

Abstract
The neural cell adhesion molecule L1 has recently been shown to be expressed in pancreatic adenocarcinoma (PDAC) cells. In this report, we demonstrate that L1 is expressed by moderately- to poorly-differentiated PDAC cells in situ, and that L1 expression is a predictor of poor patient survival. In vitro, reduced reactivity of an anti-L1 carboxy-terminus-specific antibody was observed in the more poorly differentiated fast-growing (FG) variant of the COLO357 population, versus its well-differentiated slow-growing (SG) counterpart, even though they express equivalent total L1. The carboxy-terminus of L1 mediates binding to the MAP kinase-regulating protein RanBPM and mutation of T1247/S1248 within this region attenuates the expression of malignancy associated proteins and L1-induced tumorigenicity in mice. Therefore, we reasoned that the differential epitope exposure observed might be indicative of modifications responsible for regulating these events. However, epitope mapping demonstrated that the major determinant of binding was actually N1251; mutation of T1247 and S1248, alone or together, had little effect on C20 binding. Moreover, cluster assays using CD25 ectodomain/L1 cytoplasmic domain chimeras demonstrated the N1251-dependent, RanBPM-independent stimulation of erk phosphorylation in these cells. Reactivity of this antibody also reflects the differential exposure of extracellular epitopes in these COLO357 sublines, consistent with the previous demonstration of L1 ectodomain conformation modulation by intracellular modifications. These data further support a central role for L1 in PDAC, and define a specific role for carboxy-terminal residues including N1251 in the regulation of L1 activity in PDAC cells.
Keywords: L1-CAM, Deamidation, Conformation, Differentiation
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has one of the highest mortality rates of all cancers [1, 2]. Despite this, its biology remains somewhat poorly understood. Studies have identified key factors in PDAC etiology [3], which have been incorporated into a timeline that has been in part validated using transgenic mice [4, 5]. Although such a timeline is significant in defining the onset of the disease, a similar timeline has been difficult to establish for the progression of the disease. Additionally, although almost all patients present with late-stage disease due to a lack of symptoms, several distinct presentations are observed with regard to tumor growth and dissemination [1–3], suggesting alternative mechanisms of progression and/or the presence of tumor subtypes.
The neural cell adhesion molecule L1 (L1-CAM, CD171) is a transmembrane protein of the immunoglobulin superfamily (Fig. 3a) that regulates active neural processes [6]. L1 expression has also been described in non-neural tissues and human tumor cells [7], where L1 correlates with poor prognosis and advanced disease state in several tumor types [8–10], and in serous neoplasms of the ovary L1 expression increased with tumor grade and predicted poor response to chemotherapy and shorter progression-free survival [10]. These data suggest a potential role for L1 in cancer progression. Although a prior publication failed to detect L1 in human PDAC samples [11], a subsequent report found L1 expression in moderately- and poorly-differentiated PDAC tumors, with the highest expression noted in the most poorly differentiated cells [12], similar to our findings with a larger dataset, which are reported herein.
Fig. 3.
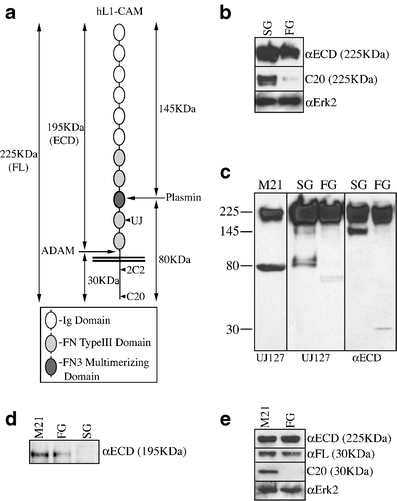
C20 reactivity correlates with differentiation/malignancy in the COLO357 system. a Schematic of the L1 domain structure and processing by proteases. Ig immunoglobulin, FN fibronectin. b Immunoblotting demonstrates that the C20 epitope is masked in the less-differentiated FG variant of COLO357, in contrast to the more-differentiated SG variant. Erk2, loading control. c Immunoblotting of whole cell lysates demonstrates that FG cells predominantly utilize ADAMs proteases to cleave L1, while SG cells predominently utilize serine proteases to cleave L1. M21, melanoma control. Relative migration shown on left in KDa. d Immunoblotting of conditioned media corroborates the data in c. Erk2, loading control. e Immunoblotting demonstrates that proteolysis does not account for loss of C20 reactivity, since the 30-KDa ADAMs fragment resolves as the same molecular weight in FG and M21 lysates, but is undetectable by C20 in the FG lysate specifically
L1’s role in regulating processes associated with invasion make it well suited for use by an aggressive tumor. Stable ectopic expression of L1 in fibroblastic and low-metastatic melanoma cells drives constitutive MAP kinase activation and induces the expression of invasion and metastasis-associated genes, thereby promoting de novo integrin utilization and concomitant enhanced migration and invasion in vitro [13]. Moreover, L1 is fully transforming and expressed at the invasive front of colon cancers in situ [14], and ectopic expression of L1 in endogenously L1-negative colon cancer cells bestows a metastatic phenotype [15]. Importantly, the cytoplasmic domain of L1 was required for this effect, although the cytoplasmic domain alone was not sufficient to drive this phenotype. A binding site for the MAP kinase activator RanBPM was recently mapped to the carboxy-terminal 28 amino acids of L1 [16]. Mutation of both T1247 and S1248 within this region abrogated L1-induced erk-dependent gene expression, cell migration, and tumor growth in HEK293 cells and SKOV3ip ovarian carcinoma cells [17]. However, this dual mutation had no effect on RanBPM binding to L1. It is not known if mutation of T1247 alone would be sufficient, since S1248A alone had no effect in this system, and the effect of T1247A alone was not tested. Moreover, no evidence was provided for regulation of this activity through post-translational modification (i.e., phosphorylation). This is important because erk2 can phosphorylate S1248 in vitro [18]. Therefore, the relevance of this double mutation to the regulation of L1 activity in cells or tissues is not clear. We found that an antibody specific to the L1 carboxy-terminus demonstrates differentiation-dependent reactivity in the COLO357 cell system. To investigate the mechanism responsible for this pattern, we utilized recombinant proteins to define the epitope of this antibody, and the corresponding regulation of erk activation and L1 tail conformation by the identified residues.
Materials and methods
Cells
Panc1 cells were originally from ATCC. COLO357 cells were from M. Korc (UCI, Irvine, CA, USA). The fast-growing COLO357 subline, fast growing (FG), was from R. Klemke (UCSD, San Diego, CA, USA). The slow-growing COLO357 subline, slow growing (SG), was from M. Vezeridis (Brown University, Providence, RI, USA). M21 human melanoma cells were derived from the UCLA-SO-M21 cell line, which was provided by Dr. DL Morton (UCLA, Los Angeles, CA, USA). Panc1, COLO357, SG, and FG cells were cultured in DMEM/10% fetal bovine serum (FBS). M21 cells were grown in RPMI/10% FBS. Panc1 cells were transfected with pCDNA3.1/Tac/L1 chimera constructs using Lipofectamine2000 (Invitrogen, San Diego, CA, USA) and stable clones were derived by zeocin resistance. Clones were assessed for Tac/L1 expression and positive clones pooled and maintained under selection.
Antibodies
αL1 Carboxy-terminus (C20), αGST (110–218) and the immunoblotting αCD25 (N19) polyclonal antibodies (pAbs) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). αCD25 monoclonal antibody (mAb) (cl.22722) used for Flow cytometry (FACS) and cluster assays was from R&D Systems (Minneapolis, MN, USA). αRanBPM pAb was from Millipore (Bedford, MA, USA). αL1 mAb 2C2 was from AbCam (Cambridge, MA, USA) and has been described in detail previously [19], αL1 mAb UJ127 was from Neomarkers/LabVision (Fremont, CA, USA), αL1 mAb 5G3 was generously provided by M. Just (eBioscience, San Diego, CA, USA). αECD pAb was generated against purified L1 ectodomain and provided by W Stallcup (The Burnham Institute, La Jolla, CA, USA). αFL pAb was generated against L1 purified from human neuroblastoma cells using a 5G3 immunoaffinity column. Both αECD and αFL have been described previously [19].
Cluster assay
Stable CD25/L1 chimera-expressing Panc1 cells were plated at 2.5 × 104/24-well and allowed to grow for 48 h prior to serum starvation overnight. The plate was placed on ice and the media replaced with ice-cold serum-free media containing 10 μg/ml αCD25 mAb. Cells were incubated 30 min on ice, washed, and fresh serum-free media replenished. Cells were warmed to 37°C for 45 min and lysates harvested and processed for immunoblotting.
Immunoprecipation
Cells were lysed in phosphate lysis buffer (PLB; 10 mM NaPO4, 100 mM NaCl, 1% TX-100, 0.4% deoxycholate, 0.1% SDS, pH 7.5) containing Complete™ Protease Inhibitor Cocktail (Roche Applied Science, Indianapolis, IN, USA) supplemented with 10 mM PMSF, 1 mM NaF and 10 mM Na3VO4, and incubated with the indicated antibody (2 μg) overnight at 4°C. Antibodies were precipitated with Protein-L sepharose4b beads (Pierce, Rockford, IL, USA), recovered by centrifugation, washed, and prepared for immunoblotting.
GST pull-down assay
GST-fusion proteins (1 μg) were incubated with the indicated Panc1 lysates (250 μg) overnight at 4°C. Glutathione-4b sepharose beads were added (10 μl) and the samples incubated 2 h at 4°C before recovery of bead-bound GST fusion proteins by centrifugation, washing and preparation for immunoblotting.
Immunoblotting
Samples were separated by reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), electroblotted to PVDF, sequentially incubated with primary and horseradish peroxidase (HRP)-conjugated secondary antibodies and visualized by ECL with PS-3 (Lumigen, Southfield, MI, USA). Gels were scanned on an Epson 4490Photo Flatbed Scanner and band intensities were densitometrically analyzed using NIH Image 1.61. Band intensities were normalized against the appropriate loading control (i.e., erk2 or GST) and compared to the indicated baseline comparison band, which was defined as 1.0.
Flow cytometry
FACS analysis and sorting was performed on a FACScalibur or FACSVantageSE, respectively (BD Biosciences, Bedford, MA, USA), at the Moores UCSD Cancer Center Flow Cytometry Shared Resource as described previously [13, 19]. Gates were set with secondary antibody controls, and propidium iodide was included to exclude dead and dying cells.
Enzyme-linked immunosorbent assay
GST-proteins were immobilized on 96-well plates and incubated with appropriate primary and HRP-conjugated secondary antibodies and detected with TMB as described previously [19]. Data was corrected for αGST signal as a measure of fusion protein adsorption to the plate. In some cases, data were confirmed using proteins captured on glutathione-coated plates (Pierce/ThermoScientific, Rockville, IL, USA) to prevent denaturing of the L1 fragments during adsorption to plastic substratum.
L1 fusion proteins
(Prokaryotic) Unless otherwise indicated, pGEX GST-fusion constructs were described previously [19], or generated by site-directed mutagenesis using the appropriate primers (Table 1) as described previously [19]. GST/L11238–1257 and GST/L11249–1257 were created by ligation of annealed and phosphorylated mini-exon primers into the EcoRI site of pGEX/6P-1. All constructs were confirmed by dideoxy sequencing at the Moores UCSD Cancer Center DNA Sequencing Shared Resource. (Eukaryotic) Inserts from the appropriate pGEX vectors were excised with EcoRI and ligated into the EcoRI site of a pCDNA3.1 (zeo) vector containing the ectodomain and transmembrane sequences of the high-affinity IL2 receptor (CD25, Tac) which had previously been inserted immediately upstream between the NheI and HindIII sites of the vector.
Table 1.
Primers used to construct recombinant proteins
| Primer type | Primer sequence |
|---|---|
| Mutagenesis | |
| −9 (1248 stop) | F 5′-GGGGCCACTTCCTAAATCAACCCTGCCGTGG-3′ |
| R 5′-CCACGGCAGGGTTGATTTAGGAAGTGGCCCC-3′ | |
| −10 (1247 stop) | F 5′-CAGGGGCCACTTAACCCATCAACCCTGCCG-3′ |
| R 5′-CGGCAGGGTTGATGGGTTAAGTGGCCCCTG-3′ | |
| −20 (1238 stop) | F 5′-AAGAAGGAGAAGGAGGCGTGAGGGGGCAATGA-3′ |
| R 5′-TCATTGCCCCCTCACGCCTCCTTCTCCTTCTT-3′ | |
| −28 (1229 stop) | F 5′-TTGGCCAGTACTAAGGCAAGAAGGAGAAGGA-3′ |
| R 5′-TCCTTCTCCTTCTTGCCTTAGTACTGGCCAA-3′ | |
| P1249A | F 5′-CAGGGGCCACTTCCGCCATCAACCCTGCCG-3′ |
| R 5′-CGGCAGGGTTGATGGCGGAAGTGGCCCCTG-3′ | |
| N1251A | F 5′-GCCACTTCCCCCATCGCCCCTGCCGTGGCCC-3′ |
| R 5′-GGGCCACGGCAGGGGCGATGGGGGAAGTGGC-3′ | |
| N1251D | F 5′-ACTTCCCCCATCGACCCTGCCGTGGCCC-3′ |
| R 5′-GGGCCACGGCAGGGTCGATGGGGGAAGT-3′ | |
| P1252A | F 5′-GCCACTTCCCCCATCAACGCTGCCGTGGCCCTA-3′ |
| R 5′-TAGGGCCACGGCAGCGTTGATGGGGGAAGTGGC-3′ | |
| E1257A | F 5′-GCCGTGGCCCTAGCATAGGAATTCCCG-3′ |
| R 5′-CGGGAATTCCTATGCTAGGGCCACGGC-3′ | |
| Mini-Exon | |
| 1238–1257 | F 5′-AATTCGCAGGGGGCAATGACAGCTCAGGGGCC |
| ACTTCCCCCATCAACCCTGCCGTGGCCCTAGAAG-3′ | |
| R 5′-AATTCTTCTAGGGCCACGGCAGGGTTGATGGGG | |
| GAAGTGGCCCCTGAGCTGTCATTGCCCCCTGCG-3′ | |
| 1249–1257 | F 5′-AATTCCCCATCAACCCTGCCGTGGCCCTAGAAG-3′ |
| R 5′-AATTCTTCTAGGGCCACGGCAGGGTTGATGGGG-3′ | |
| 1238–1257 T1247A | F 5′-AATTCGCAGGGGGCAATGACAGCTCAGGGGCCG |
| CTTCCCCCATCAACCCTGCCGTGGCCCTAGAAG-3′ | |
| R 5′-AATTCTTCTAGGGCCACGGCAGGGTTGATGGGG | |
| GAAGCGGCCCCTGAGCTGTCATTGCCCCCTGCG-3′ | |
| 1238–1257 S1248A | F 5′-AATTCGCAGGGGGCAATGACAGCTCAGGGGCCA |
| CTGCCCCCATCAACCCTGCCGTGGCCCTAGAAG-3′ | |
| R 5′-AATTCTTCTAGGGCCACGGCAGGGTTGATGGGG | |
| GCAGTGGCCCCTGAGCTGTCATTGCCCCCTGCG-3′ | |
| 1238–1257 TS1247/48AA | F 5′-AATTCGGCAATGACAGCTCAGGGGCCGCTGCCC |
| CCATCAACCCTGCCGTGGCCCTAGAAG-3′ | |
| R 5′-AATTCTTCTAGGGCCACGGCAGGGTTGATGGGG | |
| GCAGCGGCCCCTGAGCTGTCATTGCCG-3′ | |
| 1238–1257 TS1247/48EE | F 5′-AATTCGGCAATGACAGCTCAGGGGCCGAAGAAC |
| CCATCAACCCTGCCGTGGCCCTAGAAG-3′ | |
| R 5′-AATTCTTCTAGGGCCACGGCAGGGTTGATGGG | |
| TTCTTCGGCCCCTGAGCTGTCATTGCCG-3′ | |
Tissue samples and immunohistochemistry (IHC)
Samples were obtained under approved IRB protocol from the UCSD Dept. of Pathology archives. Patient demographics and tissue characterization were published previously [20]. Sections were deparaffinized, rehydrated, and renatured before staining with UJ127 and development with DAB chromagen as described previously [19].
Statistical analysis
Data shown is mean ± SD unless otherwise indicated. Antibody binding differences were analyzed by two-tailed Students t-Test. Survival was evaluated according to the method of Kaplan-Meier using a univariate log-rank test. Variables were coded as 1 for 100% L1-negative tumors and 0 for those that demonstrated evidence of L1 expression in PDAC cells.
Results
L1 is expressed by moderately- to poorly-differentiated PDAC cells in situ
We analyzed 92 human patient samples with the L1-specific UJ127 mAb and found that L1 is not expressed in normal ductal epithelium (Fig. 1a), nor well-differentiated PDAC cells (Fig. 1b). L1 is however detectable in poor- and undifferentiated/anaplastic/sarcomatoid PDAC cells (Fig. 1c), often at the tumor margin (Fig. 1d), similar to what has been reported for colon cancer [14]. Overall, our immunohistochemical analysis demonstrated that 18/22 (82%) poorly-differentiated and 4/28 (14%) moderately-differentiated PDAC tumors were L1-positive (Fig. 1e). L1 was not observed in normal (n = 20) or well-differentiated PDAC samples (n = 22). Importantly, L1 expression correlated with poor patient survival, with median survival for L1-positive patients of 6 months with a 5-year survival rate of 0%, versus median survival of L1-negative patients of 21 months with a 5-year survival of 29%. Log-Rank P < 0.001 (Fig. 1f).
Fig. 1.
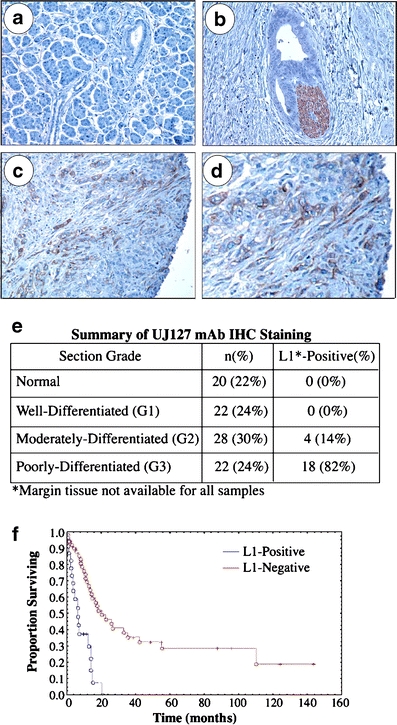
L1 is expressed by poorly differentiated PDAC cells in situ. UJ127 mAb visualized with DAB chromogen (brown). a Normal pancreas. b A well-differentiated tumor duct invading perineurally. Nerve bundle, brown. c Poorly-differentiated PDAC tumor cells at the margin. d High power image of c. e Immunohistochemical summary. f Kaplan–Meier analysis of patient survival
Loss of C20 pAb binding correlates with malignancy/differentiation in the COLO357 cell model
Sequential dissection of subcutaneous COLO357 PDAC xenograft tumors and serial repassaging of fragments in mice identified a fast-growing (FG), more malignant variant, and a slow-growing (SG), less malignant variant [21]. While the SG subline exhibits a highly differentiated epithelial phenotype in culture, including strict monolayer growth and contact inhibition, FG cells exhibit a moderate- to poorly-differentiated phenotype, including three-dimensional growth and lack of contact inhibition. When mixed in equal ratios, this population did not drift, and displayed phenotypic characteristics of the parental COLO357 cells [21]. Thus, these lines represent a unique cell system to study PDAC progression. Somewhat unexpectedly, the parental COLO357 population (Fig. 2) and both sublines (Fig. 3b) express L1 in vitro, and L1 appears to function in the growth or survival of these cells since we were unable to flow-sort stable high- and low-L1-expressing subpopulations of the COLO357, SG and FG populations. Figure 2 shows a representative experiment whereby high- and low-L1 expressing COLO357 subpopulations were flow-sorted (Fig. 2a) and 10 days later each population was resorted such that the lowest expressers were recovered from the 1Xlow-sorted population and the highest expressers from the 1Xhigh-sorted population. Two weeks later, the highest expression in the “low” population was identical to that in the “high” population (Fig. 2c), in stark contrast to their expression levels after the first sort (Fig. 2b).
Fig. 2.
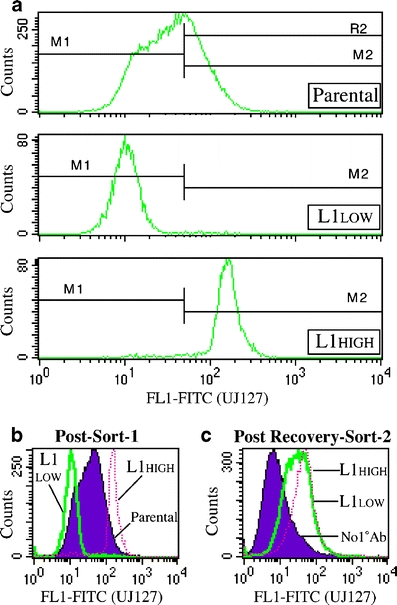
Sorted low L1-expressing Panc1 cells quickly revert to their original expression levels. a UJ127 FACS-sort profile of parental cells and derived L1-high- and L1-low-expressing subpopulations at the time of sorting. b Overlay of sorted populations onto the parental population (solid) immediately after the first sort. c FACS profiles of L1-high- and L1-low-expressing subpopulations 2 weeks after second sort. Controls are overlapped (solid)
Importantly, while the ectodomain-reactive UJ127 mAb demonstrates equivalent quantities of L1 protein in SG and FG cells, the C20 pAb directed to the L1 carboxy-terminus barely detects FG cell L1 under the denaturing conditions of immunoblotting (Fig. 3b). L1 is a known proteolytic target of both ADAMs and serine proteases [22, 23](Fig. 3a). Therefore, the possibility existed that the lack of C20 reactivity in the high molecular weight protein could represent ADAMs-mediated proteolysis of cell surface L1, yielding a C20-reactive 30KDa product and a C20-unreactive cell-associated 195KDa product lacking the cytoplasmic domain. Indeed, immunoblotting demonstrates fragments consistent with ADAMs proteolysis in FG cells, while SG cells display fragments consistent with serine proteolysis (Fig. 3c). Further corroborating these data is the fact that the 195KDa ectodomain fragment could be detected in media conditioned by FG, but not SG cells (Fig. 3d). However, immunoblotting further verifies the lack of C20 reactivity in the 30KDa fragment comprising the transmembrane and cytoplasmic sequences in FG cells specifically; M21 human melanoma cells demonstrate substantial C20 signal in the 30-KDa fragment (Fig. 3c), suggesting that obscuring of critical residues is responsible for loss of C20 reactivity in FG cells. That the cytoplasmic tail is not being further proteolyzed is demonstrated by reprobing with the αFL pAb, which was generated against the full-length protein and demonstrates equivalent amounts of identically migrating protein in both lines (Fig. 3e).
Mapping the epitope requirements of the C20 pAb
L1 contains two alternatively spliced small exons, one amino-terminal (exon2) and one in the cytoplasmic domain (exon27). Both exons are present in the neuronal isoform, and absent from the nonneuronal isoform [24]. Therefore, the possibility existed that the differential C20 binding observed in FG and SG cells might reflect differential isoform expression. Using recombinant proteins, we found that C20 detects both isoforms equivalently (Fig. 4a), demonstrating that the alternatively-spliced exon27 is not responsible for differential C20 reactivity.
Fig. 4.
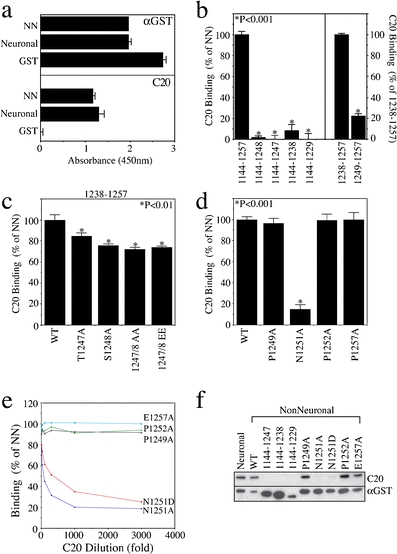
N1251 is a critical determinant of C20 binding. a ELISA of GST fusion proteins demonstrates that C20 recognizes both isoforms of L1. NN nonneuronal. b–d Truncation (b) and substitution (c,d) analysis demonstrates the requirement for the carboxy-terminal 9 amino acids, and the critical importance of N1251 for C20 binding by ELISA. e,f ELISA titration analysis (e) and immunoblotting (f) verify the data in d, extend it to N1251D, and confirm that the C20 epitope requirements are maintained under denaturing conditions
The manufacturer (Santa Cruz Biotechnology) described the C20 immunogen as being very near the carboxy-terminus of L1. Therefore, we assessed the ability of C20 to recognize sequential truncations of the GST/L1 cytoplasmic domain1144–1257 protein. We engineered −9 (L11144–1248), −10 (L11144–1247), −20 (L11144–1238), and a −28 (L11144–1229) mutant that removes the entire L1 sequence carboxy-terminal to the ankyrin-binding site [25]—this region encompasses the RanBPM binding site [16]. By enzyme-linked immunosorbent assay (ELISA), C20 binding was completely lost with the first deletion (Fig. 4b, left panel), indicating that a primary epitope requirement exists within the last nine amino acids of L1, 1249PINPAVALE1257. Presuming that the antibody might have been generated against a 20-mer encoding the absolute carboxy-terminal amino acids of L1 (hence, the name “C20”), we synthesized a protein encoding this sequence (L11238–1257), as well one encoding the last nine amino acids (L11249–1257), deletion of which completely abrogated C20 binding to the GST/L1 cytoplasmic domain protein. In contrast to the effect of deleting the 1249PINPAVALE sequence, this sequence alone only recapitulated 22% of the binding observed with L11238–1257 (Fig. 4b, right panel). This demonstrates that although residues within the nine carboxy-terminal residues are required for C20 binding, they alone are not sufficient for full binding, further suggesting the presence of additional critical residues in the previous 11 amino acid stretch (1238AGGNDSSGATS1248).
Within this sequence, T1247 and S1248 have been reported to be phosphorylated [18, 26]. Additionally, dual alanine substitution of T1247 and S1248 has been shown to disrupt MAP kinase-dependent L1-induced gene transcription and enhanced tumorigenicity in ovarian cancer cells [17]. To test the potential involvement of these residues in affecting C20 binding, we individually substituted these residues with alanine in the GST/L11238–1257 protein. These mutations caused less than 25% reduction of C20 binding versus the wild-type protein (Fig. 4c). Moreover, alanine substitution of both residues at once did not appreciably increase the effect on C20 binding, nor did mutation of both residues to glutamic acid, whose negative charge mimics that of phosphate modification. In contrast, alanine substitution of appropriate residues in the last nine amino acids (1249 PINPAVALE) of the GST/L11144–1257 protein demonstrated that N1251 is critical for C20 binding (Fig. 4d); surprisingly, no effect was observed with substitution of either surrounding proline (P1249 or P1252) or the carboxy-terminal glutamic acid (E1257). The other residues of this region constitute small, aliphatic amino acids (I1250, V1254, L1256), or alanine itself (A1253, A1255); hence, alanine substitution was not performed. It should be noted that we examined four distinct lots of C20, produced over the past 5 years, and found these epitope requirements to be maintained.
Since the N1251A mutation demonstrated such a pronounced effect on C20 binding in our recombinant proteins, we reasoned that modification of this residue might be responsible for the loss of signal noted in the FG cells. Asparagine is not subject to phosphorylation; however, asparagine deamidation has been demonstrated in various systems [for review, see 27]. Asparagine deamidation results in the production of aspartate and iso-aspartate endproducts, and iso-aspartate can be converted to aspartate by a “repair” mechanism involving protein-L-isoAsp-O-methyltransferase. However, both alanine and aspartic acid mutations of N1251 were equally effective in blocking C20 binding by ELISA (Fig. 4e) and immunoblot (Fig. 4f), suggesting that the disparate C20 reactivity with SG and FG cells are not due to different levels or activities of this enzyme and thus correspondingly differential rates of iso-aspartate to aspartate “repair”.
C20 reactivity in the L1 cytoplasmic domain reflects UJ127 reactivity in the L1 ectodomain
Previously, we found that the conformation of the L1 ectodomain is regulated by, or reflected in, the serine/threonine phosphorylation state of the L1 cytoplasmic domain in general, and the phosphorylation state of T1172 in particular [19]. To determine if the differential C20 reactivity exhibited by the SG and FG subpopulations is associated with distinct conformational states of L1 on the cell surface, we performed FACS analysis with the conformationally sensitive 5G3 mAb, which recognizes a disulfide-stabilized epitope within amino acids 57–175 spanning the end of the Ig1 and beginning of Ig2 domains at the membrane-distal amino-terminal end of L1 [19, 28]. As a reference, we used the UJ127 mAb, which recognizes a linear epitope in the membrane-proximal FN4 domain [19]. Binding of these antibodies reflects the conformation of the L1 ectodomain [19]. SG cells demonstrate identical 5G3 and UJ127 signal (Fig. 5a). In contrast, FG 5G3 reactivity is more than double UJ127 (mean fluorescence intensity 104.7 vs. 45.9). However, since 5G3 signal is identical in SG and FG cells, the actual difference is a reduction of FG UJ127 signal. This differential UJ127 epitope exposure was not limited to the live cell conditions of FACS, as both 5G3 and UJ127 immunoprecipitated similar amounts of L1 from SG cells, but UJ127 immunoprecipitated significantly less L1 than 5G3 from FG detergent lysates (Fig. 5b). Differential reactivity such as this has been linked previously to T1172 phosphorylation at the membrane-proximal end of the L1 cytoplasmic domain [19]. Consistent with more T1172-unphosphorylated L1 in the SG cells, the T1172-phosphorylation sensitive 2C2 mAb [19, 29] is much more reactive with L1 from SG cells (Fig. 5c). It should be noted that the immunoprecipitated products shown in Fig. 5b and c represent the full-length L1 species: since 2C2 recognizes the cytoplasmic domain, it is not capable of immunoprecipating the 195 kDa species, but still demonstrates the presence of a doublet that is consistent with the data shown in Fig. 5b using ectodomain-specific antibodies. Moreover, since the cells appear to shed the 195 KDa into their culture media, it is unlikely that there would be 195-KDa species that remains cell associated and therefore in the cell lysates used for immunoprecipation.
Fig. 5.
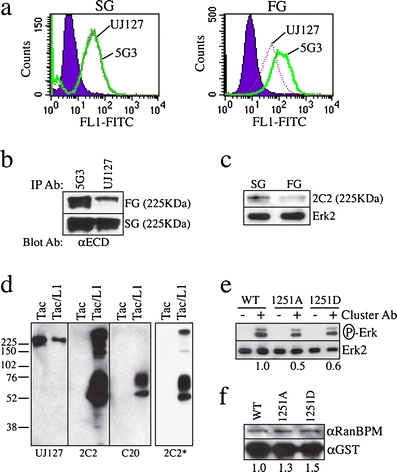
C20 reactivity correlates with the availability of the UJ127 epitope in the L1 ectodomain. a FACS analysis of cell surface L1 demonstrates reduced UJ127 reactivity in FG cells versus SG cells. b Immunoprecipitation analysis demonstrates that UJ127 immunoprecipitates less L1 than 5G3 from FG cells. c Immunoblot of the lysates from b with the 2C2 mAb, whose binding is blocked by T1172 phosphorylation. Erk2, loading control. d Reducing immunoblot of Tac-L1 constructs expressed in Panc1 cells with 2C2 or C20. *Shorter 2C2 exposure verifies that upper species are detectable at an exposure equivalent to that shown for C20 (i.e., monomer band intensities are equal). Relative migration of protein standards indicated on the left in KDa. UJ127, loading control and verification of lack of C20 reactivity with endogenous L1 in these cells. e Clustering of Tac-L1 cytoplasmic chimeras induces erk phosphorylation in an N1251-dependent manner. Erk2, loading control. f GST-pulldown analysis demonstrates that N1251 is not required for RanBPM interaction with L1. GST, loading control
The significance of these data is highlighted by the fact that the membrane–distal end of the L1 cytoplasmic domain has been shown to loop back onto the membrane–proximal end in a manner dependent on the integrity of membrane-proximal (KRSK1147) and membrane–distal (KKEK1235) dibasic motifs [29]. Thus, N1251 modification may impact aspects of L1 biology through structural modification and/or destabilization of this region, and concomitant regulation of cytoplasmic domain folding. This is potentially important because the conformation of the L1 cytoplasmic domain regulates the availability of tyrosine and threonine residues to kinases [29]. To examine the role of N1251 in regulating L1 cytoplamic domain function, we engineered chimeras encoding the ectodomain and transmembrane domain of the high-affinity IL2 receptor (IL2Rα, CD25, Tac), linked to the L1 cytoplasmic domain (Tac/L11144–1257). In isolation, Tac is a monomeric protein that lacks the ability to signal in the absence of the other heterodimer IL2R partners (i.e., IL2Rβ and IL2Rγ) [30], which are not expressed by pancreatic epithelial cells. We found that Tac/L11144–1257 multimerizes, a process that is stable enough to withstand the rigors of reducing SDS-PAGE (Fig. 5d). Importantly, whereas 2C2 recognizes these aggregates as well as the monomeric species, C20 fails to recognize the aggregate forms (Fig. 5d), demonstrating that critical determinants of C20 binding are functionally involved in mediating this multimerization process, and are thereby obscured in so doing.
As noted above, in the absence of IL2R heterodimer partners (i.e., IL2Rβ and IL2Rγ), Tac lacks the ability to signal. In response to clustering, however, chimeras such as these have been shown to signal through the intracellular fusion sequences [31]. We found that clustering of Tac/L1 cytoplasmic domain chimeras induced erk phosphorylation in a manner attenuated by mutation of N1251 to either alanine or aspartic acid (Fig. 5e). N1251 does not appear to be a mediator of RanBPM-L1 interactions, however, as mutation of N1251 did not negatively affect the binding of L1 constructs to RanBPM (Fig. 5f).
Discussion
Using a panel of 92 patient samples including normal pancreas and all grades of PDAC, we show that the neural cell adhesion molecule is expressed by moderately- and poorly-differentiated PDAC cells in situ. These data may be an underestimate, since margin tissue was not available for all samples analyzed, and we found that L1 expression was often more prominent at the tumor margin. More importantly, L1 expression correlates with poor survival in our set of PDAC patients, similar to what has been reported in colon cancer [14]. We additionally found that L1 expression is tightly controlled in PDAC cells, and FACS-sorted low-expressing subpopulations were unstable, and quickly reverted to parental levels. These data are consistent with L1 imparting a selective advantage in vitro. This proposition is actually predicted by a prior work in which L1 was identified from a library of genetic suppressor elements as required for the growth of breast cancer cells [32]. This report showed that suppression of L1, either functionally or at the level of expression, repressed growth and caused mitotic catastrophe in L1-positive breast, colon, and cervical carcinoma cells, without effect on normal breast epithelial cells or fibroblasts, indicating that L1 is required once it is expressed.
Mechanistically, our data demonstrate a loss of reactivity with the carboxy-terminus-specific antibody C20 in less differentiated FG cells, to the exclusion of their well-differentiated and less malignant counterpart SG cells. Importantly, we have found that poorly differentiated PDAC lesions demonstrate reduced C20 reactivity in situ, while C20 strongly detects nerves in the same sections (not shown). However, the polyclonal nature of C20 and the presence of parts of the C20 epitope sequence in other, non-L1-related proteins promotes some detection of L1-negative cells and structures, hampering its utility in IHC. It is important to note, however, that the lack of C20 signal in known-L1-positive PDAC cells in situ cannot be explained by this “cross-reactivity”. As such, our in situ data corraborate our in vitro data.
Since the L1 cytoplasmic domain does not contain consensus motifs for intracellular proteases (e.g., calpain), and since we accordingly do not detect proteolytic cleavage of the L1 cytoplasmic domain in a manner suggesting removal of this epitope, we propose that post-translational modification is responsible for the observed loss of reactivity. Although T1247 and S1248 near the L1 carboxy-terminus have been implicated in regulating MAP-kinase activation and gene expression associated with the L1-induced phenotype of ovarian cancer cells [17], epitope mapping instead demonstrates that modification of N1251 in the carboxy-terminal nine amino acids attenuates the binding of C20. As such, mutation of N1251 to either alanine, or one product of asparagine deamidation, aspartic acid, almost completely abrogated antibody binding. This is highly significant since glutamine–glutamate modification has been shown to regulate receptor multimerization, putatively by reducing electrostatic repulsion between helices [33], and similar mechanisms have been proposed for asparagine–aspartate conversion including the regulation of peptide hormone dimerization [34] and activation of extracellular matrix molecules [35]. Moreover, the deamidation of specific asparagines to the exclusion of neighboring asparagines has been shown to regulate protein structure and function [36]. Therefore, the relative abundance of deamidated N1251 in malignant PDAC cells might provide a regulatory mechanism for controlling L1 activity.
While it is possible that N1251 modification directly regulates erk activation, its role could also be indirect. The Neural Network Prediction program (http://npsa-pbil.ibcp.fr/) predicted that the region at the end of the L1 cytoplasmic domain forms a random coil; however, the region around N1251 contains two prolines separated by a dipeptide linker with a small aliphatic residue immediately after both prolines. Though much less constrained overall, this sequence is similar to the glycine/proline tripeptide repeat sequence of collagen molecules that provides the primary impetus for their coiled coil structure [37]. Therefore, N1251 modification might indirectly promote unfolding of the L1 cytoplasmic domain as described previously [29]. Such a mechanism may also be responsible for the dramatic phenotype observed in T1247A/S1248A mutants described previously [17]. Moreover, the regulation of specific patterns of extracellular proteolysis by these events could also indirectly lead to differential signaling through erk and other pathways as has been demonstrated previously [7, 19, 22, 23]. A model illustrating the conformational regulation of L1 activity by these events is shown in Fig. 6.
Fig. 6.
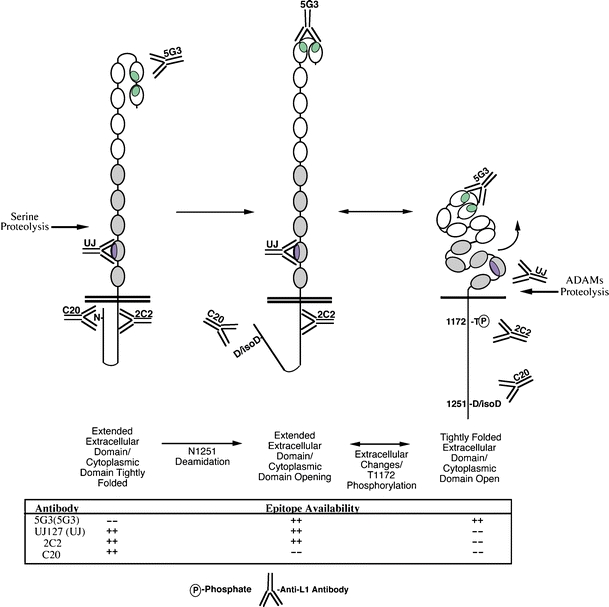
Model of L1 conformational regulation by cytoplasmic modifications. L1 has been shown to exist in distinct conformations on the cell surface [19]. These conformations have been shown to involve T1172 phosphorylation [19], and T1172 phosphorylation has been shown to be regulated by the conformation of the L1 cytoplasmic tail [29]. We propose a model whereby T1172 availability is regulated at least in part by unfolding of the L1 cytoplasmic domain that is triggered by deamidation of N1251. The ramifications of this event (e.g., proteolysis and signaling) are shown. Y tyrosine, N asparagine, D aspartate, isoD iso-aspartate
In summary, we propose a model in which modification of N1251 of L1 represents a means of regulating erk activation by the carboxy-terminal region of L1. This region has been shown previously to regulate erk activation via both RanBPM-dependent and RanBPM-independent mechanisms [16, 17]. N1251 does not appear to be involved in RanBPM binding to L1; therefore, the N1251 mechanism may be indirect, by promoting conformational alterations to the L1 cytoplasmic domain. Indeed, in this report, we show that C20 reactivity reflects the presence of a distinct conformation of the L1 ectodomain, consistent with prior reports of L1 ectodomain conformation regulation by cytoplasmic domain modification [19]. While we recognize the limitations of our studies utilizing recombinant proteins, these data are consistent with our previous observations on the flexibility of the L1 ectodomain and cytoplasmic domain, and the inter-regulated nature of each [19, 29]. We further relate our recombinant in vitro data to studies of endogenous L1 and L1 chimeras expressed in PDAC cells. Findings such as these continue to advance our understanding of L1 biology and the role of specific sequences, especially within the cytoplasmic domain, which is relatively understudied.
Acknowledgements
We thank Dr. C Behling for tissue sample procurement and analysis. S Silletti is an American Cancer Society Research Scholar supported by ACS RSG-05-116-01-CSM and NIH grants CA130104 and CA109956. Moores UCSD Cancer Center is an NCI-sponsored Comprehensive Cancer Center supported by NCI Specialized Support Grant P30 CA23100.
Conflict of interest
None to disclose
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.NCI-Pancreatic Cancer Progress Review Group . National Cancer Institute Strategic plan for addressing the recommendations of the Pancreatic Cancer Progress Review Group. PANC-PRG Implementation Plan. Bethesda: National Cancer Institute; 2002. [Google Scholar]
- 2.National Cancer Institute PDQ Database. Available online at http://www.cancer.gov/cancertopics/pdq
- 3.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 4.Aguire AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hingorani SR, Petricoin EF, III, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CVE, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 6.Burden-Gulley SM, Pendergast M, Lemmon V. The role of cell adhesion molecule L1 in axonal extension, growth cone motility, and signal transduction. Cell Tissue Res. 1997;290:415–422. doi: 10.1007/s004410050948. [DOI] [PubMed] [Google Scholar]
- 7.Silletti S, Altevogt P, Montgomery AMP. L1 cell adhesion molecule (CD171). CD171 guide for the International Workshop in Human Leukocyte Differentiation Antigens and the online “Protein Reviews on the Web” (PROW) (www.ncbi.nlm.nih.gov/prow) Prow. 2000;1:31–37. [Google Scholar]
- 8.Fogel M, Gutwein P, Mechtersheimer S, Riedle S, Stoeck A, Smirnov A, Edler L, Ben-Arie A, Huszar M, Altevogt P. L1 expression as a predictor of progression and survival in patients with uterine and ovarian carcinomas. Lancet. 2003;362:869–875. doi: 10.1016/S0140-6736(03)14342-5. [DOI] [PubMed] [Google Scholar]
- 9.Fogel M, Mechtersheimer S, Huszar M, Smirnov A, Abu-Dahi A, Tilgen W, Reichrath J, Georg T, Altevogt P, Gutwein P. L1 adhesion molecule (CD 171) in development and progression of human malignant melanoma. Cancer Letts. 2003;189:237–247. doi: 10.1016/S0304-3835(02)00513-X. [DOI] [PubMed] [Google Scholar]
- 10.Daponte A, Kostopoulou E, Kollia P, Papamichali R, Vanakara P, Hadjichristodoulou C, Nakou M, Samara S, Koukoulis G, Messinis IE. L1-CAM in ovarian serous neoplasms. Eur J Gvnaecol Oncol. 2008;29:26–30. [PubMed] [Google Scholar]
- 11.Kaifi JT, Heidtmann S, Schurr PG, Reichelt U, Mann O, Yekebas EF, Wachowiak R, Strate T, Schachner M, Izbicki JR. Absence of L1 in pancreatic masses distinguishes adenocarcinomas from poorly differentiated neuroendocrine tumors. Anticancer Res. 2006;26:1167–1170. [PubMed] [Google Scholar]
- 12.Muerkoster SS, Werbing V, Sipos B, Debus MA, Witt M, Grossmann M, Leisner D, Kotteritzsch J, Kappes H, Kloppel G, Altevogt P, Folsch UR, Schafer H. Drug-induced expression of the cellular adhesion molecule L1CAM confers anti-apoptotic protection and chemoresistance in pancreatic ductal adenocarcinoma cells. Oncogene. 2007;26:2759–2768. doi: 10.1038/sj.onc.1210076. [DOI] [PubMed] [Google Scholar]
- 13.Silletti S, Yebra M, Perez B, Cirulli V, McMahon M, Montgomery AMP. The cell adhesion molecule L1 promotes a motile and invasive phenotype via sustained MAP-kinase activation, gene transcription and induction of integrin-dependent migration. J Biol Chem. 2004;279:28880–28888. doi: 10.1074/jbc.M404075200. [DOI] [PubMed] [Google Scholar]
- 14.Gavert N, Conacci-Sorrell M, Gast D, Schneider A, Altevogt P, Brabletz T, Ben-Ze’ev A. L1, a novel target of β-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol. 2005;168:633–642. doi: 10.1083/jcb.200408051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gavert N, Sheffer M, Raveh S, Spaderna S, Shtutman M, Brabletz T, Barany F, Paty P, Notterman D, Domany E, Ben-Ze’ev A. Expression of L1-CAM and ADAM10 in human colon cancer cells induces metastasis. Cancer Res. 2007;67:7703–7712. doi: 10.1158/0008-5472.CAN-07-0991. [DOI] [PubMed] [Google Scholar]
- 16.Cheng L, Lemmon S, Lemmon V. RanBPM is an L1-interacting protein that regulates L1-mediated mitogen-activated protein kinase activation. J Neurochem. 2005;94:1102–1110. doi: 10.1111/j.1471-4159.2005.03254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gast D, Riedle S, Issa Y, Pfiefer M, Beckhove P, Sanderson MP, Arlt M, Moldenhauer G, Fogel M, Kruger A, Altevogt P. The cytoplasmic part of L1-CAM controls growth and gene expression in human tumors that is reversed by therapeutic antibodies. Oncogene. 2008;27:1281–1289. doi: 10.1038/sj.onc.1210747. [DOI] [PubMed] [Google Scholar]
- 18.Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37965–37973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- 19.Chen MM, Lee C-Y, Leland HA, Lin GY, Montgomery AM, Silletti S. Inside-out regulation of L1 conformation, integrin binding, proteolysis, and concomitant cell migration. Mol Biol Cell. 2010;21:1671–1685. doi: 10.1091/mbc.E09-10-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouvet M, Gamagami RA, Gilpin EA, Romeo O, Sasson A, Easter D, Moossa AR. Factors influencing survival after resection for periampullary neoplasms. Am J Surg. 2000;180:13–17. doi: 10.1016/S0002-9610(00)00405-0. [DOI] [PubMed] [Google Scholar]
- 21.Kajiji SM (1984) Intraneoplastic diversity in human pancreatic cancer. Dissertation, Brown University
- 22.Beer S, Oleszewski M, Gutwein P, Geiger C, Altevogt P. Metalloproteinase-mediated release of the ectodomain of L1 adhesion molecule. J Cell Sci. 1999;112:2667–2675. doi: 10.1242/jcs.112.16.2667. [DOI] [PubMed] [Google Scholar]
- 23.Nayeem N, Silletti S, Yang X, Lemmon VP, Reisfeld RA, Stallcup WB, Montgomery AM. A potential role for the plasmin(ogen) system in the posttranslational cleavage of the neural cell adhesion molecule L1. J Cell Sci. 1999;112:4739–4749. doi: 10.1242/jcs.112.24.4739. [DOI] [PubMed] [Google Scholar]
- 24.Wong EV, Schaefer AW, Landreth G, Lemmon V. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J Biol Chem. 1996;271:18217–18223. doi: 10.1074/jbc.271.30.18217. [DOI] [PubMed] [Google Scholar]
- 25.Herron LR, Hill M, Davey F, Gunn-Moore FJ. The intracellular interactions of the L1 family of cell adhesion molecules. Biochem J. 2009;419:519–531. doi: 10.1042/BJ20082284. [DOI] [PubMed] [Google Scholar]
- 26.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu T, Matsuoka Y, Shirasawa T. Biological significance of isoaspartate and its repair system. Biol Pharm Bull. 2005;28:1590–1596. doi: 10.1248/bpb.28.1590. [DOI] [PubMed] [Google Scholar]
- 28.Dahlin-Huppe K, Berglund EO, Ranscht B, Stallcup WB. Mutational analysis of the L1 neuronal cell adhesion molecule identifies membrane-proximal amino acids of the cytoplasmic domain that are required for cytoskeletal anchorage. Mol Cell Neurosci. 1997;9:144–156. doi: 10.1006/mcne.1997.0608. [DOI] [PubMed] [Google Scholar]
- 29.Chen MM, Leland HA, Lee C-Y, Silletti S. Tyrosine and serine phosphorylation regulate the conformation and subsequent threonine phosphorylation of the L1 cytoplasmic domain. Biochem Biophys Res Commun. 2009;389:257–264. doi: 10.1016/j.bbrc.2009.08.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Junghans RP, Stone AL, Lewis MS. Biophysical characterization of a recombinant soluble interleukin 2 receptor (Tac). Evidence for a monomeric structure. J Biol Chem. 1996;271:10453–10460. doi: 10.1074/jbc.271.18.10453. [DOI] [PubMed] [Google Scholar]
- 31.Meredith JE, Jr, Kiosses WB, Takada Y, Schwartz MA. Mutational analysis of cell cycle inhibition by integrin β1c. J Biol Chem. 1999;274:8111–8116. doi: 10.1074/jbc.274.12.8111. [DOI] [PubMed] [Google Scholar]
- 32.Primiano T, Baig M, Maliyekkel A, Chang B-D, Fellars S, Sadhu J, Axenovich SA, Holzmayer TA, Roninson IB. Identification of potential anticancer drug targets through the selection of growth-inhibitory genetic suppressor elements. Cancer Cell. 2003;4:41–53. doi: 10.1016/S1535-6108(03)00169-7. [DOI] [PubMed] [Google Scholar]
- 33.Surette MG, Stock JB. Role of α-helical coiled-coil interactions in receptor dimerization, signaling, and adaptation during bacterial chemotaxis. J Biol Chem. 1996;271:17966–17973. doi: 10.1074/jbc.271.30.17966. [DOI] [PubMed] [Google Scholar]
- 34.Severs JC, Froland WA. Dimerization of a PACAP peptide analogue in DMSO via asparagines and aspartic acid residues. J Pharm Sci. 2007;97:1246–1256. doi: 10.1002/jps.21116. [DOI] [PubMed] [Google Scholar]
- 35.Curnis F, Longhi R, Crippa L, Cattaneo A, Dondossola E, Bachi A, Corti A. Spontaneous formation of L-isoaspartate and gain of function in fibronectin. J Biol Chem. 2006;281:36466–36476. doi: 10.1074/jbc.M604812200. [DOI] [PubMed] [Google Scholar]
- 36.Gupta R, Srivastava OP. Effect of deamidation of asparagine 146 on functional and structural properties of human lens αB-crystallin. J Biol Chem. 2004;279:44258–44269. doi: 10.1074/jbc.M405648200. [DOI] [PubMed] [Google Scholar]
- 37.Ramachandran GN. Stereochemistry of collagen. Int J Pept Protein Res. 1988;31:1–16. doi: 10.1111/j.1399-3011.1988.tb00001.x. [DOI] [PubMed] [Google Scholar]