

Abstract
The cationic ruthenium-hydride complex [(η6-C6H6)(PCy3)(CO)RuH]+BF4− was found to be a highly regioselective catalyst for the oxidative C–H coupling reaction of aryl-substituted amides and unactivated alkenes to give ortho-alkenylamide products. The kinetic and spectroscopic analyses support a mechanism involving a rapid vinyl C–H activation followed by a rate-limiting C–C bond formation steps.
Chelate-assisted catalytic C–H activation reactions have emerged as one of the most powerful functionalization methods for arene compounds.1 Among the chelate-directed catalytic methods, oxidative C–H coupling reactions have been found to be particularly effective in forming new C–C and carbon-to-heteroatom bonds to arene compounds.1d,e Since Fujiwara’s seminal reports on the arene coupling reactions,2 oxidative C–N, C–O and C–halogen bond-forming reactions of arene compounds have been achieved by using oxygen and nitrogen chelate directing groups.3 Fagnou achieved a number of regioselective cross coupling reactions of unactivated arene compounds by using nitrogen directing groups.4 Yu recently developed a remarkably selective C–H olefination of carboxy-directed arene compounds by screening amino acid ligands for Pd catalysts.5 Late transition metal catalysts have been found to be most versatile in mediating Heck-type C–H alkenylation of heteroarene compounds, where the regioselectivity has often been found to be dictated by both steric and electronic nature of the arene substituents and chelate directing groups.6 Still, most of these oxidative C–H coupling methods require either stoichiometric amount of metal oxidants or reactive reagents, and the development of oxidative coupling methods which do not require strong oxidizing agents or reactive substrates would increase the synthetic efficiency and be beneficial from an environmental point of view. We recently discovered that the cationic ruthenium-hydride complex [(C6H6)(CO)(PCy3)RuH]+BF4− (1) is a highly effective catalyst precursor for a number of coupling reactions of arylketones and alkenes involving vinyl C–H activation.7 Herein we report a ruthenium-catalyzed oxidative C–H coupling reaction of arylamides with unactivated alkenes that does not require external oxidizing agents or additives.
 |
(1) |
Initially, the coupling reaction of an arylamide and a simple alkene was used to screen the catalyst activity (eq 1). Thus, the treatment of N,N-dimethylbenzamide (0.5 mmol) with an excess amount of cyclopentene (2.5 mmol) in the presence of a metal catalyst (5 mol %) in CH2Cl2 was analyzed by GC after 5 h of the reaction time at 80 °C.8 Among the selected ruthenium catalysts, the complex 1 exhibited uniquely high activity and selectivity for the oxidative coupling product 2a over the ortho-C–H insertion product 3a (Table S1, Supporting Information). Both CH2Cl2 and PhCl were found to be most suitable for the coupling reaction among screened organic solvents, and the formation of an equivalent amount of cyclopentane was detected in the crude reaction mixture.
The scope of the coupling reaction was explored by using the catalyst 1 (Table 1). Both secondary and tertiary arylamides were found to react smoothly with cyclic olefins to give the oxidative coupling products predominantly. The secondary amides with N-electron withdrawing group were found to promote the oxidative coupling products 2 over the insertion products 3 (entries 7–9). Cylic alkenes generally give the oxidative coupling products 2 preferentially, but cyclohexene resulted in low yield of the coupling products (<15 %). Steric and electronic environments on the amide group were found to be less important with 1,1-disubstituted and terminal olefins in yielding a mixture of 2 and 3 for these cases (entries 12–19). In contrast, the reaction with 1-hexene yielded a complex mixture of 2 and 3 that was resulted from the coupling with both terminal and internal olefins. The analogous coupling reaction of arylketones with 2-methylpropene yielded 1,3-dimethylnaphthalene product 4 along with the insertion product 3 (entries 20–23). The formation of 4 can be readily rationalized by the chelate-directed ortho-alkenylation followed by the dehydration and cyclization sequence, in light of the recently disclosed dehydrative coupling reaction of arylketones and cyclic alkenes.7b The salient feature of the coupling reaction is that the regioselective ortho-C–H oxidative olefination of arylamides has been achieved without employing any external oxidants or additives.
 |
(2) |
Table 1.
Oxidative C–H Coupling Reaction of Arylamides and Arylketones with Alkenes.a
| entry | substrate | alkene | product (ratio) | temp(°C) | yd (%) | ||
|---|---|---|---|---|---|---|---|
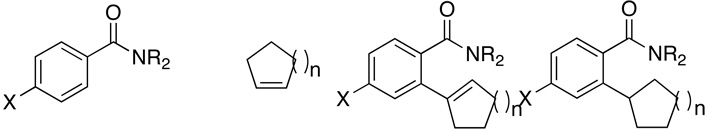 | |||||||
| 1 | X = H | R = Me, Me | n = 1 2a | (88:12) | 3a | 80 | 52 |
| 2 | X = OMe | R = Me, Me | n = 1 2b | (91:9) | 3b | 80 | 76 |
| 3 | X = Cl | R = Me, Me | n = 1 2c | (88:12) | 3c | 80 | 73 |
| 4 | X = H | R = H, Me | n = 1 2d | (95:5) | 3d | 80 | 55 |
| 5 | X = H | R = Et, Et | n = 1 2e | (87:13) | 3e | 80 | 80 |
| 6 | X = H | R = H, Bz | n = 1 2f | (92:8) | 3f | 80 | 65 |
| 7 | X = H | R = H, Ph | n = 1 2g | (100:0) | 3g | 80 | 56 |
| 8 | X = H | R = H, p-OMe-C6H4 | n = 1 2h | (100:0) | 3h | 80 | 57 |
| 9 | X = H | R = H, p-Cl-C6H4 | n = 1 2i | (100:0) | 3i | 80 | 55 |
| 10 | X = H | R = H, Bz | n = 3 2j | (87:13) | 3j | 150 | 53 |
| 11 | X = H | R = Me, Me | n = 4 2k | (95:5) | 3k | 150 | 72 |
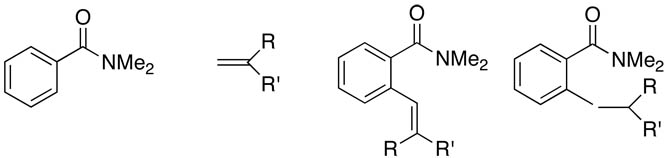 | |||||||
| 12 | R = R' = Me | 2l | (40:60) | 3l | 130 | 84 | |
| 13 | R = Me, R' = Ph | 2m | (30:70) | 3m | 150 | 82 | |
| 14 | R = H, R' = t-Bu | 2n | (30:70) | 3n | 150 | 83 | |
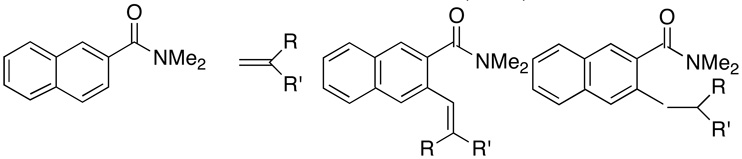 | |||||||
| 15 | R = R' = Me | 2o | (40:60) | 3o | 130 | 84 | |
| 16 | R = H, R' = t-Bu | 2p | (30:70) | 3p | 150 | 80 | |
| 17 | cyclopentene | 2q | (80:20) | 3q | 80 | 61 | |
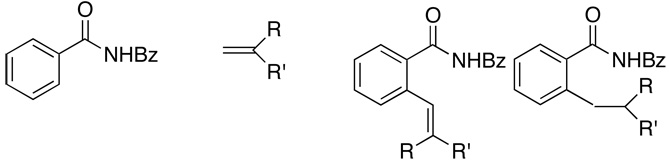 | |||||||
| 18 | R = R' = Me | 2r | (40:60) | 3r | 130 | 82 | |
| 19 | R = H, R' = t-Bu | 2s | (35:65) | 3s | 150 | 46 | |
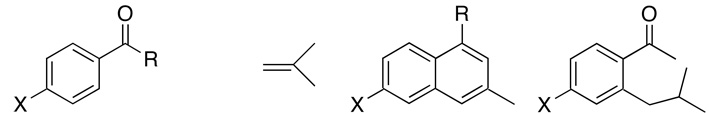 | |||||||
| 20 | X = H | R = Me | 4t | (83:17) | 3t | 130 | 64 |
| 21 | X = Me | R = Me | 4u | (61:39) | 3u | 130 | 74 |
| 22 | X = H | R = Et | 4v | (45:55) | 3v | 130 | 50 |
| 23 | X = H | R = Ph | 4w | (60:40) | 3w | 130 | 74 |
Reaction conditions: carbonyl compound (0.5 mmol), alkene (2.5 mmol), 1 (15 mg, 5 mol %), CH2Cl2 (2 mL), 5 h. PhCl (2 mL) was used as the solvent for the temperature >80 °C.
The following kinetic experiments were performed to gain mechanistic insights on the coupling reaction. To examine the H/D exchange pattern on the amide substrate, the treatment of C6D5CONEt2 (0.5 mmol) and cyclopentene (2.5 mmol) in the presence of 1 (5 mol%) in CH2Cl2 was monitored by NMR (eq 2). At 50% conversion, extensive H/D exchange pattern was observed on the ortho-arene positions of both the coupling products 2e and 3e (55% D) and the amide substrate (39% D) as well as on the unreacted cyclopentene (~5% D). Such extensive ortho-H/D exchange pattern on the amide substrate is consistent with a rapid and reversible arene C–H bond activation step. In support of this notion, a negligible isotope effect of kH/kD = 1.1±0.1 was measured from the reaction of C6H5CONEt2 and C6D5CONEt2 with cyclopentene at 80 °C (Figure S1, Supporting Information).
 |
(3) |
To discern the rate-limiting step of the coupling reaction, carbon isotope effect of the coupling reaction was measured by employing Singleton’s NMR technique at natural abundance.9 The most pronounced carbon isotope effect was observed on the ortho-carbon when the 13C ratio of recovered C6H5CONEt2 at 80% conversion was compared to that of the virgin sample (13C(recovered)/13C(virgin) at Cortho = 1.023, average of 3 runs)(eq 3). This result indicates that the C–C bond formation is the rate-limiting step of the coupling reaction.
 |
(4) |
In an effort to establish the nature of reactive species, the naphthylamide-coordinated complex 5 was prepared from the treatment of the tetranuclear complex [RuH(CO)(PCy3)]4(O)(OH)2 with N,N-dimethyl-2-naphthamide and HBF4·OEt2.10 The subsequent treatment of 5 with cyclopentene (5 equiv) at room temperature for 1 h resulted in the stable complex 6 in 82% yield (eq 4). The structure of 6 was elucidated by both solution spectroscopic and X-ray crystallographic methods. The molecular structure of 6 showed a square pyramidal arrangement on the metal center with the Ru–H in apical position. The complex 6 exhibited the same activity as the catalyst 1 toward the coupling reaction of naphthylamide and cyclopentene.
The complex 6 represents a rare example of the structurally characterized cationic Ru–H species, which is catalytically active for the chelate-assisted C–H activation reactions. Though ortho-metallated complexes have been commonly invoked as the key species for the chelate-assisted C–H activation chemistry,1 structurally well-characterized metallated ruthenium complexes were often found to be catalytically inactive for the C–H insertion reactions.11 More recently, Kakiuchi and Yu independently reported the synthesis of catalytically active ortho-metallated Ru and Pd complexes and their activity for both C–O and C–H bond activation and insertion reactions of arene compounds, respectively.12 It is also noteworthy to mention that both Ru(0)-enone and Rh(I)-olefin complexes were found to be active for the C–H insertion of arylketones.13 In our case, the successful isolation of 6 further suggests that the coupling reaction could be inhibited by the products. Indeed, the coupling reaction of N,N-diethylbenzamide and cyclopentene in the presence of 2.5 equiv of 2a showed virtually no activity under otherwise similar conditions as stipulated in eq 1 (<3% conversion).
The results provide a support for a mechanism involving vinyl C–H bond activation as depicted in Scheme 1. We propose that the initial arene exchange/π-coordination of an arylamide from 1 would form the arene-coordinated cationic Ru–H species 5, which further undergo chelate-directed ortho-C–H activation and dehydrogenation to form the ortho-metallated species 7. The subsequent vinyl C–H activation and the turnover limiting aryl-to-vinyl reductive elimination steps would form the cationic Ru–H species 8. Both carbon isotope effect study and the successful isolation of the complex 6 provide supporting evidence for the rate-limiting C–C bond formation step and the involvement of the cationic Ru–H complex 8. On the other hand, the formation of the insertion product 3 can be readily explained by invoking a direct olefin insertion and the subsequent ortho-C–H activation and reductive elimination sequence from 5. One of the major reasons why the oxidative C–H coupling path is favored over the Murai-type C–H insertion path may be due to the catalyst’s ability to promote facile vinyl C–H activation. While the formation of metal-vinyl species has been well documented in the C–H bond activation literature,14 its synthetic utility has been rarely exploited in catalytic coupling reactions.
Scheme 1.

Proposed Mechanism of the C–H Oxidative Coupling Reaction of Benzamide and Cyclopentene.
In summary, the cationic ruthenium-hydride catalyst 1 was found to be highly effective for the oxidative C–H coupling reaction of arylamides with unactivated alkenes without employing any oxidizing agents. The preliminary kinetic and spectroscopic studies suggest a mechanism involving rapid ortho-arene and vinyl C–H activation and a rate-limiting C–C bond formation steps. Current efforts are being directed to extend the synthetic utility as well as to elucidate the detailed mechanism of the coupling reaction.
Supplementary Material
Acknowledgment
Financial support from the National Institute of Health, General Medical Sciences (R15 GM55987) is gratefully acknowledged. We thank Dr. Sergey Lindeman (Marquette University) for X-ray crystallographic determination of 6.
Footnotes
Supporting Information Available Experimental procedures, spectroscopic data of organic products and the X-ray crystallographic data of 6 (53 pages, print/PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Recent reviews: Ritleng V, Sirlin C, Pfeffer M. Chem. Rev. 2002;102:1731–1770. doi: 10.1021/cr0104330. Kakiuchi F. In: Topics in Organometallic Chemistry: Directed Metallation. Chatani N, editor. Volume 24. Berlin: Springer-Verlag; 2007. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem.Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624–655. doi: 10.1021/cr900005n. Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e.
- 2.Jia C, Kitamura T, Fujiwara Y. Acc. Chem. Res. 2001;34:633–639. doi: 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]
- 3.(a) Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; (b) Wan X, Ma Z, Li B, Zhang K, Cao S, Zhang S, Shi Z. J. Am. Chem. Soc. 2006;128:7416–7417. doi: 10.1021/ja060232j. [DOI] [PubMed] [Google Scholar]; (c) Thu H-Y, Yu W-Y, Che C-M. J. Am. Chem. Soc. 2006;128:9048–9049. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]
- 4.(a) Stuart DR, Fagnou K. Science. 2007;316:1172–1175. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]; (b) Stuart DR, Villemure E, Fagnou K. J. Am. Chem. Soc. 2007;129:12072–12073. doi: 10.1021/ja0745862. [DOI] [PubMed] [Google Scholar]; (c) Campeau L-C, Schipper DJ, Fagnou K. J. Am. Chem. Soc. 2008;130:3266–3267. doi: 10.1021/ja710451s. [DOI] [PubMed] [Google Scholar]
- 5.Wang D-H, Engle KM, Shi B-F, Yu J-Q. Science. 2010;327:315–319. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Boele DKM, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:1586–1587. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]; (b) Beck EM, Grimster NP, Hatley R, Gaunt MJ. J. Am. Chem. Soc. 2006;128:2528–2529. doi: 10.1021/ja058141u. [DOI] [PubMed] [Google Scholar]; (c) Matsuura Y, Tamura M, Kochi T, Sato M, Chatani N, Kakiuchi F. J. Am. Chem. Soc. 2007;129:9858–9859. doi: 10.1021/ja071965m. [DOI] [PubMed] [Google Scholar]; (d) Cai G, Fu Y, Li Y, Wan X, Shi Z. J. Am. Chem. Soc. 2007;129:7666–7673. doi: 10.1021/ja070588a. [DOI] [PubMed] [Google Scholar]; (e) Cho SH, Hwang SJ, Chang S. J. Am. Chem. Soc. 2008;130:9254–9256. doi: 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]; (f) Tsuchikama K, Kasagawa M, Endo K, Shibata T. Org. Lett. 2009;11:1821–1823. doi: 10.1021/ol900404r. [DOI] [PubMed] [Google Scholar]; (g) Wu J, Cui X, Chen L, Jiang G, Wu Y. J. Am. Chem. Soc. 2009;131:13888–13889. doi: 10.1021/ja902762a. [DOI] [PubMed] [Google Scholar]; (h) Patureau FW, Glorius F. J. Am. Chem. Soc. 2010;132:9982–9983. doi: 10.1021/ja103834b. [DOI] [PubMed] [Google Scholar]
- 7.(a) Yi CS, Lee DW. Organometallics. 2009;28:4266–4268. doi: 10.1021/om900416k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yi CS, Lee DW. Organometallics. 2010;29:1883–1885. doi: 10.1021/om100051h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee DW, Yi CS. Organometallics. 2010;29:3413–3417. doi: 10.1021/om100468q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.See the Supporting Information for experimental details.
- 9.(a) Singleton DA, Thomas AA. J. Am. Chem. Soc. 1995;117:9357–9358. [Google Scholar]; (b) Frantz DE, Singleton DA, Snyder JP. J. Am. Chem. Soc. 1997;119:3383–3384. [Google Scholar]
- 10.(a) Yi CS, Zeczycki TN, Guzei IA. Organometallics. 2006;25:1047–1051. doi: 10.1021/om0510674. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yi CS, Zeczycki TN, Lindeman SV. Organometallics. 2008;27:2030–2035. [Google Scholar]
- 11.(a) Jazzar RFR, Mahon MF, Whittlesey MK. Organometallics. 2001;20:3745–3751. [Google Scholar]; (b) Drouin SD, Amoroso D, Yap GPA, Fogg DE. Organometallics. 2002;21:1042–1049. [Google Scholar]
- 12.(a) Ueno S, Mizushima E, Chatani N, Kakiuchi F. J. Am. Chem. Soc. 2006;128:16516–16517. doi: 10.1021/ja067612p. [DOI] [PubMed] [Google Scholar]; (b) Giri R, Lam JK, Yu J-Q. J. Am. Chem. Soc. 2010;132:686–693. doi: 10.1021/ja9077705. [DOI] [PubMed] [Google Scholar]
- 13.(a) Lu P, Paulasaari J, Jin K, Bau R, Weber WP. Organometallics. 1998;17:584–588. [Google Scholar]; (b) Lenges CP, Brookhart M. J. Am. Chem. Soc. 1999;121:6616–6623. [Google Scholar]
- 14.(a) Stoutland PO, Bergman RG. J. Am. Chem. Soc. 1985;107:4581–4582. [Google Scholar]; (b) Renkema KB, Kissin YV, Goldman AS. J. Am. Chem. Soc. 2003;125:7770–7771. doi: 10.1021/ja0289200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.