

Abstract
Purpose
Stimulation of toll-like receptor-9 (TLR9) by CpG oligodeoxynucleotides (CpG) has been shown to counteract the immunosuppressive microenvironment and to inhibit tumor growth in glioma models. Since TLR9 is located intracellularly, we hypothesized that methods that enhance its internalization may also potentiate its immunostimulatory response. The goal of this study was to evaluate carbon nanotubes (CNTs) as a CpG delivery vehicle in brain tumor models.
Experimental Design
Functionalized single-walled CNTs were conjugated with CpG (CNT-CpG) and evaluated in vitro and in mice bearing intracranial GL261 gliomas. Flow cytometry was used to assess CNT-CpG uptake and anti-glioma immune response. Tumor growth was measured by bioluminescent imaging, histology, and animal survival.
Results
CNT-CpG was nontoxic and enhanced CpG uptake both in vitro and intracranial gliomas. CNT-mediated CpG delivery also potentiated pro-inflammatory cytokine production by primary monocytes. Interestingly, a single intracranial injection of low-dose CNT-CpG (but not free CpG or blank CNT) eradicated intracranial GL261 gliomas in half of tumor-bearing mice. Moreover, surviving animals exhibited durable tumor-free remission (> 3 months), and were protected from intracranial tumor rechallenge, demonstrating induction of long-term anti-tumor immunity.
Conclusions
These findings suggest that CNTs can potentiate CpG immunopotency by enhancing its delivery into tumor-associated inflammatory cells.
Keywords: Brain neoplasm, glioma, immunotherapy, toll-like receptor-9
Introduction
Similar to other cancers, malignant gliomas, the most common and fatal primary brain neoplasm, have developed mechanisms to evade multiple effector arms of the immune system (1). One strategy to overcome this local immunosuppressive tumor microenvironment is through the activation of the innate immune system (2–4). Toll-like receptor (TLR)-family members are pattern-recognition receptors that collectively recognize lipid, carbohydrate, peptide and nucleic-acid structures that are broadly expressed by micro-organisms. Among their key functions, TLRs enhance the uptake of micro-organisms by phagocytic cells and mediate leukocyte recruitment to infected tissues (5). This broad immune activation has made TLR agonists attractive candidates for vaccine adjuvants for cancer therapy. TLRs are also expressed by brain microglia (MG) and macrophages (MP) (6), and direct intracranial (i.c.) injections of high doses of CpG oligonucleotides (CpG), a TLR9 ligand, can trigger tumor rejection and induces long-term immunity against glioma and neuroblastomas in mice (7). CpG as a single intratumoral (i.t.) injection has also been studied in patients with recurrent gliomas with tolerable toxicity and partial tumor response in a few patients (2, 8). Although higher CpG doses may improve clinical efficacy, significant inflammatory response in an already edematous brain may hinder this approach. Because TLR9 is located intracellularly, we hypothesized that methods which enhance CpG internalization by antigen-presenting cells may also potentiate its immunopotency. To examine this, carbon nanotubes (CNTs) were used as CpG carriers in gliomas.
CNTs are molecular-scale tubes of graphite carbon with unique mechanical properties that can be exploited for biomedical applications such as drug delivery (9). Although issues regarding the toxicity of CNT materials have been raised, recent studies (including ours) have reported that soluble functionalized CNTs (fCNTs) are nontoxic (10–12). Biodistribution studies in mice have shown CNT accumulation in liver and kidneys (and no uptake by the brain) with most of the CNTs eliminated within 24 h of intravenous (i.v.) injections (13–15). CNT uptake in tumor models has also been studied showing CNT size and preparation technique to be important in enhancing its uptake in systemic tumor models (13, 16, 17). We recently showed that fCNTs can be taken up by gliomas both in vitro and in vivo (12, 18) Compared to glioma cells, tumor-associated macrophages (TAMs) were far more efficient in CNT uptake as nearly half of these cells phagocytosed CNTs after a single i.t. injection (12). Furthermore, CNS injections of CNTs in normal and tumor-bearing mice were well tolerated, only eliciting a transient and self-limiting local inflammatory response (12). Overall, these studies indicated that CNTs may be suitable carriers for macromolecule, and potentially CpG, into TAMs.
Here, we demonstrate that fCNTs enhanced CpG uptake by tumor-associated phagocytic cells and resulted in their activation both in vitro and in vivo. Furthermore, in contrast to blank CNTs or free CpG, which had no anti-tumor effect, a single injection of low-dose CNT-CpG complexes eradicated i.c gliomas through activation of NK and CD8 cells. These findings demonstrate that fCNTs can improve CpG uptake into tumor-associated inflammatory cells, leading to a more robust anti-tumor response without inducing toxicity.
Materials and Methods
Reagents
Thiolated CpG (sCpG: 5’-TGACTGTAACGTTCGAGATGA-3’) and thiolated control oligodeoxynucleotides (sODN: 5’- TGACTGTAAGGTTAGAGATGA-3’) were constructed as described by Rosi et al. (19) and labeled with Cy5.5 (Lumiprobe, LLC). Anti-NK1.1 (clone PK136) was purchased from eBioscience (eBioscience Inc., San Diego, CA). Anti-CD8 Ab (clone H35) was purified as previously described (20). Control normal mouse IgG was purchased from Santa Cruz Biotechnology (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Flow Abs to mouse NK1.1 (clone PK136), CD11b (clone M1/70), CD45 (clone 30-F11), CD11c (clone N418), CD8 (clone 53–6.7) and isotype controls were purchased from BD Biosciences (San Jose, CA) or eBiosciences (San Diego, CA).
Cell lines
eGFP and luciferase-expressing GL261 glioma cell lines (GL261.gfp and GL261.luc) were generated as described before (21, 22). Primary bone marrow-derived monocytes (BMM) were harvested from normal C57BL/6 or CX3CR1GFP mice. After washing the bone marrow with cold PBS, cells were isolated and collected with Cell Strainer (BD Biosciences, San Jose, CA). The isolated BMM were then cultured in L929-conditioned DMEM medium. Red blood cells and other non-adherent cells were removed by changing the culture medium in 24 h. Cultures with more than 90% CD11b+ purity (as assessed by FACS) were used for experiments.
Single-walled carbon nanotubes construction and functionalization
Single-walled carbon nanotubes (CNTs) measuring 200–400nm in length were generated and characterized by electron microscopy as described before (18, 23). CNT funtionalization was performed using methods described by Liu et al. (24). Briefly, hipco CNTs were sonicated extensively (1h) in a solution of 1, 2-Distearoyl-Sn-Glycero-3-Phosphoethanolamine-N-[Amino (Polyethylene Glycol) 2000] (PEG) (Avanti Polar Lipids, Alabama). The supernatant solution of PEG-CNT was collected after centrifugation at 24,000g for 6 h. After removal of excess PEG molecules by Amicon centrifugal filter device (100 kDa), functionalized PEG-CNTs were conjugated with Sulfo-LC-SPDP (Thermo Fisher Scientific Inc., USA) for 1h at RT. After removal of excess Sulfo-LC-SPDP by Amicon centrifugal filter device (100 kDa) (Millipore, Billerica, Massachusetts), the CNT conjugates were quantified by SpectraMax M2 (Sunnyvale, California, USA) spectrometer with a weight extinction coefficient of 0.0465 1 mg−1 cm−1 at 808 nm. CNTs were then conjugated with sCpG or sODN through a cleavable disulfide bond at 4°C for 24h. Free sCpG was then separated from solution by Amicon centrifugal filter device (100 kDa) (Millipore, Billerica, Massachusetts) and measured with NanoDrop 1000 Spectrophotometer (Thermo Scientific). CNT-bound sCpG was quantified by subtracting the unbound sCpG from total sCpG added prior to the conjugation reaction.
RT-PCR
BMM (5×104 cells/well in 24-well plates) were incubated with sCpG (5µg/well), CNT-sCpG (CNT 2.5µg-sCpG 5µg /well), blank CNT (2.5µg/well), or CNT/sCPG mixture. At various times, cells were collected and total RNA was isolated using the Trizol system (Invitrogen Carlsbad, CA) followed by double DNase treatment and column purification using the Qiagen RNeasy Clean-up Protocol. Real-time PCR was performed in a TaqMan 5700 Sequence Detection System (Applied Biosystems, Foster City, CA) as described previously (25). PCR conditions were optimized such that a minimum of 10,000 fold range could be detected for each primer. GAPDH: 5′-GTTAGTGGGGTCTCGCTCTG-3′, 5′-GGCAAATTCAACGGCACA-3′; TNF-α: CAGACCCTCACACTCAGATCATCT, CCTCCACTTGGTGGTTTGCTA; IL-12 5′-AGAGGTGGACTGGACTCCCG-3′, 5′-AGTCTCGCCTCCTTTGTGGC-3′. IL-1β: 5′-AGGGCTGTCTGGAGTCCTC-3′, 5′-GACCAGCCGCCGCCGCAGG-3′.
Cytokine multiplex analysis
After each PCR time-point, supernatant from each sample was collected and analyzed for 20 cytokines using mouse Cytokine twenty-Plex Antibody Bead Kit (Invitrogen, Camarillo, CA) per manufacturer’s protocol. Assay plates were analyzed by a Bioplex HTF Luminex reader (Bio-Rad Laboratories, Inc., Hercules, CA). Cytokine concentrations were calculated using Bio-Plex Manager 3.1 software with a five parameter curve-fitting algorithm applied for standard curve calculations for duplicate samples. Data from detectable cytokines and chemokines was compared between each group.
Tumor implantation, treatment, and imaging
All animals were housed and handled in accordance to the guidelines of City of Hope Institutional Animal Care and Use Committee (IACUC). Intracranial tumor implantation was performed as described previously (25). GL261.luc or GL261.gfp cells were harvested by trypsinization, counted, and resuspended in PBS. Female C57BL/6 or CX3CR1GFP mice that express eGFP under control of the endogenous Cx3cr1 locus (Jackson Laboratory, Bar Harbor, ME) weighing 15–25 g were anesthetized by intraperitoneal (i.p.) administration of ketamine (132mg/kg) and xylazine (8.8mg/kg), and immobilized in a stereotactic head frame. Through a small burr hole, 3 µl of PBS containing 1 × 105 tumor cells was injected unilaterally as described before (25).
Four days after i.c. tumor implantation, mice received one i.t. injection of PBS (control, 10µl), free sCpG (5µg/10µl PBS), blank CNT (2.5 µg) mixed with free sCpG (CNT + sCpG; 5µg sCpG/10µl PBS), or sCpG conjugated to CNT (CNT-sCpG; 2.5 µg CNT/5 µg sCpG/10 µl PBS) through the initial burr hole. Tumor growth was assessed by Xenogen IVIS In Vivo Imaging System (Xenogen, Palo Alto, CA) as previously described (21).
NK and CD8 depletion studies were carried out as described (21). In these experiments mice were injected with anti-CD8, anti-NK1.1, or control IgG (200 µg/mouse, i.p.) mAb one day prior to tumor implantation and each i.t. injection. Leukocyte depletion was confirmed with FACS analysis of peripheral blood (21). For tumor rechallenge experiments, naive or GL261-bearing mice that had survived for at least three months after the initial CNT-sCpG treatment and were tumor-free by imaging, were re-challenged with i.c. GL261 (1×105 cells).
In vivo uptake studies
Tumor-bearing wt or CX3CR1GFP mice were injected with CNT bound to Cy5.5-labeled sCpG (CNT-sCpG5.5, 2.5 µg CNT/5 µg sCpG/10 µl PBS) or free sCpG5.5 (5 µg sCpG/10 µl PBS). Tumors were harvested at various time intervals and evaluated by flow cytometry as described below. For imaging, frozen brain sections were embedded in O.C.T. (Tissue-Tek) and 10 µm sections were cut using cryostat (Leica Microsystem Inc., Bannockburn, IL). Sections were mounted in Vectashield mounting medium containing 4060-diamidino-2-phenylindole (DAPI) (Vector, Burlingame, CA). Images were obtained by AX-70 fluorescent microscopy (Leica Microsystems Inc., Bannockburn, IL) and were prepared by Zeiss LSM Image Browser software.
Flow cytometry analysis
Tumors and blood samples were harvested and examined by flow cytometry as described previously (21). Cell suspensions from brain tissue were forced through a 40 µm filter. Blood samples were incubated in Gey’s buffer (pH 7.2) for 10 min. Freshly-prepared samples were resuspended in 0.1 M PBS containing 1% FBS and 2mM EDTA and incubated with FcγIII/IIR-specific Ab to block nonspecific binding. Samples were then stained with different combinations Abs or isotype controls for 1 h at 4°C and analyzed by a CyAn fluorescence cell sorter (BDIS, San Jose, CA). Inflammatory cells were gated and separated from the rest of sorted cells base on forward vs. side-scatter analysis and their staining characteristics. FlowJo 8.4.7 software (Tree Star, Inc., Ashland, OR) was used for data analysis and the proportion of each cell type was measured as percent of total inflammatory cells. Glioma MPs were gated as CD11b+/CD45high and MG as CD11b+/CD45low based on previously-described phenotype characterization (26).
Statistical analysis
Statistical comparison in all different experimental conditions was performed with the prism software using two-way analysis of variance (ANOVA) or Student’s t-test. Survival was plotted using a Kaplan-Meir survival curve and statistical significance was determined by the Log-rank (Mantel-Cox) test. A P value of less than 0.05 was considered significant.
Results
CNTs enhance CpG uptake in vitro
We first evaluated in vitro loading capacity of PEG2000-functionalized CNTs by conjugating them with sCpG. CNT conjugation capacity plateaued at 1–2 µg sCpG per µg CNT, which was consistent with other oligonucleotides (24). To evaluate sCpG uptake in vitro, BMM derived from wt or CX3CR1GFP mice were incubated with CNT bound to Cy5.5-labeled sCpG (CNT-sCpG5.5) or free sCpG5.5. sCpG5.5 uptake, which mostly localized to cytoplasmic compartments, was enhanced by CNTs (Fig. 1A and B) without inducing any additional toxicity in BMM or GL261 gliomas (Supplementary Fig. S1). CNT’s also increased sCpG uptake by GL261 gliomas, but not as strongly as BMM (Fig. 1C and D). These findings confirmed our hypothesis that intracellular CpG delivery can be enhanced with CNTs. Whether more efficient CpG uptake augmented monocyte activation was examined next.
Figure 1.
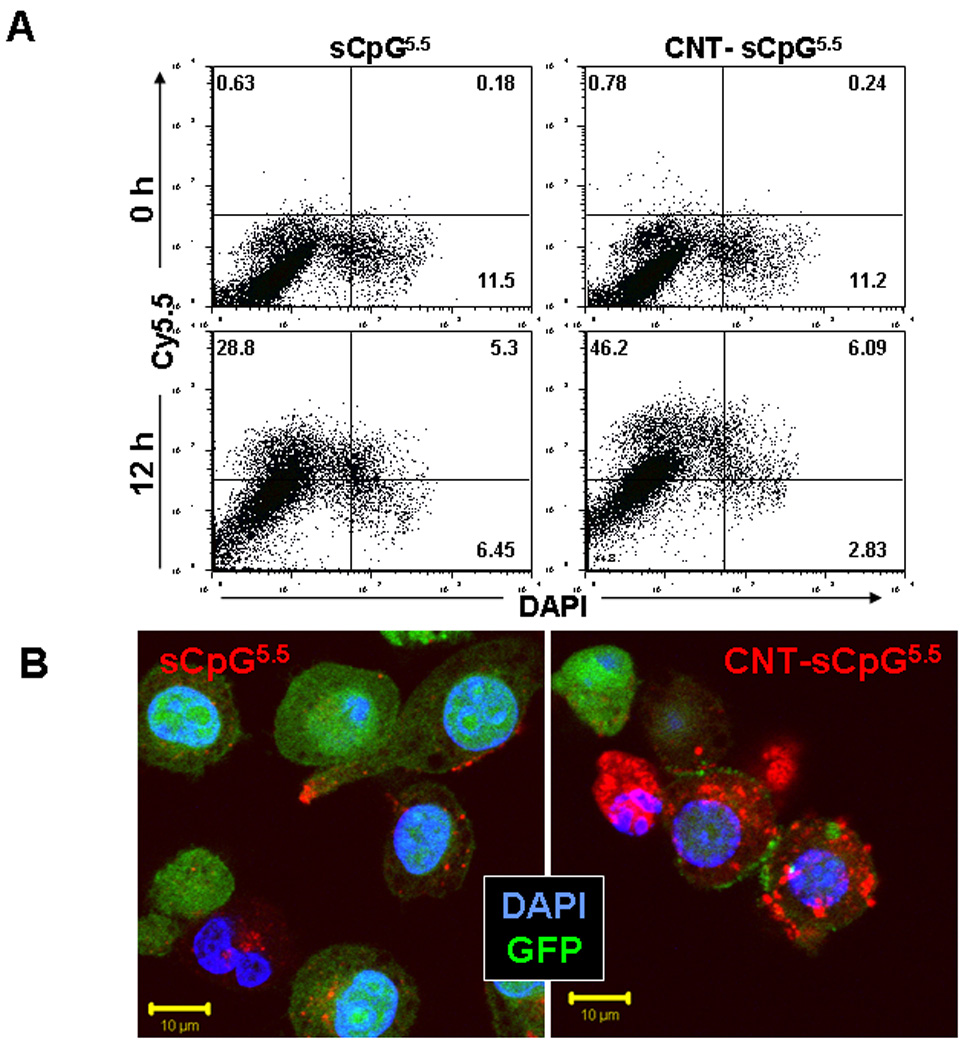
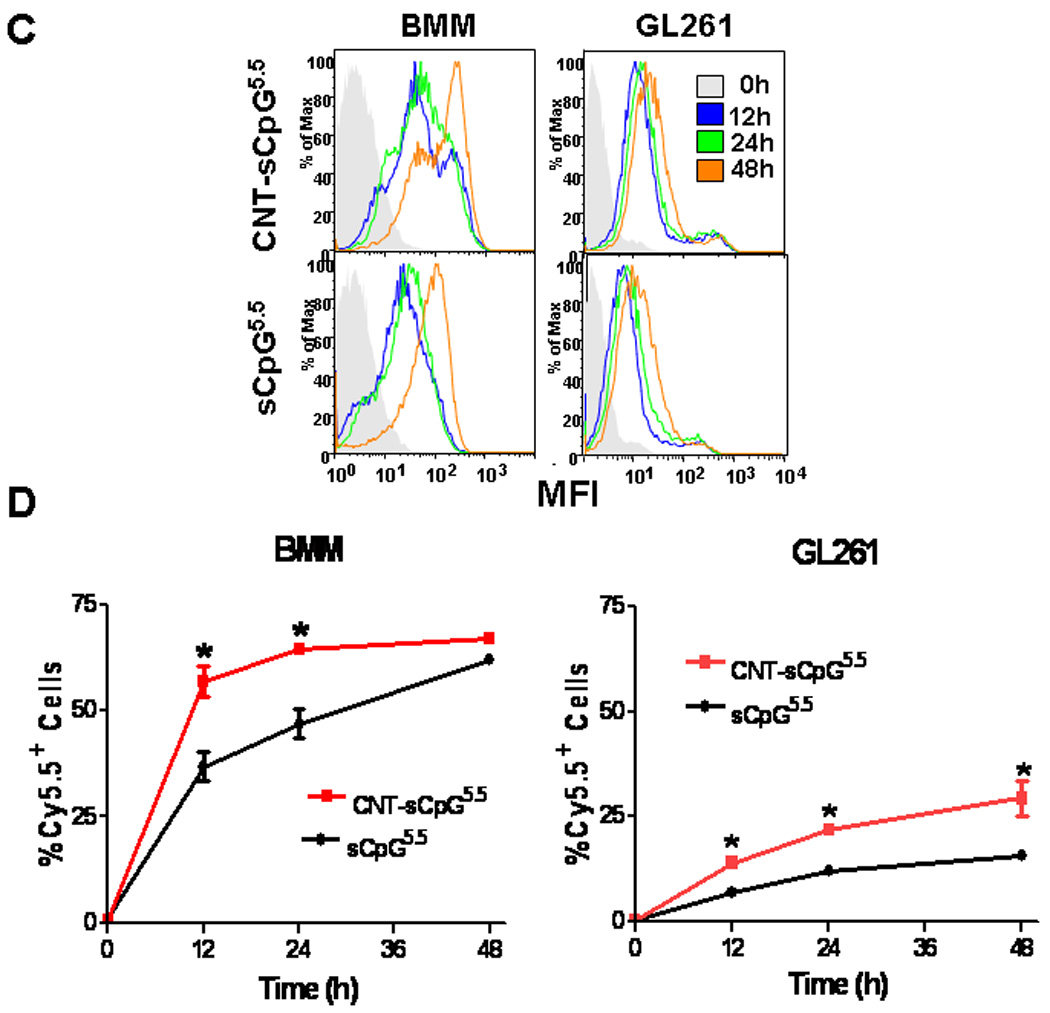
CNTs enhance CpG uptake in vitro. Primary bone marrow-derived monocytes (BMM) from wt or CX3CR1GFP mice or GL261 gliomas were incubated with PEG-functionalized single-walled CNTs conjugated to Cy5.5-labeled sCpG (CNT-sCpG5.5, 2.5 µg CNT- 5µg sCpG5.5/ml) or free sCpG5.5 (5µg/ml) and sCpG5.5 uptake was visualized by fluorescent microscopy and quantified by flow cytometry. A, dot plot demonstrating CNT-sCpG uptake by BMM to be more efficient than free sCpG5.5. B, CpG5.5 signal was stronger in BMM.gfp cells incubated with CNT-sCpG and was mostly confined to cytoplasmic compartments. C, histogram and D, time course experiments demonstrating CNT’s to also enhance sCpG5.5 uptake by GL261 gliomas, but not as efficiently as in BMM. Data is representative of three separate experiments. (n=3 samples /point, ±SEM); *, P<0.05, t-test.
CNT-mediated CpG delivery potentiates monocyte activation in vitro
CNT-sCpG was incubated with BMM and cytokine expression evaluated by qRT-PCR and ELISA. The thiolation process that was employed to link CpG with CNT appeared to suppress CpG pro-inflammatory function (Supplementary Fig. S2). Nevertheless, when equivalent doses of sCpG were delivered with CNT, a significant upregulation of IL-12 and TNF-α expression was seen (Fig. 2). CNT-sCpG also enhanced release of chemokines, but not VEGF, by BMM (Fig. 2). Thus, CNT appeared to not only improve CpG uptake, but also enhanced its proinflammatory function in BMM. We next tested the efficacy of CNTs as a potential CpG carrier and immunotherapy agent in i.c. gliomas.
Figure 2.
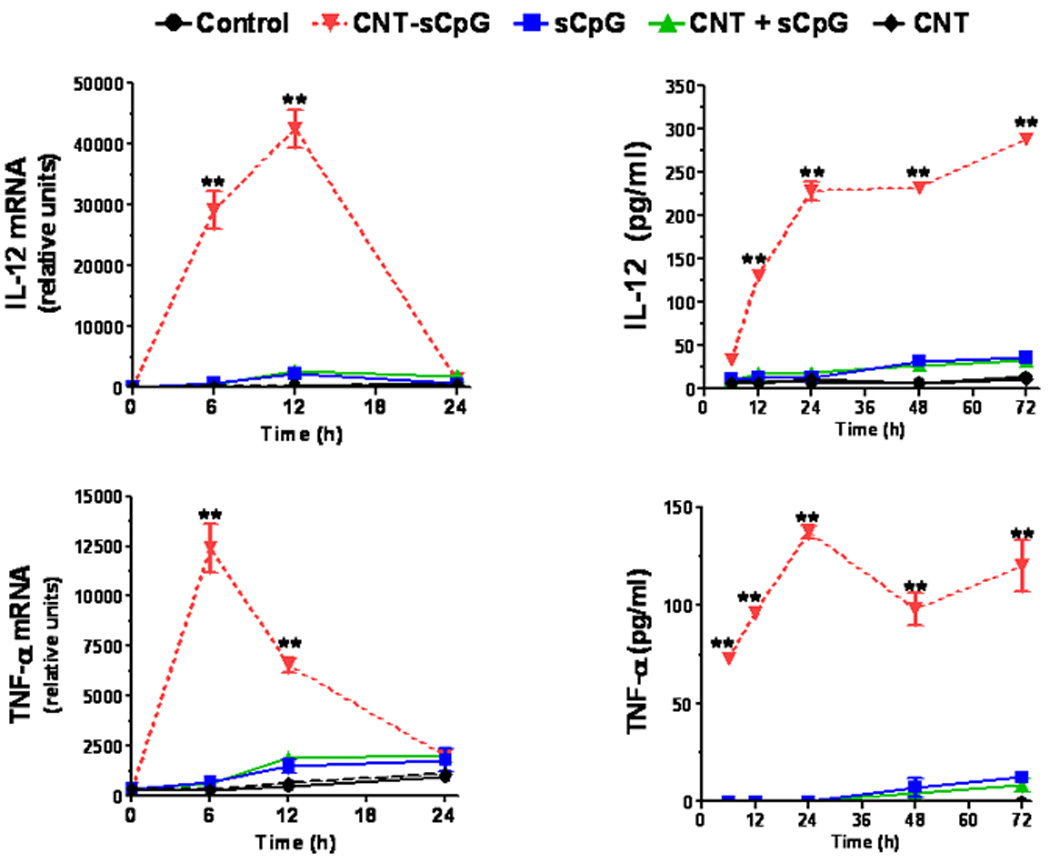
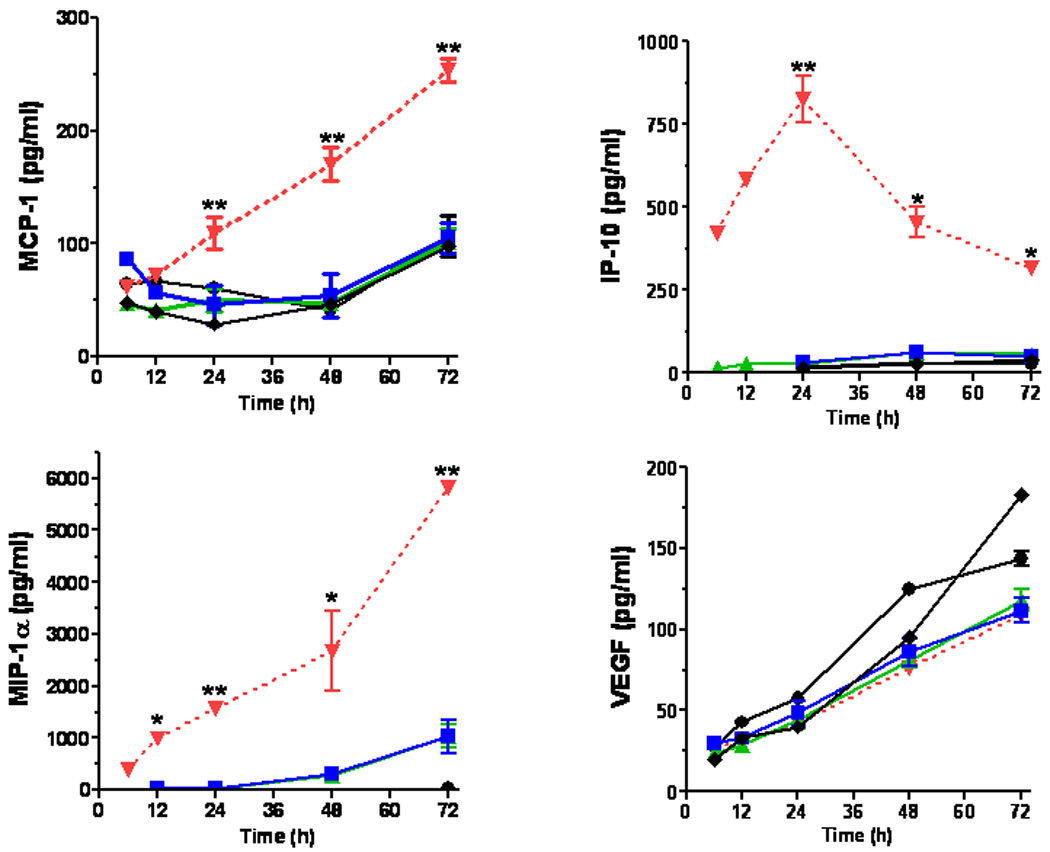
CNT-mediated CpG delivery potentiates monocyte activation in vitro. BMM (5×104 cells/well in 24-well plates) were incubated with sCpG (5µg/well), CNT-sCpG (CNT 2.5µg-sCpG 5µg /well), blank CNT (2.5µg/well), or CNT/sCpG mixture (CNT + sCpG) and cytokine/chemokine expression evaluated by qRT-PCR and ELISA. CNTs-CpG significantly upregulated the expression of proinflammatory cytokines and chemokines, but not VEGF, by BMM. Data is representative of two separate experiments. (n=3 samples /point, ±SEM); *, P<0.05, **, P<0.001 for comparisons to control group (ANOVA).
CNTs enhance CpG uptake by tumor-associated inflammatory cells
To evaluate CNT-sCpG uptake in vivo, two different i.c. GL261 glioma models were used. To assess uptake by tumor-associated inflammatory cells, GL261 cells were implanted into CX3CR1GFP mice. Although in these transgenic mice eGFP is expressed in MG, MPs, and other myeloid-derived cells, our flow cytometry studies (not shown) have indicated that the majority (more than 70%) of eGFP-expressing cells in this glioma model are MPs (CD11b+, CD45high) and MG (CD11b+, CD45low) based on previously-described phenotype characterization (26). In the second model, GL261.egfp cells were i.c. implanted into wt mice in order to measure sCpG uptake by tumor cells. Four (GL261 model) or ten days (slower-growing GL261.egfp model) after i.c. tumor implantation, tumors were injected with CNT-sCpG or sCpG, and tumor GFP+ cells were sorted and examined for Cy5.5 uptake at various time points (Fig. 3A and B). Consistent with the in vitro experiments, CNTs enhanced sCpG uptake by tumor-associated inflammatory cells, and to a lesser (non-significant) degree, by gliomas. Interestingly, sCpG clearance was slower in CNT-sCpG5.5-injected mice as both sCpG5.5 particles and sCpG5.5+ MG/MP persisted in the brains of these animals for more than seven days after the initial injection (Fig. 3A and C). This suggested that either clearance of CNT-sCpG by inflammatory cells was slower than free sCpG, and/or, robust chemokine release in CNT-sCpG group prevented migration of sCpG5.5+ inflammatory cells away from injection site. To further evaluate CNT-sCpG+ cell types, i.c. GL261 tumors in wt mice were injected with PBS (control), CNT-sCpG5.5, free sCpG5.5, or CNT/sCpG5.5 mixture, and phenotypes of Cy5.5+ inflammatory cells characterized by FACS. As expected, CNT promoted CpG uptake by MG/MP, NK, and DC cell (but not CD8 cells, not shown) (Fig. 4A and B). Although 20–30% of tumor leukocytes internalized CNT-sCpG5.5 particles within two days, the actual proportion of cells that were sCpG5.5+ locally was probably much higher because cells from the entire tumor (and not just the injection site) were analyzed in these experiments. Also, to assess which cell types were the prominent carriers of CNT-sCpG, and to check if the decline in sCpG5.5+ cells in the CNT-sCpG5.5 group at four days was due to the migration of these cells away from the injection site or trafficking of circulating sCpG5.5-negative cells into the tumor, we measured the total number of each cell type for all treatment groups. Neither sCpG nor sCpG/CNT treatments induced a significant change in tumor leukocyte infiltration (not shown). However, in the CNT-sCpG-treated mice, total and Cy5.5+ cells rapidly increased within two days of injection before stabilizing. Furthermore, CD11b+ cells (i.e. MG and MP) were the most frequent inflammatory cell and CNT-sCpG+ cell type within tumors. These findings suggested that: 1) CNT delivery enhanced CpG-induced innate immune response to gliomas, and 2) the decline in the proportion of CpG5.5+ inflammatory cells was most likely due to both influx of circulating cells into tumors and migration of CNT-sCpG-carrying cells out of the tumor environment. Whether this inflammatory response resulted in tumor rejection was evaluated next.
Figure 3.
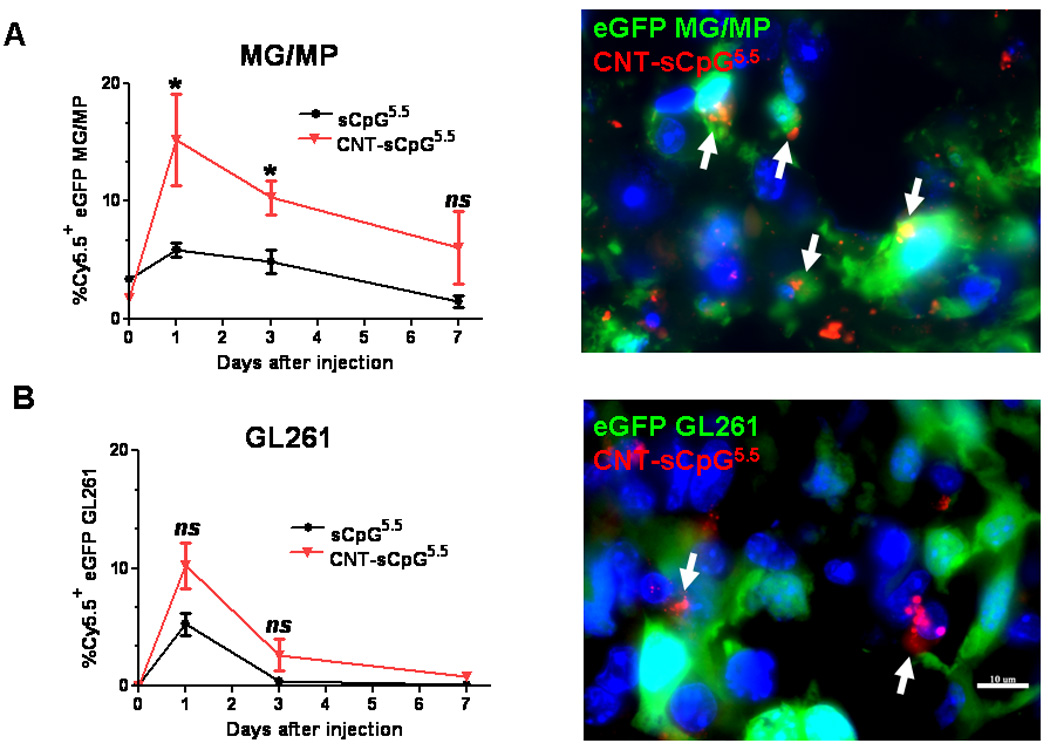
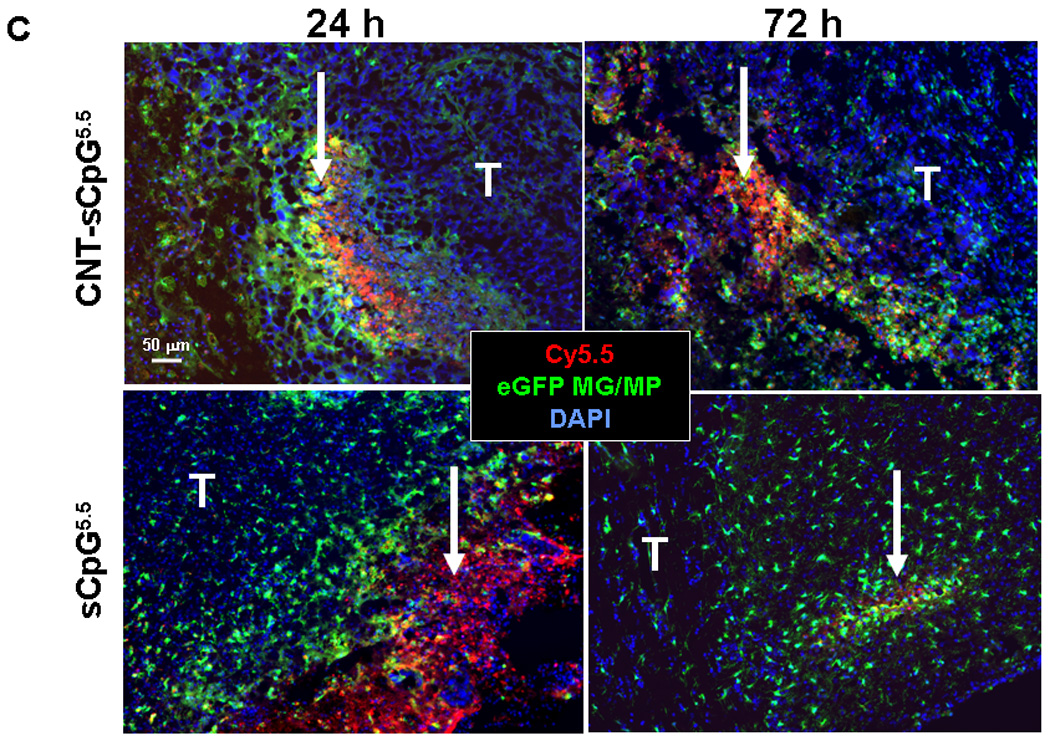
CNTs promote CpG uptake by tumor-associated inflammatory cells. A, Four days after i.c. implantation of GL261 cells into CX3CR1GFP mice, tumors were injected with CNT-sCpG (2.5 µg CNT-5 µg sCpG/10 µl PBS) or sCpG, (5µg/10µl PBS) and tumor-associated GFP+ cells (mostly microglia and macrophages, MG/MP) examined for Cy5.5 uptake at various time-points (left). Most of the intracellular CNT-sCpG (arrows) was seen in GFP-expressing MG/MP (right). B, GL261.egfp cells were implanted i.c. into wt mice in order to measure sCpG uptake by tumor cells. Ten days later, tumors were injected with CNT-sCpG or sCpG (same concentrations as A), and GFP+ tumor cells examined for Cy5.5 uptake at various time-points (left). Most of the CNT-sCpG (arrows) was seen in non-GFP-expressing cells (right). C, Representative fluorescent micrographs of brains from A demonstrating persistence of sCpG5.5+ MG/MP at the injection sites (arrows) in animals treated with CNT-sCpG5.5. (n=3 mice/time-point ±SEM); *, P<0.05, t-test; T: tumor, ns: not significant.
Figure 4.
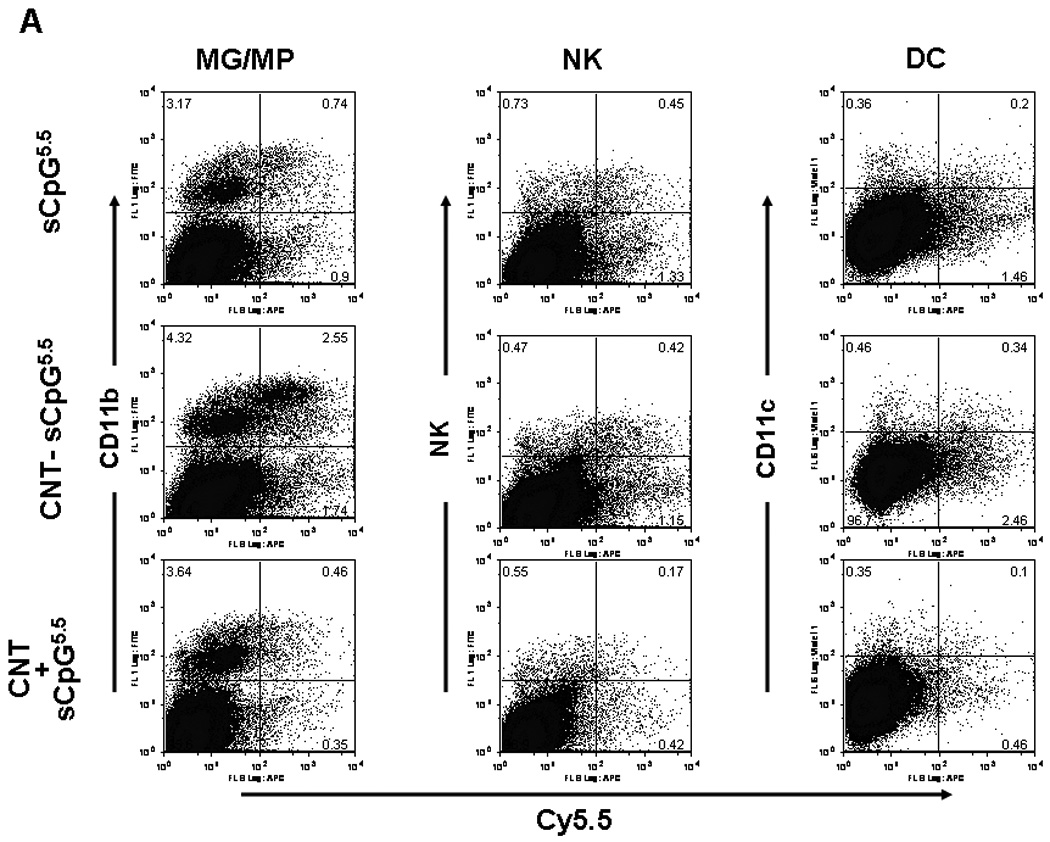
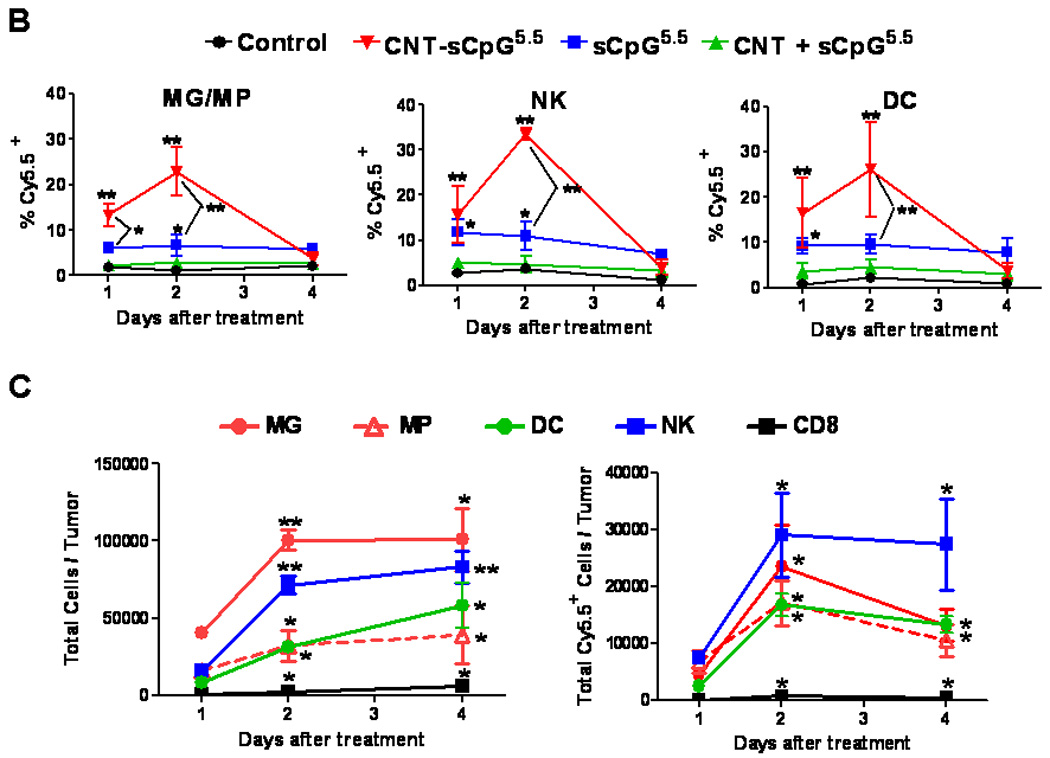
CNT-sCpG uptake by glioma-associated inflammatory cells. GL261 tumors in wt mice were injected with PBS (control), CNT-sCpG5.5, free sCpG5.5 or unconjugated CNT/sCpG5.5 mixture at the same concentrations as Fig. 3 and phenotypes of Cy5.5+ inflammatory cells characterized by FACS at different time intervals. A, Dot plots of tumor-inflammatory cells illustrating stronger CNT-sCpG5.5 uptake by tumor-associated microglia and macrophages (MG/MP), NK, and dendritic cells (DC) as compared to free sCpG or CNT/sCpG. B, The proportion of CNT-sCpG5.5-positive MG/MP, NK and DC increased within 24 hr of CNT-sCpG injection. C, Injection of CNT-sCpG increased total and CNT-sCpG5.5-positive inflammatory cells into tumors. Representative data from two experiments is shown. (n=3 mice/time-point ±SEM); *, P<0.05, **, P<0.001 for comparisons to control group in B and to 24 hr time-point in C (ANOVA).
CNT-sCpG improves survival of glioma-bearing mice
To evaluate CNT-sCpG immunotherapy, mice bearing four day-old i.c. GL261.luc gliomas were given a single i.t. injection of PBS (control), free sCpG, blank CNT mixed with free sCpG, or sCpG conjugated to CNT. The CNT/sCpG dose combination was selected based on the CNT conjugation capacity of sCpG (discussed above) and our recent observation indicating CpG to effectively eradicate gliomas at this dose (i.e. 5µg), albeit after multiple injections (21). As seen in previous in vivo studies (21), a single low-dose injection of free CpG had no anti-tumor effect (Fig. 5A and B). In contrast, CNT-sCpG not only delayed tumor growth, but also cured 50–60% of mice with established gliomas. Blank CNT by itself, or CNT conjugated with control oligodeoxynucleotide (CNT-sODN), had no effect on tumor growth (supplementary Fig. S3). The exact mechanism for this improved sCpG therapy with CNT delivery is unclear but may have been due to: 1) higher sCpG uptake by tumor-associated TLR9+ inflammatory cells (such as MG/MP), 2) slower intra- or extra-cellular sCpG release and clearance by CNTs (depot effect), and/or 3) migration of CNT-carrying tumor inflammatory cells into lymphoid organs where other immune cells are activated.
Figure 5.
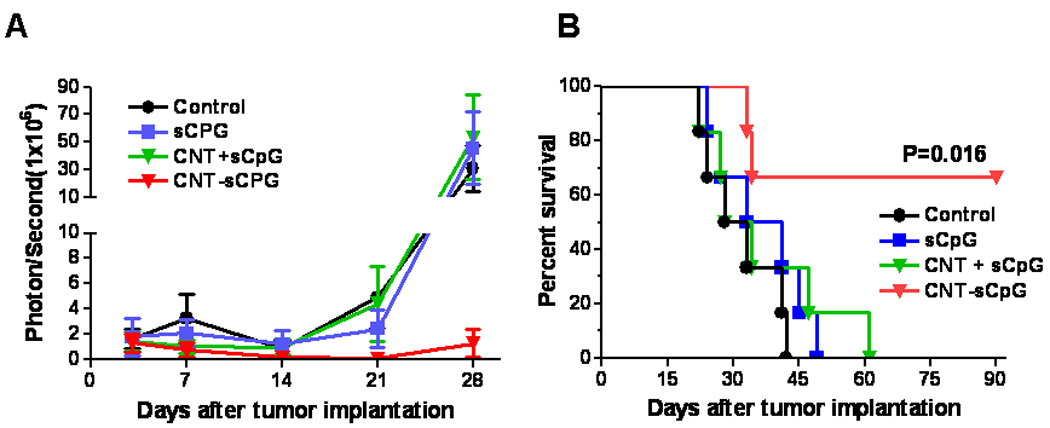
CNT-sCpG eradicates established gliomas. Mice bearing four day-old i.c. GL261.luc gliomas (n=6) were given a single i.t. injection of PBS (control), free sCpG (5µg/10µl PBS), blank CNT (2.5 µg) mixed with free sCpG (CNT + sCpG; 5µg CpG/10µl PBS), or sCpG conjugated to CNT (CNT-sCpG; 2.5 µg CNT-5 µg sCpG/10 µl PBS). A, Intracranial tumor burden was assessed by biophotonic imaging. B, Kaplan-Meier analysis demonstrates improved survival of mice treated with a single injection of CNT-sCpG.
Effect of CNT-sCpG on tumor-associated inflammatory response
In order to investigate inflammatory cellular responses to CNT-sCpG therapy, mice bearing 4 day-old i.c. tumors were treated with PBS, sCpG, CNT-sCpG and CpG-sODN and local and systemic inflammatory changes analyzed by FACS (supplementary Fig. S4). Although all agents increased CNS MG/MP, CD8, and NK cell responses, only CNT-sCpG caused a significantly higher infiltration of MG/MP over free sCpG. Furthermore, only CNT-sCpG-treated mice demonstrated a sustained elevation of circulating NK cells. These observations suggested that both MG/MP and NK cells may have played a role in CNT-sCpG anti-tumor response. To better characterize which cell types were responsible for tumor rejection, leukocyte depletion studies were performed next.
Anti-glioma effect of CNT-sCpG is mediated by both CD8 and NK cells
To verify if the anti-glioma effect of CNT-sCpG was solely due to activated NK cells (as suggested by an increase in circulating NK cells), depleting doses of CD8 or NK-specific Abs were given one day prior to both tumor implantation and CNT-sCpG treatment. The CNT-sCpG anti-tumor effect was completely abrogated by either NK or CD8 depletion (Fig. 6A), indicating that both cell types played a role in tumor rejection.
Figure 6.
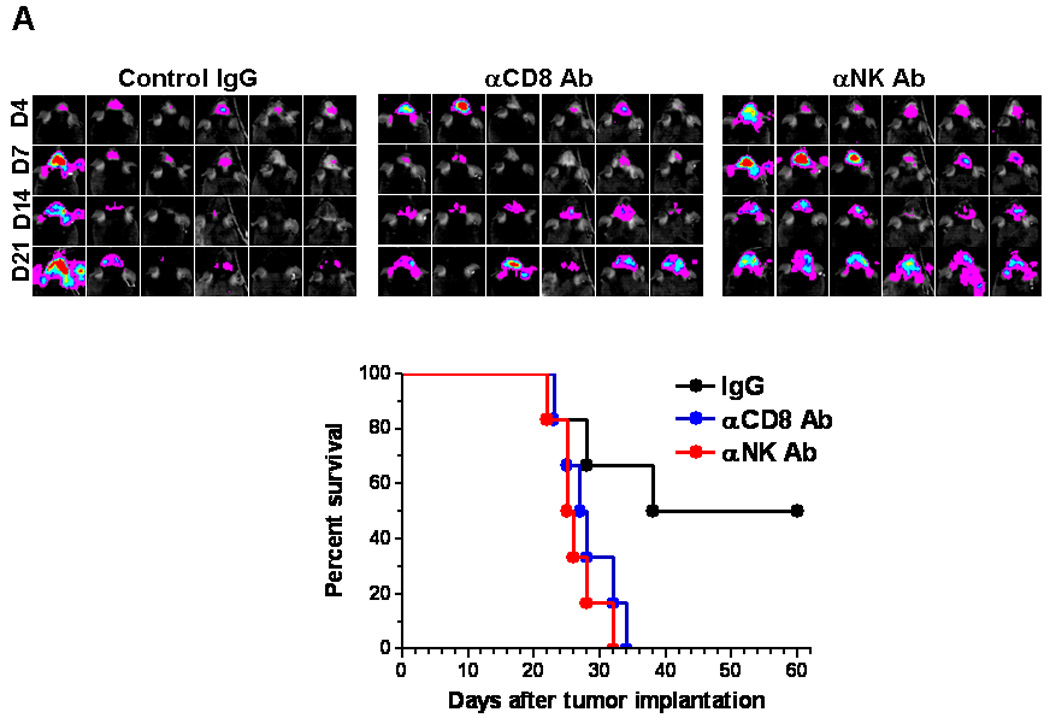
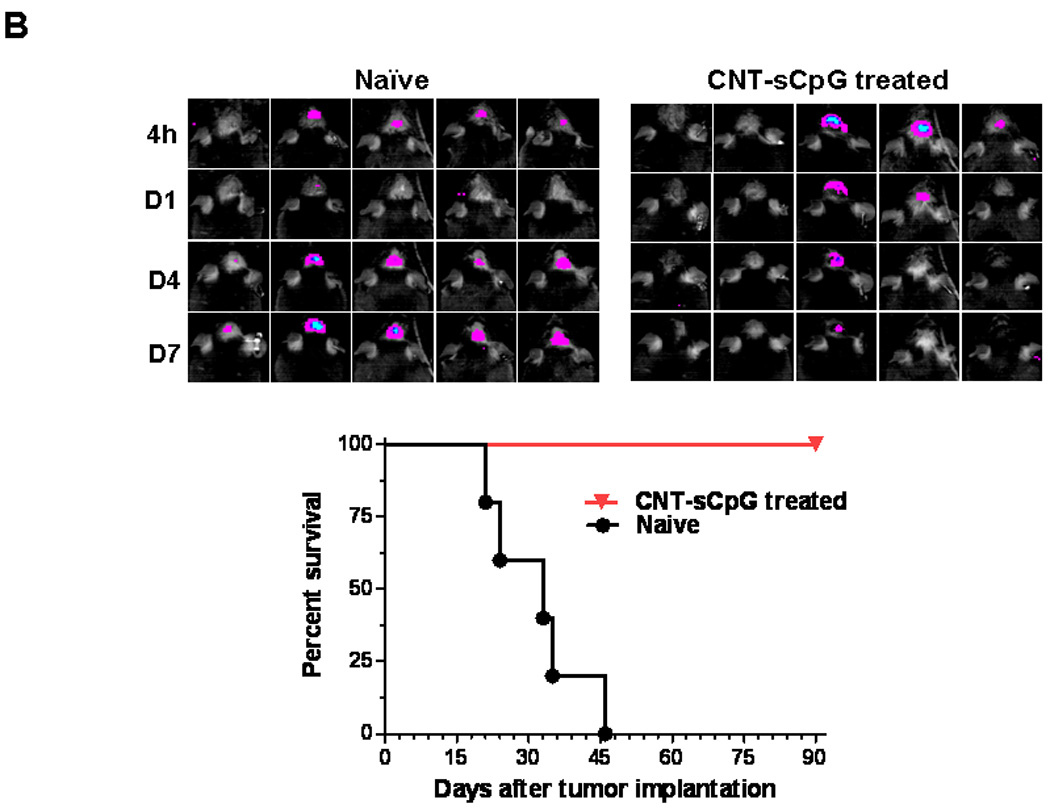
CNT-sCpG-treated mice develop immunity against tumor rechallenge. A, Naïve mice (n=6/group) were depleted of CD8 or NK cells by i.p. injection of relevant mAb (αCD8 and αNK) or control IgG (200 µg/injection) one day prior to both tumor implantation and CNT-sCpG (2.5 µg CNT/5 µg CpG/10 µl PBS) treatment. Intracranial tumor burden was assessed by biophotonic imaging at day 4, 7, 14, and 21 post tumor implantation (top panel). NK and CD8-depleted mice exhibited more rapid tumor growth and lower survival rates (bottom panel). B, Normal naïve mice and CNT-sCpG-treated GL261-bearing mice (n=5/group) that had survived for at least three months after the initial tumor inoculation were rechallenged with an i.c. injection of GL261 glioma (1×105). Intracranial tumor burden was assessed at 4 h, days 1, 4, and 7 after tumor implantation.
Finally, to determine if CNT-sCpG treatment induced immunity against gliomas, CNT-sCpG-treated GL261-bearing mice that had survived for at least three months, along with naïve mice, were rechallenged with an i.c. injection of GL261 glioma. These “cured” mice developed full immunity against GL261 rechallenge (Fig. 6B).
Discussion
As potent stimulators of both innate and adaptive immune systems, CpG ODNs are currently being examined for cancer immunotherapy (5). Anti-tumor function of CpG has also been studied in brain tumors. Although initial reports by Carpentier et al. demonstrated an 88% cure rate and long-term immunity in rats with i.c. gliomas (27, 28), others have shown variable CpG anti-glioma responses ranging from no efficacy (29), partial response (4), and complete tumor eradication (30). One explanation for these contrasting reports may be differences in animal models and CpG constructs. Nevertheless, results from early-stage human clinical trials utilizing convection-enhanced CpG therapy in patients with recurrent malignant gliomas have been disappointing with only modest anti-tumor response in a few patients (2, 8). Although increasing the CpG dose may further enhance its pro-inflammatory effect, high i.c. CpG could exacerbate brain edema and result in CNS toxicity by activating TLR9-independent pathways (31). Alternatively, pro-inflammatory CpG response can be augmented with repeated injections of low-dose CpG or nanoparticles delivery systems (21, 32). Both lipid and nonlipid-based nanoparticles have been shown to promote CpG uptake into TLR9-rich endosomes to enhance its immunostimulatory activity in vitro and in vivo. (32–37). Here we demonstrate that functionalized CNTs may also be used to enhance CpG uptake by tumor-associated phagocytic cells as an immunotherapy approach. Although the use of CNTs for CpG delivery has been examined in vitro before (9, 38, 39), this is the first report demonstrating the anti-tumor efficacy of CNT-CpG complexes in vivo. Our data indicate that improved delivery of CpG into endosomes by CNTs and “slow-clearance” of CNT-sCpG complexes in the CNS both played a role in augmenting CpG immunotherapy in this model.
Located in the endosomal compartment, TLR9 was initially shown to specifically respond to unmethylated CpG motifs, such as those present in bacterial DNA. As a result, synthetic CpGs were designed to contain repeat unmethylated CpG motifs to enhance TLR9 engagement, and phosphorothioate linkages to prevent breakdown (40). Recent work, however, suggests that DNA recognition by TLR9 depends on the 2’ deoxyribose phosphate backbone and that neither phosphorothioate linkage nor specific sequences (i.e. CpG motifs) are necessary to induce the immune response (41, 42). These findings have challenged the dogma that unmethylated CpG motifs constitute the “foreign signature” that triggers TLR9. Instead, phosphorothioate linkages and CpG motifs appear to increase CpG stability, aggregation, and uptake into cells. Thus, any approach that stabilizes and improves CpG uptake into endosomal compartments may enhance its stimulatory functions. Accordingly, nanoparticles have been employed to augment CpG uptake and function (32–37). Lipid nanoparticles, for example, can not only improve CpG uptake and immunopotency (32), but as demonstrated recently, are critical in sequestering TLR9 migration from endoplasmic reticulum to the late endosomal compartment where it can interact with CpG (35). Although we did not correlate subcellular colocalization of CNT-CpG with TLR9 in this study, rapid CNT-CpG sequestration into cytoplasmic compartments seen here suggests that CNTs were responsible for augmenting CpG prostimulatory function by facilitating its uptake.
CNTs can enter cells through several mechanisms including phagocytosis, diffusion, or receptor-mediated endocytosis (43). This internalization process not only depends on the target cell type, but also on a variety of CNT characteristics like size, clustering, and surface charge. Here, we noted CNTs to enhance CpG uptake by both monocytes and gliomas in vitro. CNT uptake, however, was more efficient in phagocytic BMM and resulted in upregulation of pro-inflammatory cytokines/chemokines. Similarly, in i.c. tumors, CNT-mediated CpG uptake was more efficient in TAMs suggesting active phagocytosis/endocytosis of CNT-sCpG complexes by these antigen-presenting cells. In fact, MG and MP collectively accounted for the highest number of CNT-sCpG positive cells in tumors. Early rise in tumor MG/MP, followed by sustained elevation of circulating NK cells indicated that stimulation of TAM’s into M1 phenotype by CNT-sCpG may have triggered induction of anti-glioma NK and CD8 cells. However, since NK cells also internalized CNTs very efficiently, it’s not clear if their stimulation was entirely dependent on TAM activation or due to CNT-sCpG uptake. Future studies will evaluate the differential contribution of MG/MP and NK cells to CNT-sCpG immune responses.
Another potential mechanism that may have accounted for enhanced CpG immunopotency is the unique CNT-sCpG biodistribution within the CNS. Because of the blood-brain barrier and first-pass clearance by liver and lungs, CNTs typically do not penetrate normal brain tissue after i.v. injections (13–15). Biodistribution of CNTs following i.c. injections has not been studied before. But recently, we demonstrated that fCNTs are efficiently phagocytosed by TAMs in i.c. gliomas where they can remain for nearly one week after direct i.t. injections (12). Similarly, CNT-sCpG complexes studied here remained within i.c. tumors longer than free CpG. Slow clearance of CNT-sCpG complexes from the CNS may have provided a “depot” effect, allowing enough time for migration of inflammatory cells into the tumor environment and inducing a stronger anti-glioma effect.
Toxicity has been raised as a potential limitation of CNT biomedical application. While CNTs can be biodegraded through enzymatic catalysis (44), pristine (non-modified/non-functionalized) CNTs have been shown to persist for several months raising concerns of potential negative health effects (45). CNT toxicity, however, is strongly related to physicochemical properties (such as structure, length, surface area, degree of metallic contamination, and surface coating) and can be minimized with functionalization techniques (11, 46). PEG-functionalized CNTs, for example, have been shown to be nontoxic, and cleared from biliary and renal pathways within 2 months of i.v. injection in mice (47). Consistent with our previous report, fCNTs studied here were non toxic after i.c. injections in these short-term experiments, thereby supporting their utilization in cancer immunotherapy.
CNTs may have various potential advantages over other nanomaterials in biomedical applications. fCNTs can be used to deliver siRNA into ‘hard-to-transfect’ cells such as MPs and T cells that are usually inert to conventional liposomal transfection agents (48). They can also be used as contrast agents in multimodality optical imaging, and since they do not contain heavy metals, have a safer chemical composition as compared to gold/silver-containing quantum dots (49). Finally, the high optical absorbance of CNTs can be used in photothermal therapy, which may potentially be combined with CNT-based chemotherapy and gene therapy of cancer (24, 50).
In summary, we demonstrated CNT delivery system to significantly enhance CpG immunotherapy, eradicate i.c. gliomas at low doses, and induce immunity against tumor rechallenge without inducing toxicity. Our findings have direct implications to the development of CNT-based treatments for malignant brain tumors. Considering that fCNTs can be used to carry other oligonucleotiedes such as siRNA (51), this strategy can be employed to overcome local immunosuppressive microenvironment through modulations of tumor inflammatory responses.
Statement of Translational Relevance
The prognosis of patients with malignant gliomas, the most common type of primary brain neoplasm, remains dismal even after aggressive multimodality treatment. Although immunotherapy is being investigated as an adjunct treatment, the ability of gliomas to escape immune response will continue to be a significant obstacle to this strategy. One approach to overcome the local immunosuppressive tumor microenvironment is the activation of the innate immune system by toll-like receptor (TLR) agonists such as CpG oligonucleotides (CpG). Because TLR9, CpG receptor, is located intracellularly, we hypothesized that methods that enhance CpG internalization may also potentiate its immunostimulatory response. Here, we demonstrate that Carbon nanotubes (CNTs) enhanced CpG uptake by tumor-associated phagocytic cells and resulted in their activation both in vitro and in vivo. Furthermore, a single injection of low-dose CNT-CpG complexes eradicated intracranial gliomas through activation of NK and CD8 cells. These findings demonstrate that CNTs are nontoxic vehicles that can improve CpG uptake into tumor-associated inflammatory cells, leading to a more robust anti-tumor response. These findings have direct application to the design of future CpG immunotherapy trials.
Supplementary Material
Acknowledgements
The authors thank Dr. Harish Manohara (Jet Propulsion Laboratory, Pasadena, CA) for providing the carbon nanotubes, Dr. Piotr Swiderski for generating thiolated CpG constructs, Dr. Brian Armstrong for assisting with confocal microscopy, and Dr. Simon Lacey and Shu Mi (Clinical Immunobiology Correlative Studies Laboratory) for conducting the Luminex studies.
Grant Support: This work was supported by R21CA131765-01A2 (BB), P01-CA030206 and CA077544 (DJD), James S. McDonnell Foundation (BB) and ThinkCure Foundation (to BB and DJD). The City of Hope Flow Cytometry Core was equipped in part through funding provided by ONR N00014-02-1 0958, DOD 1435-04-03GT-73134, and NSF DBI-9970143.
Literature Cited
- 1.Gomez GG, Kruse CA. Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene therapy & molecular biology. 2006;10:133–146. [PMC free article] [PubMed] [Google Scholar]
- 2.Carpentier A, Laigle-Donadey F, Zohar S, et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006 Aug;:60–66. doi: 10.1215/S1522851705000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalpke AH, Schafer MK, Frey M, et al. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- 4.El Andaloussi A, Sonabend AM, Han Y, Lesniak MS. Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia. 2006;54:526–535. doi: 10.1002/glia.20401. [DOI] [PubMed] [Google Scholar]
- 5.Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev. 2009;61:195–204. doi: 10.1016/j.addr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 7.Carpentier AF, Auf G, Delattre JY. CpG-oligonucleotides for cancer immunotherapy : review of the literature and potential applications in malignant glioma. Front Biosci. 2003;8:e115–e127. doi: 10.2741/934. [DOI] [PubMed] [Google Scholar]
- 8.Carpentier A, Metellus P, Ursu R, et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 2010;12:401–408. doi: 10.1093/neuonc/nop047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianco A, Hoebeke J, Kostarelos K, Prato M, Partidos CD. Carbon nanotubes: on the road to deliver. Curr Drug Deliv. 2005;2:253–259. doi: 10.2174/1567201054367959. [DOI] [PubMed] [Google Scholar]
- 10.Pulskamp K, Diabate S, Krug HF. Carbon nanotubes show no sign of acute toxicity but induce intracellular reactive oxygen species in dependence on contaminants. Toxicol Lett. 2007;168:58–74. doi: 10.1016/j.toxlet.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Schipper ML, Nakayama-Ratchford N, Davis CR, et al. A pilot toxicology study of single-walled carbon nanotubes in a small sample of mice. Nat Nano. 2008;3:216–221. doi: 10.1038/nnano.2008.68. [DOI] [PubMed] [Google Scholar]
- 12.VanHandel M, Alizadeh D, Zhang L, et al. Selective uptake of multi-walled carbon nanotubes by tumor macrophages in a murine glioma model. J Neuroimmunol. 2009;208:3–9. doi: 10.1016/j.jneuroim.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 13.McDevitt MR, Chattopadhyay D, Kappel BJ, et al. Tumor targeting with antibody-functionalized, radiolabeled carbon nanotubes. J Nucl Med. 2007;48:1180–1189. doi: 10.2967/jnumed.106.039131. [DOI] [PubMed] [Google Scholar]
- 14.Singh R, Pantarotto D, Lacerda L, et al. Tissue biodistribution and blood clearance rates of intravenously administered carbon nanotube radiotracers. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3357–3362. doi: 10.1073/pnas.0509009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Wang J, Deng X, et al. Biodistribution of carbon single-wall carbon nanotubes in mice. J Nanosci Nanotechnol. 2004;4:1019–1024. doi: 10.1166/jnn.2004.146. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z, Cai W, He L, et al. In vivo biodistribution and highly efficient tumour targeting of carbon nanotubes in mice. Nat Nanotechnol. 2007;2:47–52. doi: 10.1038/nnano.2006.170. [DOI] [PubMed] [Google Scholar]
- 17.Villa CH, McDevitt MR, Escorcia FE, et al. Synthesis and Biodistribution of Oligonucleotide-Functionalized, Tumor-Targetable Carbon Nanotubes. Nano letters. 2008 doi: 10.1021/nl801878d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kateb B, Van Handel M, Zhang L, Bronikowski MJ, Manohara H, Badie B. Internalization of MWCNTs by microglia: possible application in immunotherapy of brain tumors. Neuroimage. 2007;37 Suppl 1:S9–S17. doi: 10.1016/j.neuroimage.2007.03.078. [DOI] [PubMed] [Google Scholar]
- 19.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 20.Daftarian P, Song GY, Ali S, et al. Two distinct pathways of immuno-modulation improve potency of p53 immunization in rejecting established tumors. Cancer Res. 2004;64:5407–5414. doi: 10.1158/0008-5472.CAN-04-0169. [DOI] [PubMed] [Google Scholar]
- 21.Alizadeh D, Zhang L, Brown CE, Farrukh O, Jensen MC, Badie B. Induction of anti-glioma NK cell response following multiple low-dose intracerebral CpG therapy. Clin Cancer Res. 2010;16:3399–3408. doi: 10.1158/1078-0432.CCR-09-3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alizadeh D, Zhang L, Hwang J, Schluep T, Badie B. Tumor-associated macrophages are predominant carriers of cyclodextrin-based nanoparticles into gliomas. Nanomedicine. 2010;6:382–390. doi: 10.1016/j.nano.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manohara HM, Wong EW, Schlecht E, Hunt BD, Siegel PH. Carbon nanotube Schottky diodes using Ti-Schottky and Pt-Ohmic contacts for high frequency applications. Nano Lett. 2005;5:1469–1474. doi: 10.1021/nl050829h. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Tabakman SM, Chen Z, Dai H. Preparation of carbon nanotube bioconjugates for biomedical applications. Nat Protoc. 2009;4:1372–1382. doi: 10.1038/nprot.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Alizadeh D, Van Handel M, Kortylewski M, Yu H, Badie B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia. 2009;57:1458–1467. doi: 10.1002/glia.20863. [DOI] [PubMed] [Google Scholar]
- 26.Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000;46:957–961. doi: 10.1097/00006123-200004000-00035. discussion 61-2. [DOI] [PubMed] [Google Scholar]
- 27.Carpentier AF, Chen L, Maltonti F, Delattre JY. Oligodeoxynucleotides containing CpG motifs can induce rejection of a neuroblastoma in mice. Cancer Res. 1999;59:5429–5432. [PubMed] [Google Scholar]
- 28.Carpentier AF, Xie J, Mokhtari K, Delattre JY. Successful treatment of intracranial gliomas in rat by oligodeoxynucleotides containing CpG motifs. Clin Cancer Res. 2000;6:2469–2473. [PubMed] [Google Scholar]
- 29.Wu A, Oh S, Gharagozlou S, et al. In vivo vaccination with tumor cell lysate plus CpG oligodeoxynucleotides eradicates murine glioblastoma. J Immunother. 2007;30:789–797. doi: 10.1097/CJI.0b013e318155a0f6. [DOI] [PubMed] [Google Scholar]
- 30.Grauer OM, Molling JW, Bennink E, et al. TLR ligands in the local treatment of established intracerebral murine gliomas. J Immunol. 2008;181:6720–6729. doi: 10.4049/jimmunol.181.10.6720. [DOI] [PubMed] [Google Scholar]
- 31.Sanjuan MA, Rao N, Lai KT, et al. CpG-induced tyrosine phosphorylation occurs via a TLR9-independent mechanism and is required for cytokine secretion. J Cell Biol. 2006;172:1057–1068. doi: 10.1083/jcb.200508058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson KD, de Jong SD, Tam YK. Lipid-based delivery of CpG oligonucleotides enhances immunotherapeutic efficacy. Adv Drug Deliv Rev. 2009;61:233–242. doi: 10.1016/j.addr.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 33.Bourquin C, Anz D, Zwiorek K, et al. Targeting CpG oligonucleotides to the lymph node by nanoparticles elicits efficient antitumoral immunity. J Immunol. 2008;181:2990–2998. doi: 10.4049/jimmunol.181.5.2990. [DOI] [PubMed] [Google Scholar]
- 34.Chikh G, de Jong SD, Sekirov L, et al. Synthetic methylated CpG ODNs are potent in vivo adjuvants when delivered in liposomal nanoparticles. Int Immunol. 2009;21:757–767. doi: 10.1093/intimm/dxp044. [DOI] [PubMed] [Google Scholar]
- 35.de Jong SD, Basha G, Wilson KD, et al. The immunostimulatory activity of unmethylated and methylated CpG oligodeoxynucleotide is dependent on their ability to colocalize with TLR9 in late endosomes. J Immunol. 184 doi: 10.4049/jimmunol.0802442. 6092-102. [DOI] [PubMed] [Google Scholar]
- 36.Malyala P, O'Hagan DT, Singh M. Enhancing the therapeutic efficacy of CpG oligonucleotides using biodegradable microparticles. Adv Drug Deliv Rev. 2009;61:218–225. doi: 10.1016/j.addr.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Schreiber HA, Prechl J, Jiang H, et al. Using carbon magnetic nanoparticles to target, track, and manipulate dendritic cells. J Immunol Methods. 356:47–59. doi: 10.1016/j.jim.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krishnamachari Y, Salem AK. Innovative strategies for co-delivering antigens and CpG oligonucleotides. Adv Drug Deliv Rev. 2009;61:205–217. doi: 10.1016/j.addr.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Partidos CD, Hoebeke J, Wieckowski S, et al. Immunomodulatory consequences of ODN CpG-polycation complexes. Methods. 2009;49:328–333. doi: 10.1016/j.ymeth.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 40.Wagner H. The sweetness of the DNA backbone drives Toll-like receptor 9. Curr Opin Immunol. 2008;20:396–400. doi: 10.1016/j.coi.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 41.Haas T, Metzger J, Schmitz F, et al. The DNA sugar backbone 2' deoxyribose determines toll-like receptor 9 activation. Immunity. 2008;28:315–323. doi: 10.1016/j.immuni.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 42.Yasuda K, Rutz M, Schlatter B, et al. CpG motif-independent activation of TLR9 upon endosomal translocation of "natural" phosphodiester DNA. Eur J Immunol. 2006;36:431–436. doi: 10.1002/eji.200535210. [DOI] [PubMed] [Google Scholar]
- 43.Raffa V, Ciofani G, Vittorio O, Riggio C, Cuschieri A. Physicochemical properties affecting cellular uptake of carbon nanotubes. Nanomedicine (Lond) 2010;5:89–97. doi: 10.2217/nnm.09.95. [DOI] [PubMed] [Google Scholar]
- 44.Allen BL, Kichambare PD, Gou P, et al. Biodegradation of Single-Walled Carbon Nanotubes through Enzymatic Catalysis. Nano Letters. 2008;8:3899–3903. doi: 10.1021/nl802315h. [DOI] [PubMed] [Google Scholar]
- 45.Chou C-C, Hsiao H-Y, Hong Q-S, et al. Single-Walled Carbon Nanotubes Can Induce Pulmonary Injury in Mouse Model. Nano Letters. 2008;8:437–445. doi: 10.1021/nl0723634. [DOI] [PubMed] [Google Scholar]
- 46.De La Zerda A, Zavaleta C, Keren S, et al. Carbon nanotubes as photoacoustic molecular imaging agents in living mice. Nat Nano. 2008;3:557–562. doi: 10.1038/nnano.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z, Davis C, Cai W, He L, Chen X, Dai H. Circulation and long-term fate of functionalized, biocompatible single-walled carbon nanotubes in mice probed by Raman spectroscopy. Proc Natl Acad Sci U S A. 2008;105:1410–1415. doi: 10.1073/pnas.0707654105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Z, Winters M, Holodniy M, Dai H. siRNA delivery into human T cells and primary cells with carbon-nanotube transporters. Angew Chem Int Ed Engl. 2007;46:2023–2027. doi: 10.1002/anie.200604295. [DOI] [PubMed] [Google Scholar]
- 49.Zavaleta C, de la Zerda A, Liu Z, et al. Noninvasive Raman spectroscopy in living mice for evaluation of tumor targeting with carbon nanotubes. Nano Lett. 2008;8:2800–2805. doi: 10.1021/nl801362a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakravarty P, Marches R, Zimmerman NS, et al. Thermal ablation of tumor cells with antibody-functionalized single-walled carbon nanotubes. Proc Natl Acad Sci U S A. 2008;105:8697–8702. doi: 10.1073/pnas.0803557105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Jamal KT, Toma FM, Yilmazer A, et al. Enhanced cellular internalization and gene silencing with a series of cationic dendron-multiwalled carbon nanotube:siRNA complexes. FASEB J. 2010 doi: 10.1096/fj.09-141036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.