei. The numerical value for percent stained (PS) is determined by using Image software (http://rsb.info.nih.gov/ij/).
Protein subcellular localization
Cos7 cells were cultured in 4-well chamber slides (Lab-Tek) and transfected with NF-kB p65 along with T/E fusions Type III+72 or VI+72, which have the V5 tag (4), or control vector (pCMV-Tag2B). Cells were fixed 48h after transfection with 4% formaldehyde for 15 min and incubated with 0.5% bovine serum albumin for 30 min. Antibodies (anti-V5 from Invitrogen, anti-p65 and anti-phospho-Ser536 from Cell Signaling) were applied to cells overnight at 4° C. FITC-conjugated goat anti-mouse secondary antibody (1:500 dilution) was used for detection of fusion proteins, and TRITC-conjugated goat anti-rabbit antibody (1:500 dilution) was used for total p65 or phospho-Ser536 p65 staining. Both secondary antibodies were purchased from Sigma-Aldrich, Inc. For nuclear staining, 1ml of mounting solution was applied with DAPI (Vectashield, Vector) following manufacturer's instructions. Slides were mounted and observed under Nikon Eclipse E400 microscope connected with Nuance Multispectral Imaging System. Cells with positive fluorescence staining in 10 random fields at 200× magnification field were counted. This experiment was repeated twice.
Tissue Microarrays
Prostate cancer tissue microarrays have been described previously (15–18). Immunohistochemistry was performed as described above for the VCaP tumors. Arrays stained with anti-phospho-Ser536 were scanned and staining quantitated using a multiplicative staining index of intensity (0–3) and extent (0–3) of staining yielding a 10 point staining index (0–9) as described in previous publications (15–18). The arrays were also stained with anti-ERG antibody as described by Park et al (19). Cancer nuclei were scored as positive or negative and cancers were considered positive if ERG staining was seen in cancer cells in any tissue core. In almost all cases where positive staining was seen it involved the majority of cancer cells in all cores. No staining of benign epithelial nuclei was identified although strong staining of endothelial cells was noted as described previously (19) which was useful as an internal positive control.
NFκB Signaling Pathway PCR array
Human NFκB Signaling Pathway PCR Array from SA Biosciences was used to analyze cDNAs from PNT1a cells overexpressing T/E fusion Type III+72, Type VI+72 or vector control following the manufacturer's protocol. Individual PCR primer pair mixes for gene F2R, FOS, TLR3, TLR4 and CCL2 were purchased from SA Biosciences and were used for quantitative RT-PCR as described previously (20). This experiment was repeated twice.
shRNA against TLR4
Two shRNAs targeting TLR4 were purchased from Open Biosystems, (V2LHS_171352 (shRNAa) and V2LHS_171350 (shRNAb)). Negative control shRNA was also purchased from Open Biosystems. Cells were transfected with shRNA plasmids against TLR4 or negative control shRNA as described previously (21). TLR4 mRNA levels were evaluated by quantitative RT- PCR as described above. This experiment was repeated twice.
RESULTS
Upregulation of NF-κB transcriptional activities by T/E fusion isoforms
To explore mechanisms by which T/E fusions affect PCa initiation and progression, we performed a series of luciferase reporter assays with a variety of reporter constructs and T/E fusion gene constructs (data not shown). Among the promoters tested, NF-κB transcriptional activities were the most highly upregulated. Briefly, we transiently transfected a composite NF-κB promoter construct linked to a luciferase reporter gene into the immortalized normal prostate epithelial PNT1a cell line with the plasmids expressing the III+72 fusion gene isoform, the VI+72 isoform, control empty vector plasmid or VI+72 deletion constructs missing key ERG domains. The Type III+72 and VI+72 isoforms enhanced NF-κB transcription more than 2-fold and approximately 3-fold, respectively, compared to the control plasmid at 24 hrs after transfection (Fig 1) and this difference was statistically significant from control for both fusion gene isoforms (p<0.02, t-test). Similar results were seen at 8–72 hrs after transfection (data not shown). Control transfections with VI +72 expression constructs with deletion of the ETS or CAE domain had activities that were even lower than control (Fig 1), suggesting a possible dominant negative activity directed against basal ERG expressed in PNT1a cells. Similar results were seen in 293T cells, although the induction was more robust (Supplemental Figure S1).
Figure 1. The TMPRSS2/ERG fusion gene increases NF-κB transcrptional activity.

PNT1a cells were transiently co-transfected with a NF-κB reporter construct and plasmids expressing the III+72 T/E fusion gene, the VI+72 T/E fusion gene, the Type VI+72 expression construct with deletion of the ETS or central activation (CAE) domains and vector controls (CON). Well were transfected in triplicate and the experiment was repeated three times. Standard deviations are shown. Both the Type III +72 and VI+72 expression constructs were statistically significantly increased over controls (p=.02 and p=.004, respectively, t-test).
Induction of p65 phospho-Ser536 by T/E fusion isoforms
Phosphorylation of p65 has been shown to enhance p65 transcriptional activity and phosphorylation at Ser536 regulates NF-κB p65 activation, nuclear localization, protein-protein interactions and transcriptional activity (10, 22–23). We therefore examined phosphorylation of p65 at Ser536 in stably selected PNT1a cells expressing T/E fusion gene isoforms. Both fusion gene expressing cell lines displayed significantly increased phosphorylation of the NFκB p65 subunit on Serine 536 (Fig 2A) with unchanged total p65 protein. Of note, the VI +72 cell line had a higher level of p65 phospho-Ser536 than cells expressing the III+72 isoform. VCaP cells with stable knockdown of the T/E fusion gene by ShRNA that we have generated previously (see ref (4)) showed a marked decrease in levels of p65 phospho-Ser536. In previous studies we used these shRNA and vector control VCaP cell lines for tumor progression studies following orthotopic injection and shown decreased tumor progression in shRNA expressing cells (4). We performed immunohistochemistry on nine VCaP orthotopic tumors with stable knockdown of the fusion gene versus ten vector controls using anti-p65 phospho-Ser536 antibody and quantitated percentage of cells with positive nuclear staining in each tumor. The tumors with stable T/E fusion knockdown had a significantly lower p65 phospho-Ser536 staining percentage by image analysis (36.9 Vs 48.8%, p<0.002, t-test). All staining was nuclear, with no cytoplasmic staining. Of note, VCaP cells, the only PCa cell line that expresses the T/E fusion gene, is also the only PCa cell line that expresses significant levels of Ser536 phosphorylated p65 (Fig 2B). Therefore, T/E fusions enhance NF-κB activities through increased phosphorylation at the p65 Ser536 site.
Figure 2. The TMPRSS2/ERG fusion gene increases phosphorylation of p65 Ser 536.

A. Western blots of extracts of PNT1a cells expressing Type III+72, Type VI+72 or vector control with anti-p65, anti-p65 phospho-Ser536 or β-actin antibodies (left); Similar Western blots oh VCaP cells expressing a fusion gene specific shRNA or vector controls (right). B. Western blot of PNT1a and multiple PCa cell lines with anti-p65, anti-p65 phospho-Ser536 or β-actin antibodies. C and D. Cos7 cells were transfected with NF-κB p65 along with Type III+72 or VI+72 fusion gene expression constructs or control vector (pCMV-Tag2B). TRITC-conjugated goat anti-rabbit antibody was used to detect total p65 (C) or phospho-Ser536 p65 (D) after incubation with anti-p65 or anti-p65 Ser536 antibody. Total positive nuclei per 10 high power fields (200×) is shown as mean +/− standard deviation of 10 fields. Statistically significant differences from controls by t-test are indicated by asterisks (p<.01).
Increased nuclear p65 phospho-Ser536 staining when overexpressing T/E fusion isoforms
We next determined the localization of T/E fusions and their regulation of p65 phosphorylation using fluorescence labeling techniques. The inactive form of NF-κB is localized in the cytoplasm and mainly consists of multiple subunits including the DNA-binding p50 and p65 subunits and an inhibitory subunit IkB, which is bound to p65 and masks the nuclear localization sequence and its release initiates the activation of NF-κB and its subsequent translocation to the nucleus. Cos7 cells were transfected with NF-κB p65 along with T/E fusions Type III+72 or VI+72, which have a V5 tag (4), or control vector (pCMV-Tag2B). FITC-conjugated goat anti-mouse secondary antibody was used for detection of fusion proteins, and TRITC-conjugated goat anti-rabbit antibody was used for total p65 or phospho-Ser536 p65 staining. As expected, T/E fusion isoforms were seen exclusively in the nucleus with similar total expression level for each fusion type (data not shown). A similar number of cells with total nuclear p65 staining was observed in both fusion groups and the control group (Figure 2C). A significantly higher number of p65 phospho-Ser536 expressing cells were found following cotransfection with fusion gene expressing constructs compared to the control group (Figure 2D).
Expression of NF-κB p65 phospho-Ser536 in prostate cancer
To determine if NF-κB p65 Ser536 phosphorylation is relevant to human PCa we carried out an immunohistochemical analysis of 371 clinically localized PCas using tissue microarrays. Expression was quantitated as described previously (15–18, 24–27) based on a multiplicative index of the average staining intensity (0–3) and extent of staining (1–3) in the cores yielding a 10 point staining index (0–9). Examples of strong (index 9), moderate (index 6), weak (index 3) and no (index 0) staining are shown in Figure 3A. The mean staining index was 4.5 and 85% of cancers showed some staining (i.e. >0). NF-κB p65 Ser536 phosphorylation was weakly correlated with Gleason score (r2 =.122, p=0.02) but not with other pathological variables. Compared to cancers with no expression, cancers with p65 phospho-Ser536 had a significantly decreased time to recurrence (p=.005, Log rank) as shown in the Kaplan-Meier plot in Figure 3B. Cox regression was significant by univariate (hazard ratio 3.83, 95% confidence interval 1.41–10.43, p=.0086) and multivariate (hazard ratio 3.64, 95% confidence interval 1.13–11.73, p=.03) analysis. Thus the presence p65 Ser536 phosphorylation is common in PCa and influences clinical outcome.
Figure 3. Expression of p65 phospho-Ser536 in prostate cancer using tissue microarrays.

A. Immunohistochemistry using anti-p65 Ser536 antibody was performed using a prostate cancer tissue microarray was performed and quantiated using a 10 point quantitation scale as described in Materials and Methods. Examples of staining with staining indices of 9, 6, 3 and 0 are shown. Original maginification 400×, dash equals 200 microns. B. Kaplan-Meier plot of recurrence free survival following radical prostatectomy of cancers with no staining with anti-p65 Ser536 antibody versus cancers with staining. C. Association of ERG expression with 65 phospho-Ser536 IHC. Cases with the indicated staining indices for p65 phospho-Ser536 by IHC 0, 1–3 (low), 4–6 (moderate) and 7–9 (high) were scored for ERG expression by IHC and the fraction of cases with ERG expression, which is highly correlated with expression of the T/E fusion gene was determined. The distribution of ERG positive cases between groups was significantly different than chance (p<.0001, Chi-square)
We next explored the association of NF-κB p65 Ser536 phosphorylation expression in PCa with the expression of ERG using the protocol described by Park et al (19) to detect ERG expression by immunohistochemistry in the same tissue microarray analyzed for p65 phospho-Ser536 expression. The presence of any level of ERG expression in tumor cells as detected by this antibody is highly correlated with the presence of the T/E fusion gene (19). We therefore dichotomized staining into positive and negative for ERG. Overall, 364 of 371 cancers informative for p65 phospho-Ser536 staining could also be evaluated for ERG expression. In the vast majority of cases ERG staining was strong and in all cases was confined to cancer nuclei, with no staining of benign epithelium. Strong staining of endothelial cells was noted as described previously (19), which represents expression of endogenous ERG in these cells. A total of 164 cases were positive, representing 45% of evaluable cases, consistent with prior reported rates of T/E fusion gene expression in surgically treated PCa. ERG positive cancers tended to have a shorter time to recurrence following radical prostatectomy (Supplemental Figure S2) but the difference between ERG positive and negative cases was not statistically significant (p=.312, Log rank). We then compared the expression index for p65 phospho-Ser536 in fusion gene positive and fusion gene negative cases. The expression index was significantly higher in ERG expressing PCa (mean 5.29, median 6.0) versus ERG negative PCa (mean 3.98, median 3.0) by Mann-Whitney test (p<.001). Of note, there was no significant difference in the matched benign prostate tissues from ERG positive and negative cancers in p65 phospho-Ser536 expression. We also examined the fraction of cases that were ERG positive at various levels of p65 phospho-Ser536 expression. As seen in Fig 3C, the percentage of ERG positive cases was only 17% in cases with no p65 phospho-Ser536 staining (staining index 0) and this increased to 60% in cases with strong staining (index 7–9). The differences seen were highly statistically significant (p<.001, Chi-square). To confirm the reproducibility of the NF-κB phospho-Ser536 scoring system when grouped in this manner a second observer scored 270 individual tissue cores. Only 4 of 270 cores had a difference in scoring that resulted in change from one scoring group to another, indicating that this scoring system is highly reproducible. Overall the data strongly support the concept that ERG can significantly enhance phosphorylation of p65 at Ser536 in vivo, although other factors clearly play a role since ERG negative cases do have phosphorylation at this site, although at significantly lower levels. We then analyzed recurrence free survival on four groups of cases grouped by p65 phospho-Ser536 and ERG expression status. (Supplemental Figure S3). The ERG positive and negative cases that did not express p65 phospho-Ser536 were not statistically significantly different than each other. Among the cases expressing phospho-Ser536 the ERG positive cases tended to have earlier recurrence than the ERG negative cases, although this difference was not statistically significant (p=.34, Log rank). The phospho-Ser536 positive/ERG positive cases recurred earlier that both groups of phospho-Ser536 negative cases (p=.047 and .025, Log rank). This data indicates that NF-κB Ser536 phosphorylation as a result of ERG expression impacts disease aggressiveness.
NF-κB pathway inhibition decreases proliferation of VCaP cells
To confirm the importance of the NF-κB pathway in ERG expressing PCa cell lines we treated VCaP cells with PS1145, an inhibitor of IKK (28). As shown in Figure 4, PS1145 significantly inhibited VCaP proliferation (p<.001, day 7, t-test), confirming that NF-κB pathway plays an important role in proliferation in PCa cells expressing the T/E fusion gene.
Figure 4. Inhibition of the NF-κB pathway inhibits VCaP proliferation.
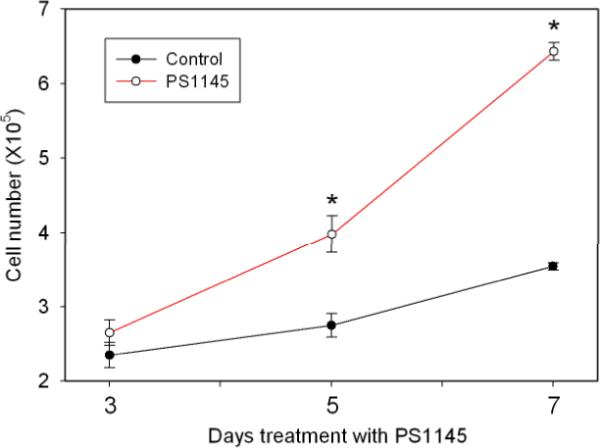
VCaP cells were plated and then treated with 20 uM PS1145, an IKK inhibitor or vehicle control and cells counted using a Coulter counter after the indicated number of days of treatment. Cells were plated in triplicate. The experiment was repeated three times. The mean +/− standard deviation is shown; statistically significant differences by t-test are indicated by asterisks.
Genes in NF-κB pathway upregulated by T/E fusion isoforms
To further explore the activation of the NF-KB pathway by the fusion isoforms, we used a SuperArray NF-κB PCR Array to analyze RNAs from vector control PNT1a cells and stably transfected PNT1a cells expressing the III+72 or VI+72 isoforms. Genes which were upregulated by 3-fold or more in one or both fusion gene expressing cells lines were then confirmed using standard quantitative RT-PCR. Results are shown in Fig 5. The five upregulated genes meeting this criterion were F2R (Thrombin receptor; PAR1), Toll-like receptor 3 (TLR3), Toll-like receptor 4 (TLR4), FOS, and CCL2. The most significantly upregulated gene, particularly by the Type VI+72 isoform, was TLR4.
Figure 5. NF-κB pathway genes upregulated by T/E fusion gene isoforms.

RNAs from vector control PNT1a cells and stably transfected PNT1a cells expressing the III+72 or VI+72 fusion gene isoforms were analyzed using a SuperArray NFκB PCR Array. Genes which were upregulated by 3-fold or more in one or both fusion gene expressing cells lines were then confirmed using standard quantitative RT-PCR.
Increased NF-kB p65 phospho-Ser536 by T/E fusion isoforms is mediated through TLR4
Since the overexpression of T/E fusion isoforms can upregulate TLR4 expression and there is evidence that TLR4 can increase phosphorylation of p65 Ser536 when activated (11–12), we hypothesized that NF-kB transcription activated by T/E fusions is mediated through TLR4 pathway. Therefore, we carried out a knockdown experiment to determine if p65 phospho-Ser536 level can be affected by decreased TLR4 expression in fusion gene expressing cells. We tested two ShRNA plasmids targeting TLR4 in PNT1a cells expressing the Type VI+72 isoform, which showed the highest TLR4 expression at RNA level, by transient transfection. The knockdown efficiencies for the two shRNA plasmids are 53% and 40% respectively by quantitative RT-PCR (Fig 6A). Dramatically decreased phospho-p65 Ser536 was seen in both transfectants by Western blot (Fig 6B). Therefore, our in vitro data supports our hypothesis that NF-kB p65 phospho-Ser536 upregulation by T/E fusion isoforms is mediated at least in part through TLR4.
Figure 6. Knockdown of Toll-like receptor-4 decrease p65 Ser536 phosphorylation.

A. Expression of TLR4 as determined by quantitative RT-PCR in PNT1a cells stably expressing the VI+72 fusion gene isoform transiently transfected with two plasmids expressing shRNas targeting TLR4 or control plasmid. B. Western blot of protein extracts on transiently transfected PNT1a cells described above using anti-p65 phospho-Ser536 antibody. β-actin is a loading control.
DISCUSSION
We report here for the first time that T/E fusion isoforms can activate NF-κB transcriptional activity through increased p65 Ser536 phosphorylation. Clinical and preclinical observations have shown that NF-kB plays an important role in PCa growth, survival, angiogenesis, tumorigenesis and metastatic progression (29–33). To date, investigations in PCa have focused on expression and translocation of p50/p65 subunits. Our data indicates that p65 phosphorylation also plays a role in modulating PCa progression and is increased in fusion gene expressing cells. Furthermore, our data indicates that TLR4 plays an important role in increasing p65 Ser536 phosphorylation in fusion gene expressing cells. Of note, ETS factor binding sites in the TLR4 promoter may play an important role of TLR4 mRNA expression (34), raising the possibility that TLR4 may be a direct target of the T/E fusion gene.
TLR4 is well known for its role in immune cells, in particular, its significant role in response to lipopolysacharide (LPS) produced by Gram (−) bacteria (11). DU145 and PC3 PCa cell lines also express TLR4 (35–36). Knockdown of TLR4 in PC3 cells with siRNA significantly decreased invasion, survival and tumorigenicity (35). Finally, it has been shown that specific sequence variants of TLR4 are associated with risk of PCa (37). TLR4 is a potent regulator of NF-κB signaling and has been shown to increase p65 Ser536 phosphorylation when activated (11–12). It should be noted that while TLR4 is activated by LPS it can also be activated by endogenous ligands such as hylauronic acid, fibronectin and heparin sulfate (38) which are abundant in the tumor microenvironment. It has been suggested that activation of TLR4 by LPS released from infection of the prostate by Gram (–) bacteria may promote tumorigenicity (39). Symptomatic prostatitis occurs in ~10% of men while the prevalence of asymptomatic prostatitis in unknown. There is a correlation between inflammation and the detection of bacterial RNA in the PCa tissues (40). Whether endogenous ligands or LPS or both activate TLR4 in fusion gene expressing cells is an important question. If LPS does indeed play a role this is clinically important in that it indicates that a potentially treatable infectious process may promote PCa initiation and/or progression.
TLR3 (Toll-like receptor 3) is also increased in response to fusion gene expression. TLR3 is expressed by both LNCaP and PC3 cell lines (41). TLR3 responds to dsRNA or poly I:C (12). Of note, activation of TLR3 can activate NF-κB and MAPK but can also induce apoptosis and has been shown to do so in LNCaP cells (41). Thus it is possible that the observed induction of TLR3 may be exploited therapeutically in PCa if indeed TLR3 activation induces apoptosis rather than tumor promotion. Such direct anti-tumor effects would synergize with the known ability of dsRNA to promote anti-tumor immunity (42).
PAR1 (thrombin receptor) is a G-protein coupled receptor that has previously been shown to be overexpressed in human PCa (43–44) and is among the genes we found to be upregulated. Of note, PAR1 is strongly expressed and biologically active in VCaP cells (44). PAR1 is activated by thrombin and Factor 10a. The uPA pathway and MMPs (upregulated by the T/E fusion gene) can enhance PAR1 activation by enhancing thrombin formation in the tumor microenvironment. PAR1 can activate NF-κB in PCa cells (43) as well other cell types. Interestingly, a functional ETS element has been shown to be present at position −506 in the PAR1 promoter (45), raising the possibility that PAR1 may be direct transcriptional target of the T/E fusion gene protein.
CCL2 (MCP-1) is a chemokine which is a potent regulator of PCa cell migration and proliferation and acts, at least in part, via activation of the PI3 kinase/AKT pathway (46) and is a well known direct transcriptional target of NF-κB (47). CCL2 is expressed by endothelial cells within the tumor microenvironment but can also be expressed by tumor cells directly (48). Of note, VCaP cells express CCL2 and secrete it into the media (48). Furthermore, the CCL2 receptor CCR2 is expressed by PCa cells (48). CCL2 has been shown to significantly enhance growth of PCa bone metastases, the most common site of PCa metastasis and may act synergistically with PAR1 (see above) to promote migration (49).
Finally, we noted a significant upregulation of FOS by the T/E fusion gene. c-JUN is upregulated by the fusion gene and c-JUN can bind c-FOS. It is known that AP-1 can synergize with NF-KB to promote expression of specific target genes including proangiogenic genes such as Il-8 and VEGF (50). Thus increased FOS expression may enhance the tumor promoting activities of the NF-KB pathway.
In summary, our study has identified a unique mechanism by which TMPRSS2/ERG fusion genes contribute to PCa tumorigenesis by upregulating the NF-kB pathway. We hypothesize that in a subset of PCa, T/E fusions can upregulate TLR4, and then activated TLR4 increases NF-kB transcriptional activities, resulting in expression of genes favoring tumor cell growth and invasion activated. This process can be enhanced by exposure of fusion gene expressing cells to Gram (−) bacteria, which releases LPS, and/or exposure to endogenous proteins in the tumor microenvironment. These findings have implications for prevention and treatment of PCa by treatment Gram (−) prostatic infections and targeting the NF-κB pathway.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge the technical assistance of DeeAnne Killeen with immunohistochemistry studies and Anna Frolov with statistical analysis.
Grant Support: This work was supported by grants from the Department of Defense Prostate Cancer Research program (DOD W81XWH-08-1-0055; JW), the National Cancer Institute to the Baylor Prostate Cancer SPORE (P50CA058204) and the University of Michigan Prostate Cancer SPORE (P50CA069568), the Dept of Veterans Affairs Merit Review program (MI) and by the use of the facilities of the Michael E. DeBakey VAMC.
REFERENCES
- 1.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 2.Clark J, Merson S, Jhavar S, et al. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene. 2007;26:2667–73. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Cai Y, Ren C, Ittmann M. Expression of Variant TMPRSS2/ERG Fusion Messenger RNAs Is Associated with Aggressive Prostate Cancer. Cancer research. 2006;66:8347–51. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Cai Y, Yu W, et al. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer research. 2008;68:8516–24. doi: 10.1158/0008-5472.CAN-08-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carver BS, Tran J, Chen Z, et al. ETS rearrangements and prostate cancer initiation. Nature. 2009;457:E1. doi: 10.1038/nature07738. discussion E2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klezovitch O, Risk M, Coleman I, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2105–10. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomlins SA, Laxman B, Varambally S, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia (New York, N.Y. 2008;10:177–88. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun C, Dobi A, Mohamed A, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene. 2008;27:5348–53. doi: 10.1038/onc.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shukla S, MacLennan GT, Fu P, et al. Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia. 2004;6:390–400. doi: 10.1593/neo.04112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an I{kappa}B{alpha}-independent NF-{kappa}B pathway. The Journal of biological chemistry. 2005;280:34538–47. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- 11.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–51. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 12.Kenny EF, O'Neill LA. Signalling adaptors used by Toll-like receptors: an update. Cytokine. 2008;43:342–9. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Cai Y, Wang J, Li R, et al. GGAP2/PIKE-a directly activates both the Akt and nuclear factor-kappaB pathways and promotes prostate cancer progression. Cancer Res. 2009;69:819–27. doi: 10.1158/0008-5472.CAN-08-2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Stockton DW, Ittmann M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin Cancer Res. 2004;10:6169–78. doi: 10.1158/1078-0432.CCR-04-0408. [DOI] [PubMed] [Google Scholar]
- 15.Agoulnik IU, Vaid A, Bingman WE, 3rd, et al. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005;65:7959–67. doi: 10.1158/0008-5472.CAN-04-3541. [DOI] [PubMed] [Google Scholar]
- 16.Agoulnik IU, Vaid A, Nakka M, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006;66:10594–602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- 17.Ayala G, Thompson T, Yang G, et al. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin Cancer Res. 2004;10:6572–8. doi: 10.1158/1078-0432.CCR-04-0477. [DOI] [PubMed] [Google Scholar]
- 18.Dai H, Li R, Wheeler T, et al. Pim-2 upregulation: biological implications associated with disease progression and perinueral invasion in prostate cancer. Prostate. 2005;65:276–86. doi: 10.1002/pros.20294. [DOI] [PubMed] [Google Scholar]
- 19.Park K, Tomlins SA, Mudaliar KM, et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia. 2010;12:590–8. doi: 10.1593/neo.10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dakhova O, Ozen M, Creighton CJ, et al. Global gene expression analysis of reactive stroma in prostate cancer. Clin Cancer Res. 2009;15:3979–89. doi: 10.1158/1078-0432.CCR-08-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Yu W, Cai Y, Ren C, Ittmann MM. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression. Neoplasia. 2008;10:847–56. doi: 10.1593/neo.08450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doyle SL, Jefferies CA, O'Neill LA. Bruton's tyrosine kinase is involved in p65-mediated transactivation and phosphorylation of p65 on serine 536 during NFkappaB activation by lipopolysaccharide. The Journal of biological chemistry. 2005;280:23496–501. doi: 10.1074/jbc.C500053200. [DOI] [PubMed] [Google Scholar]
- 23.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends in biochemical sciences. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Haqq C, Li R, Khodabakhsh D, et al. Ethnic and racial differences in prostate stromal estrogen receptor alpha. Prostate. 2005;65:101–9. doi: 10.1002/pros.20272. [DOI] [PubMed] [Google Scholar]
- 25.Li R, Erdamar S, Dai H, et al. Cytoplasmic accumulation of glycogen synthase kinase-3beta is associated with aggressive clinicopathological features in human prostate cancer. Anticancer Res. 2009;29:2077–81. [PubMed] [Google Scholar]
- 26.Li R, Erdamar S, Dai H, et al. Forkhead protein FKHR and its phosphorylated form p-FKHR in human prostate cancer. Hum Pathol. 2007;38:1501–7. doi: 10.1016/j.humpath.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li R, Wheeler T, Dai H, et al. High level of androgen receptor is associated with aggressive clinicopathologic features and decreased biochemical recurrence-free survival in prostate: cancer patients treated with radical prostatectomy. Am J Surg Pathol. 2004;28:928–34. doi: 10.1097/00000478-200407000-00013. [DOI] [PubMed] [Google Scholar]
- 28.Yemelyanov A, Gasparian A, Lindholm P, et al. Effects of IKK inhibitor PS1145 on NF-kappaB function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene. 2006;25:387–98. doi: 10.1038/sj.onc.1209066. [DOI] [PubMed] [Google Scholar]
- 29.Muenchen HJ, Lin DL, Walsh MA, Keller ET, Pienta KJ. Tumor necrosis factor-alpha-induced apoptosis in prostate cancer cells through inhibition of nuclear factor-kappaB by an IkappaBalpha “super-repressor”. Clin Cancer Res. 2000;6:1969–77. [PubMed] [Google Scholar]
- 30.Suh J, Rabson AB. NF-kappaB activation in human prostate cancer: important mediator or epiphenomenon? Journal of cellular biochemistry. 2004;91:100–17. doi: 10.1002/jcb.10729. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Altuwaijri S, Deng F, et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. The American journal of pathology. 2009;175:489–99. doi: 10.2353/ajpath.2009.080727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Huang X, Olumi AF. Repression of NF-kappaB and activation of AP-1 enhance apoptosis in prostate cancer cells. International journal of cancer. 2009;124:1980–9. doi: 10.1002/ijc.24139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fradet V, Lessard L, Begin LR, et al. Nuclear factor-kappaB nuclear localization is predictive of biochemical recurrence in patients with positive margin prostate cancer. Clin Cancer Res. 2004;10:8460–4. doi: 10.1158/1078-0432.CCR-04-0764. [DOI] [PubMed] [Google Scholar]
- 34.Roger T, Miconnet I, Schiesser AL, et al. Critical role for Ets, AP-1 and GATA-like transcription factors in regulating mouse Toll-like receptor 4 (Tlr4) gene expression. Biochem J. 2005;387:355–65. doi: 10.1042/BJ20041243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hua D, Liu MY, Cheng ZD, et al. Small interfering RNA-directed targeting of Toll-like receptor 4 inhibits human prostate cancer cell invasion, survival, and tumorigenicity. Mol Immunol. 2009;46:2876–84. doi: 10.1016/j.molimm.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 36.Gatti G, Quintar AA, Andreani V, et al. Expression of Toll-like receptor 4 in the prostate gland and its association with the severity of prostate cancer. Prostate. 2009;69:1387–97. doi: 10.1002/pros.20984. [DOI] [PubMed] [Google Scholar]
- 37.Chen YC, Giovannucci E, Lazarus R, et al. Sequence variants of Toll-like receptor 4 and susceptibility to prostate cancer. Cancer Res. 2005;65:11771–8. doi: 10.1158/0008-5472.CAN-05-2078. [DOI] [PubMed] [Google Scholar]
- 38.Arumugam TV, Okun E, Tang SC, et al. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32:4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 39.Kundu SD, Lee C, Billips BK, et al. The toll-like receptor pathway: a novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate. 2008;68:223–9. doi: 10.1002/pros.20710. [DOI] [PubMed] [Google Scholar]
- 40.Sandhu JS. Prostate cancer and chronic prostatitis. Current urology reports. 2008;9:328–32. doi: 10.1007/s11934-008-0056-6. [DOI] [PubMed] [Google Scholar]
- 41.Paone A, Starace D, Galli R, et al. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-alpha-dependent mechanism. Carcinogenesis. 2008;29:1334–42. doi: 10.1093/carcin/bgn149. [DOI] [PubMed] [Google Scholar]
- 42.Seya T, Matsumoto M. The extrinsic RNA-sensing pathway for adjuvant immunotherapy of cancer. Cancer Immunol Immunother. 2009;58:1175–84. doi: 10.1007/s00262-008-0652-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tantivejkul K, Loberg RD, Mawocha SC, et al. PAR1-mediated NFkappaB activation promotes survival of prostate cancer cells through a Bcl-xL-dependent mechanism. J Cell Biochem. 2005;96:641–52. doi: 10.1002/jcb.20533. [DOI] [PubMed] [Google Scholar]
- 44.Chay CH, Cooper CR, Gendernalik JD, et al. A functional thrombin receptor (PAR1) is expressed on bone-derived prostate cancer cell lines. Urology. 2002;60:760–5. doi: 10.1016/s0090-4295(02)01969-6. [DOI] [PubMed] [Google Scholar]
- 45.Li F, Baykal D, Horaist C, et al. Cloning and identification of regulatory sequences of the human thrombin receptor gene. J Biol Chem. 1996;271:26320–8. doi: 10.1074/jbc.271.42.26320. [DOI] [PubMed] [Google Scholar]
- 46.Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem. 2008;283:25057–73. doi: 10.1074/jbc.M801073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roebuck KA, Carpenter LR, Lakshminarayanan V, et al. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J Leukoc Biol. 1999;65:291–8. doi: 10.1002/jlb.65.3.291. [DOI] [PubMed] [Google Scholar]
- 48.Loberg RD, Day LL, Harwood J, et al. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia. 2006;8:578–86. doi: 10.1593/neo.06280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loberg RD, Tantivejkul K, Craig M, Neeley CK, Pienta KJ. PAR1-mediated RhoA activation facilitates CCL2-induced chemotaxis in PC-3 cells. J Cell Biochem. 2007;101:1292–300. doi: 10.1002/jcb.21252. [DOI] [PubMed] [Google Scholar]
- 50.Bancroft CC, Chen Z, Dong G, et al. Coexpression of proangiogenic factors IL-8 and VEGF by human head and neck squamous cell carcinoma involves coactivation by MEK-MAPK and IKK-NF-kappaB signal pathways. Clin Cancer Res. 2001;7:435–42. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.