

Abstract
Clusterin is a ubiquitously expressed glycoprotein with multiple binding partners including IL-6, Ku70, and Bax. Clusterin blocks apoptosis by binding to activated Bax and sequestering it in the cytoplasm, thereby preventing Bax from entering mitochondria, releasing cytochrome c, and triggering apoptosis. Because increased clusterin expression correlates with aggressive behavior in tumors, clusterin inhibition might be beneficial in cancer treatment. Our recent findings indicated that, in neuroblastoma cells, cytoplasmic Bax also binds to Ku70; when Ku70 is acetylated, Bax is released and can initiate cell death. Therefore, increasing Ku70 acetylation, such as by using histone deacetylase inhibitors, may be therapeutically useful in promoting cell death in neuroblastoma tumors. Since clusterin, Bax, and Ku70 form a complex, it seemed likely that clusterin would mediate its anti-apoptotic effects by inhibiting Ku70 acetylation and blocking Bax release. Our results, however, demonstrate that while clusterin level does indeed determine the sensitivity of neuroblastoma cells to histone deacetylase inhibitor-induced cell death, it does so without affecting histone deacetylase-inhibitor-induced Ku70 acetylation. Our results suggest that in neuroblastoma, clusterin exerts its anti-apoptotic effects downstream of Ku70 acetylation, likely by directly blocking Bax activation.
Keywords: Apoptosis, Clusterin, Neuroblastoma
Introduction
Despite its discovery more than 25 years ago in ram rete testis fluid as the factor responsible for clustering of Sertoli cells [1], the physiologic significance of clusterin (CLU) expression in a range of other tissues is still unclear. CLU is present in nearly all mammalian cells and in plasma, breast milk, urine, and cerebrospinal fluid [2]. CLU expression has been implicated as a factor in a variety of biological processes such as spermatogenesis, lipid transport, cell differentiation, cell death, and cell survival, which at least partially accounts for the multiplicity of names given to this protein, including ApoJ (apolipoprotein J) [3], TRPM-2 (testosterone-repressed prostatic message-2) [4], SGP-2 (sulfated glycoprotein 2) [5], XIP8 (ionizing radiation-induced protein 8) [6], SP 40-40 (serum protein 40,40) [7], CLI (complement-lysis inhibitor) [8], gp80, glycoprotein III, and T64 [2, 9]. CLU is encoded by a single gene located on human chromosome 8 [10]. There are two isoforms that result from alternate translation start sites. One is a glycosylated, secretable form that is translated from the full-length CLU message using the first AUG initiation codon. This isoform includes a leader sequence directing ER synthesis and secretion as well as sites for glycosylation. It is detected as two bands on immunoblots that represent a 60-kDa uncleaved protein (p-CLU) and a 40-kDa product resulting from cleavage of the leader sequence (m-CLU) [11]. The second isoform of CLU is translated using the second AUG codon of the CLU mRNA, and the product lacks a leader sequence, is not secreted, and is not glycosylated. This form of CLU can be detected in the nucleus (n-CLU) [12].
Increased CLU expression is related to neoplastic progression in some cancers. The strongest evidence linking high levels of CLU to carcinogenesis comes from studies of breast and prostate cancer [13, 14]. Based in part on differential expression levels between prostate cancer and normal prostate tissue as well as on data identifying CLU as an anti-apoptotic (pro-cell survival) factor, clinical trials evaluating antisense RNA specific for CLU (OGX-011) are underway in patients with prostate cancer [15]. Tumor cell apoptosis is proposed as the therapeutic mechanism for anti-CLU therapy. It is of note, however, that depending on the tumor type studied, CLU expression can have paradoxical anti-tumor effects [16, 17].
CLU interacts with the pro-apoptotic multi-domain Bcl-2 family protein Bax and Bax–Ku70 protein complexes [18]. CLU–Bax binding inhibits Bax activation and mitochondrial translocation and cell death [19]. Recently, we showed that histone deacetylase (HDAC) inhibitor (HDACI)-induced cell death in neuroblastoma (NB) is Bax-dependent and occurs as a result of HDACI-induced disruption of Bax binding to Ku70 protein [20]. In NB, HDACI treatment leads to increased Ku70 acetylation and, because Ku70-Bax binding is acetylation sensitive, Bax is released from Ku70, resulting in Bax-dependent apoptosis. Since CLU is expressed in NB cells [17], its binding to Bax and Ku70–Bax raises the possibility that CLU inhibits apoptosis by modulating Ku70 acetylation. Here we test the hypothesis that CLU suppresses Ku70 acetylation, stabilizing the Ku70–Bax complex and preventing apoptosis. We conducted overexpression as well as CLU depletion experiments to test this model. Although CLU prevents apoptosis in NB, our results show that CLU does not affect Ku70 acetylation in NB cells. These results suggest that the anti-apoptotic effects of CLU are independent of modulation of Ku70 acetylation.
Materials and methods
Cell culture and cell transfection
Human neuroblastoma cell lines SH-EP1, SH-SY5Y, IMR32, GOTO, SK-N-AS, and LA1-5S cultured in modified Eagle’s medium (MEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin were maintained at 37°C in a humidified 5% CO2 incubator. SH-SY5Y cells were transiently transfected for viability assay with either pIRES-hyg Vector or pIRES-sCLU or pIRES-nCLU using nucleofector kit V (Amexa) as per the manufacturer’s protocol. SH-SY5Y cells expressing transient or stable full-length CLU protein (f-CLU) or the empty vector were generated by transfecting the cells with vector or full-length CLU (f-CLU). Stable CLU expressing clone was selected using 200 µg/ml of hygromycin.
Cell viability assay
N-type SH-SY5Y and S-type SH-EP1 human NB cell lines as well as the SH-SY5Y cell lines expressing either stable full-length CLU or empty vector were treated with varying concentrations of Trichostatin A (TSA), and viability was determined after 24 and 48 h by MTT assay as previously described [20]. The viabilities of CLU-knocked down SH-SY5Y and SH-EP1 cells were similarly measured. All experiments were carried out in triplicate and the average values and the standard deviations were calculated.
siRNA mediated silencing and quantitative real-time PCR
SH-SY5Y cells and SH-EP1 cells were plated at a density of ∼2 × 106 cells per 10-cm plate 24 h before transfection. The following day, the cells were transfected either with smart pool CLU siRNA or the scrambled non-targeting siRNA (Dharmacon Inc.) using nucleofector kit V (Amexa) as per the manufacturer’s instruction. Mock transfection as well as the non-targeting siRNA transfection served as controls. The knockdown of CLU was confirmed 72 h after transfection by immunoblot analysis using anti-CLU antibodies.
For quantitative real-time PCR analysis, total RNA was prepared from SH-SY5Y and SH-EP1 cells. Samples were then reverse transcribed and the levels of expression of CLU gene before and after TSA treatment were quantified using ABI prism 7700 sequence detector (Applied Biosystems). CLU and the internal control 18S RNAs were amplified using Taqman probes labeled with FAM from Applied Biosystems. The standard curve for CLU and 18S was prepared using serial dilution of known concentrations of CLU cDNA (0.5, 2.5, 12.5, 62.5, 312.5, and 1562.5 pg). From the Ct values, the quantity of CLU was calculated after normalizing for the internal control 18S. For treatment with TSA, fold activation was calculated using comparative Ct method and untreated cells were kept as one and the fold activation of the TSA treatment was then calculated at 8 and 16 h after treatment.
Immunoblot analysis and immunoprecipitation
For the immunoblot analysis, whole cell extracts were prepared from SH-SY5Y, SH-EP1, IMR32, GOTO, SK-N-AS, and LA1-5S human NB cell lines. In the case of NB tumor, the samples were first homogenized in RIPA buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 1% NP-40; 1 mM EGTA; 0.05% SDS) and then the extract was prepared. The proteins were separated by SDS–PAGE, transferred to PVDF membrane, and then immunoblotted for CLU. The following antibodies were used for western blot analyses: CLU (C-18) and Ku70 (M-19) from Santa Cruz, and α-tubulin (T-4026) from Sigma, and Bax NT (06-499) from Millipore. The presence of protein was visualized by using Enhanced Chemiluminescence Plus. Co-immunoprecipitation of endogenous Bax before and after TSA treatment was performed in CHAPS buffer according to the protocol described by Sawada et al. [21].
Oligonucleotide microarray hybridization and analysis
Preparation of cDNA from total RNA, hybridization, scanning, and image analysis were performed according to the manufacturer’s protocol and as previously described [22]. We used commercially available oligonucleotide arrays (HG_U133A; Affymetrix, Santa Clara, CA, USA) containing 22,283 probe sets. Each probe set typically contains 11 perfectly matched 25-base long probes (PMs) as well as 11 mismatched probes (MMs) that differ from the PM probes at the central base. Using publicly available software [23], we computed the average of the PM-MM differences for each probe set on each array after discarding the largest and smallest 20% of the PM-MM differences, and scaled the data for each array to give a mean of 1,500 units. The data was then quantile normalized to make the values of 99 evenly spaced quantities agree across all of the arrays. We log-transformed values using log (max(x + 50;0) + 50). Conservative average fold differences were computed based on the differences in log-transformed data.
Tissue microarray construction
Tissue specimens were obtained from the University of Michigan Department of Pathology archives under the approval of the Institutional Review Board. Tissue arrays were constructed using three 1.0-mm cores taken from 48 paraffin-embedded, formalin-fixed neuroblastic tumors (32 NB, 10 intermixed and nodular ganglioneuroblastomas, and six ganglioneuromas), five normal adrenal glands, and 20 other neural crest-derived (melanoma, pheochromocytoma) and unrelated neoplasms (breast and colon adenocarcinoma, Ewing sarcoma). The histologic classification of archived tumor specimens was available, but all specimens were blindly reviewed by a single pathologist (JAJ) and classified according to current standards using the International Neuroblastoma Pathology Classification [24]. Core sampling was performed to ensure optimal representation of both neuroblastic and stromal components were present. Completed array blocks were cured at 37°C for at least 24 h, then heated at 52°C for 15 min and rapidly cooled in an ice water bath. Multiple 5-μm tissue sections were cut from each block with every tenth section stained with hematoxylin/eosin for morphologic assessment and comparison to adjacent immunostained levels.
Protein expression analysis
Immunohistochemistry was performed using commercially available monoclonal antibodies and standard manufacturers’ protocols on a Ventana automated stainer. Briefly, slides were incubated with anti-CLU (C-18, Santa Cruz) at a 1:1,000 dilution following a 20-min pretreatment in high pH buffer at 95°C. Signals were evaluated for tumoral distribution (neuroblastic cells versus Schwannian stromal cells), cellular distribution (nuclear versus cytoplasmic), and intensity (semiquantitatively graded as 0, 1+, 2+, or 3+). The neuroblastic component was defined as any or all of the following: neuroblasts, immature gangliocytoid cells, and mature ganglion cells; the stromal component was defined as spindled cells with scant clear to eosinophilic cytoplasm embedded in a loose connective tissue matrix resembling Schwann cells or perineurium. Specimens excluded from analysis included those tumor array cores with insufficient tissue or missing sections, those that were necrotic, and those with insufficient neuroblastic or stromal components for evaluation. For each evaluated specimen, the mean neuroblastic component staining and the mean stromal component staining were calculated for all three cores.
Results
CLU mRNA expression in a panel of NB cell lines identifies differential expression between N- and S-type cell lines
NB tumors are composed of a mixed population of cells, including neuroblastic (N-type) and stromal (S-type) cells [24, 25]. Results in Fig. 1a show expression data on a subset of genes that identify N-type versus S-type NB cells. Vimentin, collagen type IV, and fibronectin, known stromal markers [26], were expressed at significantly higher levels in S-type cell lines, whereas neurofilament and chromogranin A, markers of neuroblastic cells [27], were expressed at higher levels in N-type cells. Analysis revealed marked differences between N-type and S-type cells in their levels of CLU messenger RNA; S-type cells expressed higher levels of CLU RNA than N-type cell lines. These results suggest that differences in CLU expression between N- and S-type cells in culture may be responsible for biologic differences, such as their differential sensitivity to treatment with HDACIs [28].
Fig. 1.
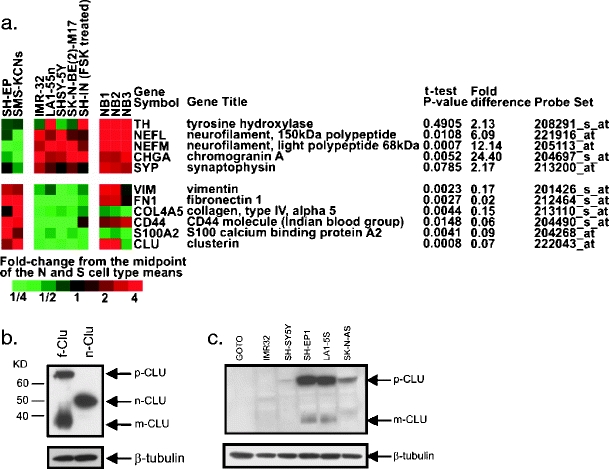
CLU is differentially expressed in S-type NB cells. a Expression of CLU and selected RNAs previously shown to be specific for neuroblastic (top panel) or Schwannian stromal cells (bottom panel) in a panel of N-type [IMR-32, LA1-55n, SH-SY5Y, SK-N-BE(2)-M17, and SH-IN (FSK-treated)] and S-type (SH-EP and SMS-KCNs) NB cell lines as well as in three human NB tumor specimens (NB1, NB2, and NB3). Color indicates fold difference of each sample from the midpoint of the two means for S- and N-type cells, with red indicating higher and green indicating lower expression. P values are from two-tailed t tests, and the fold difference indicates the ratio of average N-type to S-type expression values. b SH-SY5Y cells are transfected with a full-length CLU (f-Clu) construct expressing precursor form (p-CLU) and mature form (m-CLU) of CLU. Both forms are localized in the cytoplasm. Nuclear form of CLU (n-CLU) is expressed from a CLU expression vector starting at the second methionine of the CLU sequence. Note the differences of the positions of each CLU on a SDS–polyacrylamide gel. c Immunoblot analyses of three N-type (IMR32, SH-SY5Y, and GOTO) and three S-type (SH-EP1, SK-N-AS, and LA1-5S) cell lines show endogenous expression of secreted form of CLU and mature form in S-type and not in N-type NB cell lines
Cytoplasmic forms of CLU protein are expressed in NB tumor tissue and NB cell lines
Basal CLU protein expression was evaluated in S-type and N-type cell lines to determine if differential CLU protein expression correlates with differential CLU mRNA expression and to define the isoforms expressed in NB cells. N-type (GOTO, IMR32, and SH-SY5Y) and S-type (SH-EP1, LA1-5S, and SK-N-AS) cells were analyzed by immunoblotting. To distinguish between different CLU forms, we transfected SH-SY5Y cells with an expression vector encoding Flag-tagged full-length CLU (f-Clu) that generates both p-CLU (precursor form) and m-CLU (mature form) and a vector that encodes CLU starting at the second ATG codon that results in n-CLU (n-CLU, Fig. 1b). With these FLAG-epitope detected forms as standards, results shown in Fig. 1c indicate that S-type NB cell lines express much more of the p-CLU and m-CLU isoforms compared to N-type cell lines where CLU protein expression is barely detectable. Within each group of cells, p-CLU and m-CLU are present in similar levels. n-CLU is not detected in any of these cells. Thus, basal CLU protein expression differs between culture phenotypes and correlates with differences in RNA expression.
To determine CLU protein expression in NB tumors, tissue homogenates were prepared from tumor tissue and immunoblotted to detect CLU. While the p-CLU is clearly identified in five of the nine NB tumor samples (Fig. 2a), it is not clear whether these tumors contain the nuclear form of CLU or the mature form of CLU (m-CLU) or both. NB tumors are composed of histologically distinct areas described as either neuroblastic rich regions or Schwannian/stromal area. Given the in vitro differences in expression between S-type and N-type cells, we quantified CLU protein expression in NB tumors according to neuroblastic or Schwannian/stromal region by probing a NB tumor tissue microarray with anti-CLU antibodies. This array contains tumor tissue representing 48 unique NB tumors (Fig. 2b). In apparent contrast to results in NB cell lines, CLU protein expression is greater in neuroblastic than in stromal tumor regions. Indeed, all tumors had high (2+ or 3+) CLU expression in neuroblastic regions. CLU expression in Schwannian/stromal regions was more variable; however, in the majority (29 out of 38), CLU stromal expression was low (0 or 1+ staining). Given this result, the same microarray was probed for vimentin and S100, two other proteins whose genes are differentially expressed in vitro with S-type expression greater than N-type (see Fig. 1). For each, expression in NB tumor tissue was nearly exclusive to stromal regions (data not shown). Based on these results, we conclude that cells comprising both neuroblastic and Schwannian/stromal regions in NB tumors can express CLU. Since CLU is expressed in cells from both tumor regions, mechanistic experiments were designed to evaluate the function of CLU in both S- and N-type cells in vitro.
Fig. 2.
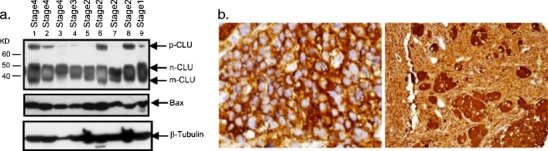
Clusterin is highly expressed in the neuroblastic, but not stromal, components of neuroblastic tumors. a The NB tissue is obtained from our tissue core at the University of Michigan, with one stage I, four stage II, one stage III, and three stage IV tumors. b Immunohistochemistry was performed with antibodies specific for CLU as described in the text. Strong cytoplasmic staining is seen in the small neuroblasts of a poorly differentiated NB (left) and the larger, more mature-appearing ganglion cells of an intermixed ganglioneuroblastoma (right). Staining prevalence and intensity were scored semiquantitatively from 0 to 3+, with separate scores assigned to the histologically distinct neuroblastic and stromal cells. Categorization of expression revealed that whereas the N-type components of all but one tumor had at least 2+ expression (mean 2.95), most S-type components had CLU present at less than 1+ levels (mean 0.95)
HDACI treatment induces cytosolic CLU protein expression
HDACIs increase acetylation of Ku70 protein. In NB cells, this disrupts Ku70:Bax binding and releases activated Bax to kill cells. Since CLU sequesters activated Bax, and binds Ku70 and Bax:Ku70 protein complexes with unknown effects on Ku70 acetylation, CLU expression may be a factor limiting sensitivity of NB cells to HDACI therapy. Since S-type cells in vitro are resistant to HDACI-induced Ku70 acetylation, Bax activation, and cell death (whereas N-type cells are responsive to this mechanism), finding high levels of CLU protein in S-type cells provides support for this hypothesis.
To test this, we first determined if HDACI treatment affects CLU expression in three N-type NB cell lines (GOTO, IMR32, and SH-SY5Y) and three S-type NB cell lines (SH-EP1, LA1-5S, and SK-N-AS). In all N-type cells, basal levels of CLU are low, but both the m and p forms are clearly increased by TSA (1 μM, 24 h) treatment (Fig. 3a). S-type cells have high basal CLU, and after TSA treatment levels of the m and p forms are modestly increased (1.3 times basal level). Even after maximal effects of TSA treatment are accounted for (Fig. 3b), the overall protein level achieved in GOTO and IMR32 cells in culture remains significantly lower than the basal levels in all S-type cells. However, the CLU expression in SH-SY5Y cells is high after TSA treatment compared to that of the S-type cells. In parallel with the increase in CLU protein, TSA treatment induces a corresponding increase in CLU mRNA levels in N-type cells. RT–PCR-quantified CLU mRNA after TSA treatment showed increased mRNA levels in SH-SY5Y cells 8 and 16 h after TSA treatment (Fig. 3c). CLU message level in SH-EP1 cells was not significantly increased in response to TSA treatment. We also tested two other HDAC inhibitors, SAHA and MS-275, which also indicated increased CLU level in SH-SY5Y cells, but to a lesser extent in SH-EP1 cells (Fig. 3d). Taken together, these results mean that in addition to basal CLU expression, HDACI-induced CLU expression may be a factor modulating the effectiveness of this class of drugs against NB.
Fig. 3.
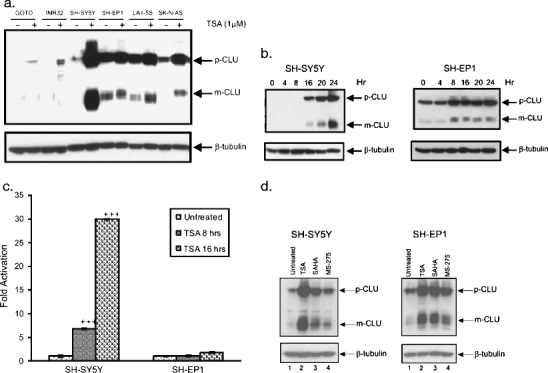
Clusterin expression is increased with HDACI treatment. a NB N-type (IMR32, SH-SY5Y, and GOTO) and S-type (SH-EP1, SK-N-AS, and LA1-5S) cell lines were treated with 1 μM TSA for 24 h before immunoblotting with anti-CLU antibody. β-Tubulin was used as a loading control. b SH-SY5Y and SH-EP1 cell lines were treated with 1 μM TSA and cell lysates were collected at different time points (4, 8,16, 20, and 24 h), and the expression of CLU was analyzed by immunoblotting. c mRNA level CLU is present in S-type SHEP1 cells whereas it is induced after 1 μM TSA treatment in N-type SH-SY5Y cells both after 16 and 24 h. d SH-SY5Y cells and SH-EP1 cell lines were treated with 1 μM of TSA, SAHA, or MS-275 for 24 h, and the expression of CLU was analyzed by immunoblotting
We tested whether increased CLU expression occurs when NB cells are exposed to other cytotoxic treatments. SH-SY5Y and SH-EP1 were treated with doxorubicin, VP-16, cisplatin, or irradiation (15 Gy). CLU expression was not increased with any of the other treatments (Fig. 4), suggesting that in NB cells, CLU expression is selectively increased by HDACIs.
Fig. 4.
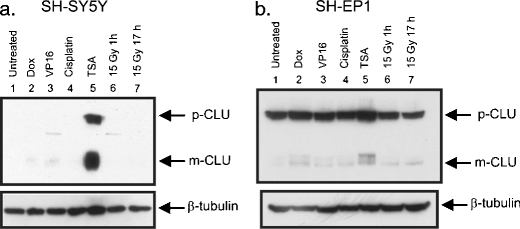
CLU is induced by HDACI but not by other stressors in NB cells. Both N-type SH-SY5Y (a) and S-type SH-EP1 (b) cell lines were treated for 24 h with TSA (1 μM), cisplatin (10 μg/ml), doxorubicin (Dox) (0.5 μg/ml), etopicide (VP16) (10 μg/ml), or radiation (15 Gy for 1 or 17 h as indicated). The presence of CLU was analyzed by immunoblot using CLU-specific antibodies
CLU limits HDACI-induced cell death without inhibiting HDACI-induced Ku70 acetylation
To determine whether basal CLU expression affects sensitivity of HDACIs, various amounts of a vector expressing full-length CLU cDNA (f-CLU) expression vector were transfected into SH-SY5Y cells. In the transfected cells, basal levels of p-CLU and m-CLU are increased (data not shown). While TSA reduces the viability of the NB cells in a dose-dependent manner, increasing amounts of transfected CLU reduces the sensitivity to TSA (Fig. 5a). Since CLU is known to interact with activated Bax, we tested whether expression of CLU will block Bax entering into the mitochondria. Results shown in Fig. 5b indeed demonstrate that stable expression of CLU in SH-SY5Y reduces the appearance of Bax in mitochondria after TSA treatment.
Fig. 5.
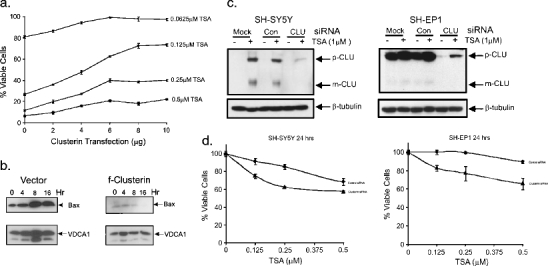
Knocking down CLU sensitizes NB cells to TSA-induced cell death. a Various amounts of CLU expression vectors (as indicated) were transfected into SH-SY5Y cells. The cells were treated with different concentration of TSA for 24 h as shown. Cell viability is determined by MTT assay. Results are expressed as the percentage of viable cells compared with vehicle-treated controls (mean ± SD; n = 3). b Stable SH-SY5Y cell line expressing full-length CLU or vector were treated with TSA (1 μM) for 0, 4, 8, and 16 h as indicated. Mitochondria were isolated; Bax level was determined by immunoblotting. VDCA1 is used as the loading control for mitochondria fraction. c SH-SY5Y cells or SH-EP1 cells were transfected with scrambled siRNA or CLU-specific siRNA. Forty-eight hours after transfection, cells were treated with TSA (1 μM, 24 h). Cells in mock sample received no transfection. Immunoblot shows effective knockdown of CLU. d SH-SY5Y cells or SH-EP1 cells were transfected with CLU-specific siRNA as shown in (c). The cells were treated with varying concentrations of TSA for 24 h as shown. Cell viability was determined by MTT assay. Results are expressed as the percentage of viable cells compared with vehicle-treated controls (mean ± SD; n = 3)
We have further tested the effects that CLU knockdown has on sensitivity to TSA-induced cell death. We knocked down SH-SY5Y cells and SH-EP1 cells with CLU-specific siRNA. As shown in Fig. 5c, CLU siRNA successfully prevented the TSA-induced increased CLU level in SH-SY5Y cells and reduced basal levels of CLU in SH-EP1 cells. Indeed, knocking down CLU in both SH-SY5Y and SH-EP1 cells sensitized TSA-induced killing (Fig. 5d).
Since CLU binds to both activated Bax [19] and also to Ku70:Bax complexes [18], we envisioned two alternative mechanisms by which CLU can modulate the therapeutic response to HDACIs, either by maintaining Ku70:Bax binding or sequestering activated Bax. To distinguish between these models, we evaluated binding between Ku70 and Bax before and after HDACI treatment. Figure 6a shows that in SH-SY5Y cells, with low levels of CLU prior to HDACI treatment, Bax binds to Ku70. However, after HDACI treatment, the level of Ku70 binding to Bax decreases, as shown previously [20, 28]. Interestingly, CLU overexpression does not alter binding between Ku70 and Bax before or after TSA treatment (Fig. 6a). Furthermore, TSA treatment only slightly reduces the binding between CLU and Ku70 (Fig. 6b).
Fig. 6.
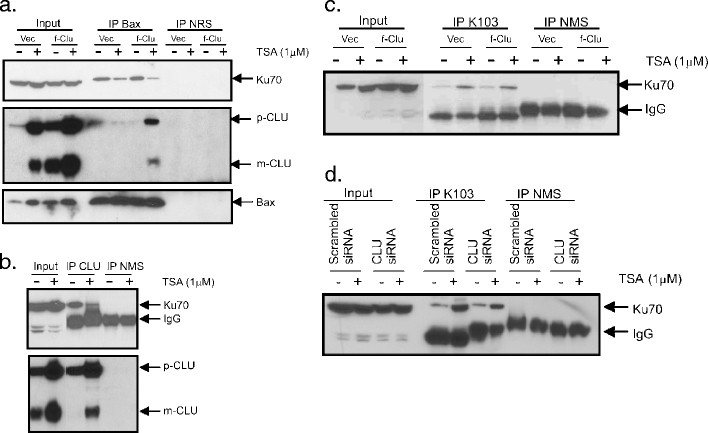
CLU does not affect Ku70 acetylation or Ku70–Bax binding in NB cells. a Stable SH-SY5Y cell line expressing full-length CLU or vector were treated with TSA (1 μM, 24 h) and cytoplasmic extracts were immunoprecipitated using Bax-specific antibodies or normal rabbit serum (NRS) as control. The immunoprecipitates were separated by SDS–PAGE and probed with Ku70, CLU, or Bax antibodies. b SH-SY5Y cells were treated with vehicle or TSA (1 μM, 24 h). CLU was immunoprecipitated using CLU-specific antibodies or normal mouse serum (NMS). The immunoprecipitates were separated by SDS–PAGE and probed with Ku70 or CLU antibodies. c Stable SH-SY5Y cells expressing full-length CLU or vector were treated with TSA (1 μM, 24 h). Cytoplasmic extracts were immunoprecipitated using an anti-acetyl-lysine antibody (K103). The immunoblot was probed with Ku70 as shown. d CLU siRNA or scramble siRNA-treated SH-SY5Y cells were treated with TSA (1 μM, 24 h). Cytoplasmic extracts were immunoprecipitated using anti-acetyl-lysine antibody (K103). The immunoblot was probed with Ku70 antibody
Since CLU does not prevent HDACI-induced disruption of Ku70:Bax complexes, we predicted that it does not affect Ku70 acetylation in response to TSA. To test this, acetylated proteins were immunoprecipitated from cytoplasmic extracts prepared from SH-SY5Y cells transfected with f-CLU that were either HDACI treated or untreated. Acetylated proteins were resolved on SDS–PAGE and immunoblotted with Ku70-specific antibody. As shown in Fig. 6c, CLU expression did not reduce levels of acetylated Ku70. In an orthogonal approach, we used CLU-specific siRNA transfection to knock down CLU in SH-SY5Y cells (Fig. 6d). When Ku70 acetylation was determined in cells treated with TSA, no differences were detected between CLU knockdown and control cells.
Discussion
HDACIs have the potential to be an effective therapy for NB. Since these agents are not broadly cytotoxic but instead act selectively against particular cell types, understanding key elements of the mechanism(s) by which they kill tumor cells is critical to apply these drugs against tumors that are most likely to respond. At present, it is not known whether all NB tumors or which particular subset of NB tumors are likely to be responsive to HDACI therapy. The results from this study showing that CLU protein expression limits HDACI-induced NB tumor cell apoptosis identify CLU as a molecular marker associated with resistance to HDACIs and present an opportunity to combine therapeutic approaches designed to decrease CLU expression together with HDACI-based treatment to more effectively treat NB tumors. Our results support a model in which CLU functions as an anti-apoptotic factor in NB cells. We have shown that the anti-apoptotic forms of CLU (p-CLU and m-CLU) are exclusively expressed in NB cell lines, but in NB tumors, it is not clear whether m-CLU or n-CLU (the apoptotic form) is expressed. While there is a differential expression of basal CLU in NB cells (high in S-type but low in N-type NB cells), inhibiting HDACs induces expression of CLU in N-type, but only a modest increase is observed in S-type NB cells. This increase of CLU expression reduces Bax-mediated cell death as overexpression of CLU reduces HDACI-induced cell death activity, whereas knocking down CLU in NB cells facilitates HDAC-induced cell death.
We have previously shown that NB tumor cells are variably sensitive to HDACIs. Specifically, S-type cells are resistant whereas N-type cells are sensitive [20, 28]. In sensitive N-type cells, HDACI treatment increases Ku70 protein acetylation at two lysine residues, 539 and 542. These residues regulate Ku70 binding to Bax, a potent inducer of apoptosis. Acetylation at these sites is necessary for HDACI-induced NB cell death. Mechanistically, HDAC inhibition increases Ku70 acetylation disrupting Ku70:Bax binding which in turn leads to Bax activation and Bax-dependent cell death.
Since CLU expression is significantly higher in HDACI-resistant S-type NB cell lines compared to sensitive N-type lines, and that CLU protein binds Ku70, Bax, and Ku70:Bax complexes, the significance of CLU expression in terms of regulating Ku70 acetylation was investigated. We initially reasoned that since CLU is proposed to be anti-apoptotic in NB cells, and that CLU is shown to interact with Ku70 and Bax in cells, one potential model would be that CLU suppressed Bax-dependent cell death by binding to Ku70 and inhibiting HDACI-induced Ku70 acetylation. However, our results do not support this model. Our results showed that CLU interacts with Ku70 at basal level in N-type NB cells. TSA treatment increases the binding between CLU and Bax but reduces the binding between Ku70 and CLU. Overexpression of CLU does not alter this pattern, suggesting that CLU level increase, either by TSA or by overexpression, does not affect CLU and Ku70 binding. Furthermore, we have shown that while TSA treatment induces Ku70 acetylation in N-type NB cells, CLU overexpression or CLU depletion by siRNA did not affect the TSA-induced increase of Ku70 acetylation. These results suggest that the anti-apoptotic effect of CLU in TSA-induced NB cell death occurs downstream of Ku70 acetylation, perhaps, as described previously, by CLU binding to activated Bax, reducing Bax-dependent activation.
Collectively, our results indicate that high level of CLU in NB cells will serve as a biomarker to predict resistance of drugs that induce Bax activation. Because HDACIs are generally non-toxic to other cells, targeting CLU in NB cells can be used as a strategy to increase NB sensitivity to HDACIs.
Acknowledgments
This work was supported in part by the National Institutes of Health grants DK067102 (R.P.S.K.), the Janette Ferrantino Hematology Research Fund (V.P.C.), the Ravitz Foundation (V.P.C.), Hope Street Kids Foundation (C.S.), and the A. Alfred Taubman Medical Research Institute (V.P.C., R.P.S.K., A.W.O.). This work used the Sequencing Core of the Michigan Diabetes Research and Training Center, which was funded by National Institute of Diabetes and Digestive and Kidney Diseases Grant NIH5P60 DK20572.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Blaschuk O, Burdzy K, Fritz IB. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J Biol Chem. 1983;258(12):7714–7720. [PubMed] [Google Scholar]
- 2.Jenne DE, Tschopp J. Clusterin: the intriguing guises of a widely expressed glycoprotein. Trends Biochem Sci. 1992;17(4):154–159. doi: 10.1016/0968-0004(92)90325-4. [DOI] [PubMed] [Google Scholar]
- 3.de Silva HV, et al. Apolipoprotein J: structure and tissue distribution. Biochemistry. 1990;29(22):5380–5389. doi: 10.1021/bi00474a025. [DOI] [PubMed] [Google Scholar]
- 4.Miyake H, et al. Testosterone-repressed prostate message-2 is an antiapoptotic gene involved in progression to androgen independence in prostate cancer. Cancer Res. 2000;60(1):170–176. [PubMed] [Google Scholar]
- 5.Sensibar JA, et al. Prevention of cell death induced by tumor necrosis factor alpha in LNCaP cells by overexpression of sulfated glycoprotein-2 (clusterin) Cancer Res. 1995;55(11):2431–2437. [PubMed] [Google Scholar]
- 6.Yang CR, et al. Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc Natl Acad Sci USA. 2000;97(11):5907–5912. doi: 10.1073/pnas.97.11.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirszbaum L, et al. Molecular cloning and characterization of the novel, human complement-associated protein, SP-40, 40: a link between the complement and reproductive systems. EMBO J. 1989;8(3):711–718. doi: 10.1002/j.1460-2075.1989.tb03430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenne DE, Tschopp J. Molecular structure and functional characterization of a human complement cytolysis inhibitor found in blood and seminal plasma: identity to sulfated glycoprotein 2, a constituent of rat testis fluid. Proc Natl Acad Sci USA. 1989;86(18):7123–7127. doi: 10.1073/pnas.86.18.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sylvester SR, et al. Localization of sulfated glycoprotein-2 (clusterin) on spermatozoa and in the reproductive tract of the male rat. Biol Reprod. 1991;45(1):195–207. doi: 10.1095/biolreprod45.1.195. [DOI] [PubMed] [Google Scholar]
- 10.Rizzi F, Bettuzzi S. The clusterin paradigm in prostate and breast carcinogenesis. Endocr Relat Cancer. 2010;17(1):R1–R17. doi: 10.1677/ERC-09-0140. [DOI] [PubMed] [Google Scholar]
- 11.Pucci S, et al. Modulation of different clusterin isoforms in human colon tumorigenesis. Oncogene. 2004;23(13):2298–2304. doi: 10.1038/sj.onc.1207404. [DOI] [PubMed] [Google Scholar]
- 12.Leskov KS, et al. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J Biol Chem. 2003;278(13):11590–11600. doi: 10.1074/jbc.M209233200. [DOI] [PubMed] [Google Scholar]
- 13.Cappelletti V, et al. Patterns and changes in gene expression following neo-adjuvant anti-estrogen treatment in estrogen receptor-positive breast cancer. Endocr Relat Cancer. 2008;15(2):439–449. doi: 10.1677/ERC-07-0274. [DOI] [PubMed] [Google Scholar]
- 14.Miyake H, et al. Acquisition of chemoresistant phenotype by overexpression of the antiapoptotic gene testosterone-repressed prostate message-2 in prostate cancer xenograft models. Cancer Res. 2000;60(9):2547–2554. [PubMed] [Google Scholar]
- 15.Chi KN, et al. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J Natl Cancer Inst. 2005;97(17):1287–1296. doi: 10.1093/jnci/dji252. [DOI] [PubMed] [Google Scholar]
- 16.Zellweger T, et al. Overexpression of the cytoprotective protein clusterin decreases radiosensitivity in the human LNCaP prostate tumour model. BJU Int. 2003;92(4):463–469. doi: 10.1046/j.1464-410X.2003.04349.x. [DOI] [PubMed] [Google Scholar]
- 17.Chayka O, et al. Clusterin, a haploinsufficient tumor suppressor gene in neuroblastomas. J Natl Cancer Inst. 2009;101(9):663–677. doi: 10.1093/jnci/djp063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trougakos IP, et al. Intracellular clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70–Bax protein complex. Clin Cancer Res. 2009;15(1):48–59. doi: 10.1158/1078-0432.CCR-08-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, et al. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol. 2005;7(9):909–915. doi: 10.1038/ncb1291. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian C, et al. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102(13):4842–4847. doi: 10.1073/pnas.0408351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawada M, et al. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003;5(4):320–329. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
- 22.Giordano TJ, et al. Organ-specific molecular classification of primary lung, colon, and ovarian adenocarcinomas using gene expression profiles. Am J Pathol. 2001;159(4):1231–1238. doi: 10.1016/S0002-9440(10)62509-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shedden K, et al. Comparison of seven methods for producing Affymetrix expression scores based on False Discovery Rates in disease profiling data. BMC Bioinform. 2005;6:26. doi: 10.1186/1471-2105-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada H, et al. The International Neuroblastoma Pathology Classification (the Shimada system) Cancer. 1999;86(2):364–372. doi: 10.1002/(SICI)1097-0142(19990715)86:2<364::AID-CNCR21>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Shimada H, et al. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer. 1999;86(2):349–363. doi: 10.1002/(SICI)1097-0142(19990715)86:2<349::AID-CNCR20>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 26.Ross RA, et al. Human neuroblastoma I-type cells are malignant neural crest stem cells. Cell Growth Differ. 1995;6(4):449–456. [PubMed] [Google Scholar]
- 27.Ross RA, Biedler JL, Spengler BA. A role for distinct cell types in determining malignancy in human neuroblastoma cell lines and tumors. Cancer Lett. 2003;197(1–2):35–39. doi: 10.1016/S0304-3835(03)00079-X. [DOI] [PubMed] [Google Scholar]
- 28.Subramanian C, et al. CREB-binding protein is a mediator of neuroblastoma cell death induced by the histone deacetylase inhibitor trichostatin A. Neoplasia. 2007;9(6):495–503. doi: 10.1593/neo.07262. [DOI] [PMC free article] [PubMed] [Google Scholar]