

Abstract
Prenatal systemic inflammation has been implicated in neurological diseases, but optimal animal models have not been developed. We investigated whether a partial genetic deletion of glial cell line‐derived neurotrophic factor (Gdnf +/−) increased vulnerability of dopamine (DA) neurons to prenatal lipopolysaccharide (LPS). LPS [0.01 mg/kg intraperitoneal (i.p.)] or saline was administered to wild‐type (WT) or Gdnf +/− pregnant mice on gestational day 9.5. Male offspring were examined at 3 weeks, 3 and 12 months of age. There was a progressive degeneration of tyrosine hydroxylase (TH)‐positive neurons in the substantia nigra (SN) with age in Gdnf +/− but not in WT mice, with no observed effects on locus coeruleus (LC) noradrenergic neurons or DA neurons of the ventral tegmental area. Inflammatory markers were elevated in SN of LPS treated offspring, with exacerbation in Gdnf +/− mice. Intracellular accumulation of α‐synuclein (α‐syn) immunoreactivity in DA neurons of SN was observed in all groups of Gdnf +/− and in WT mice with prenatal LPS, with altered distribution between pars reticulata (pr) and pars compacta (pc). The findings suggest that prenatal LPS leads to accelerated neuropathology in the SN with age, and that a partial loss of GDNF exacerbates these effects, providing a novel model for age‐related neuropathology of the nigrostriatal DA system.
Keywords: aging, dopamine, endotoxin, growth factors, microglia, neuroinflammation, striatum, substantia nigra
INTRODUCTION
The sporadic nature of many neurodegenerative diseases, such as Parkinson's disease (PD) (85), suggests that a combination of a genetic predisposition with an infectious agent or neurotoxin increases vulnerability of the nigrostriatal dopamine (DA) system to neurodegeneration or aging (37). The brain is especially susceptible to extrinsic or intrinsic irritants during fetal development, with the effects sometimes manifesting as adult‐onset diseases (2). This developmental pathological process is termed “Fetal Basis of Adult Disease” (FeBAD) (45). A well‐studied example of FeBAD is schizophrenia, in which the interaction of a genetic predisposition and early‐life‐environment is implicated in disease etiology (82). Although the “dual‐hit” interplay between environmental and genetic factors has been proposed for other neurological disorders, such as PD 13, 43, 80, the process has not been fully explored. Recent interest in developing appropriate animal models to test this dual‐hit hypothesis prompted experiments in which the DA neurotoxin, 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), was administered to α‐syn, Parkin, or DJ‐1 transgenic mouse models 71, 72. These “fusion models” integrate genetic alterations with environmental factors, producing several pathological processes in the same animal model 71, 72. We recently contributed a novel model for the “dual‐hit” hypothesis, by combining a reduction of glial cell line‐derived neurotrophic factor (GDNF; Gdnf +/− mice) with young‐adult methamphetamine (METH) exposure 7, 8. Young‐adult METH exposure in the experiment produced long‐lasting and progressive effects on DA systems, which were more prominent in the Gdnf +/− mice than their wild‐type (WT) littermates, suggesting an enhanced vulnerability to neurotoxins. The enhanced vulnerability of Gdnf +/− mice to METH during early adulthood suggests the possibility that they might also be more responsive to challenges during fetal development which is the focus of the current study. In this study, we examined whether exposure to an inflammogen during fetal development of the DA system accelerated DA cell loss with aging and whether this effect was exacerbated by GDNF reduction in Gdnf +/− mice.
Pro‐inflammatory cytokines, such as tumor necrosis factor alpha (TNFα) and interleukin (IL)‐1β, along with markers for microglial activation, neuroinflammation and oxidative stress are elevated in substantia nigra (SN) of patients with PD 74, 76. The SN has an unusually high density of microglia compared with other brain regions 55, 60, suggesting that neurons in this region may be more susceptible to inflammatory events (55). Maternal intrauterine infection induces pro‐inflammatory cytokines (93), such as TNFα, IL‐β(16) and IL‐6 (31), which can cause preterm delivery and/or central nervous system (CNS) disorders in the offspring (24). Lipopolysaccharide (LPS), a bacterial endotoxin which activates microglia (77), administered to pregnant rats produced dose‐responsive increases in the expression of TNFα and IL‐1β mRNA, and increased astroglial cells in the hippocampus and the cortex of fetal rat brain 16, 49. In vivo experiments established that LPS decreases tyrosine hydroxylase (TH) expression and DA content in the SN when administered either prenatally 18, 64 or postnatally 20, 68 to rats. In vitro experiments on ventral mesencephalic cultures 13, 20, 64 have shown that LPS administration reduces levels of neurotrophic factors in the rat brain (38). Although systemic injections of LPS in young‐adult mice are reported to produce prolonged increases in brain levels of TNFα, progressive loss of DA neurons in SN (90) and elevated apoptosis in the SN (78), possible interactive effects of DA growth factor availability and prenatal LPS exposure have not been fully explored.
Both in vivo and in vitro studies establish GDNF as a potent neurotrophic factor for SN DA neurons 3, 10, 11, 15, 19, 20, 23, 28, 47, 54, 62, 63, 68, 95, 102, 106, and also that GDNF is essential for postnatal development and maintenance of SN DA neurons in rodents 33, 34. Furthermore, the reduced levels of GDNF mRNA and protein in SN neurons of PD patients (21) suggest that this growth factor is necessary to prevent the progressive loss of DA neurons characterizing the disease. GDNF infusion into the striatum or SN can rescue DA neuronal degeneration in rodents and non‐human primates during the aging process 25, 44, 58 and following neurotoxin exposure 11, 25, 28, 44, 56, 58, 63, 105. Recent studies from our laboratory indicate that age‐related loss of DA neurons and accompanying motor dysfunction is exacerbated in Gdnf +/− mice, differing from WT controls around 12 months of age (6). Further, striatal DA terminals in Gdnf +/− mice are especially vulnerable to neurotoxins, such as METH (7). Gdnf +/− mice exhibited a heightened inflammatory response in the SN pars compacta (SNpc) to METH exposure, suggesting that inflammation might contribute to the increased vulnerability of SN DA neurons to neurotoxicity noted for Gdnf +/− mice. Although there is one report that lenti‐viral delivery of GDNF does not protect against DA cell loss in a mouse model for α‐syn over‐expression, our recent experiment suggests that GDNF reduction increases α‐syn mRNA expression in the striatum (7), prompting us to examine α‐syn distribution patterns in the current study. The aims of the present study were therefore to determine if prenatal LPS exposure would increase inflammatory markers and α‐syn expression; thereby increasing degeneration of DA neurons in the SN and if the GDNF reduction in Gdnf +/− mice would enhance these effects.
MATERIALS AND METHODS
Animals
Young‐adult Gdnf +/− mice were obtained from Dr. Barry Hoffer at the National Institutes on Drug Abuse (NIDA), and a breeding colony was expanded at The Medical University of South Carolina (MUSC) within the animal core of our program project grant (AG023630). This strain of Gdnf +/− mice were created by replacing part of the third exon that encodes GDNF protein with a cassette expressing the selectable marker neomycin phosphotransferase, as described previously in detail 33, 34, 86. The Gdnf +/− mice exhibit a continuous 40%–50% loss of GDNF protein levels in the brain at all ages examined here (3 weeks–12 months), as demonstrated in our previous studies (8). Mice were maintained under a 12:12 h light:dark cycle with access to mouse chow and water ad libitum and at an ambient temperature of 20°C–22°C. All experiments were performed in accordance with the Society for Neuroscience Statement for the Use of Animals in Research and were approved by the local Institutional Animal Care and Use Committee.
Maternal treatment
Fifty timed pregnant females (32 Gdnf +/− and 18 WT) were used. Pregnant females were randomly selected to receive an intraperitoneal (i.p.) injection of either saline or LPS (Sigma Inc., St. Louis, MO, LPS from Esherichia coli 026:B6; product number L8274; Lot 117K4028, 300 000 endotoxin units per mg) on gestational day 9.5 at a dose of 0.01 mg/kg body weight in sterile saline. Control animals received the same volume of saline. This injection paradigm into two different genotypes resulted in four different groups at each age time‐point. Only males were used in the present study because of possible interaction between the LPS treatment and estrogen levels in middle‐aged female mice. To ensure that the prenatal effect of endotoxin was not due to alterations in rearing of the pups by the LPS‐treated dam, some of the litters (n = 9) from LPS treatment groups were transferred to saline‐injected foster mothers within a day after birth. These offspring were housed with their foster mother until weaning. The offspring were separated at the age of 3 weeks, weighed, and a small piece of tail was cut to perform genotyping as described above. A maximum of four mice were housed per cage.
Tissue processing and immunohistochemistry
The brain tissues from mice of the four groups, WT‐saline, Gdnf +/− saline, WT‐LPS and Gdnf +/− LPS from all three ages (3 weeks = 0.7 months, 3 months and 12 months) were collected at different time‐points depending on availability of mice and were prepared for histological analysis. One male pup from each litter was chosen randomly for morphological analysis allowing us to treat each litter as n = 1 to avoid interference of inter‐litter variability in the study. The mice were anesthetized using isoflurane and decapitated. The brain was dissected and the left hemisphere was immediately immersed in 4% paraformaldehyde (in 0.1 M phosphate buffer (PB) at pH 7.4) for 48 h and then transferred into 30% sucrose at 4°C. Forty‐five µm coronal cryosections were obtained through the striatum and midbrain into 24 well plates containing PB. Serial sections, that is, every sixth from the striatum and every third from the midbrain were processed for immunohistochemical localization of tyrosine hydroxylase (TH, 1:1000; Pel‐Freez, Rogers, AR, anti‐rabbit, batch P40101‐0), Iba‐1 (pan marker for microglia; 1:1000, Wako Pure Chemical Industries Ltd, Richmond, VI, batch no. 019‐19741), TNFα (rabbit anti‐mouse, 1:1000; Biosource International Inc., Camarrillo, CA, cat# AMC3012) or F4/80 (1:1000; Abd Serotec, Raleigh, NC, cat# MCA497GA), using our routine immunohistochemistry protocol (112). In brief, free‐floating sections were treated with H2O2 (30%), methanol (100%) and 0.01 M tris buffered saline (TBS; pH 7.6; 1:2:7, respectively) for 15 minutes to quench endogenous peroxidase activity. After two 10 minutes washes with TBS, sections were incubated for 20 minutes with sodium m‐periodate (0.1 M) in TBS. Sections were washed in TBS and incubated for 30 minutes in 10% normal goat serum (NGS, Sigma‐Aldrich, St. Louis, MO) in TBST [TBS + 0.25% Triton X‐100 (Fisher Scientific, Pittsburgh, PA, USA)] to block nonspecific binding sites, followed by 48‐h incubation with the TH antibody or Iba‐1 in 3% NGS‐TBST at 4°C. Sections were then incubated for 1 h with a goat anti‐rabbit biotinylated secondary antibody (1:200, Vector Labs, Burlingame, CA) followed by 1‐h incubation with the avidin‐biotin complex (ABC Kit, Vector Laboratories, Burlingame, CA). The reaction was developed using 3′3′ diaminobenzidine (DAB, Sigma‐Aldrich, St. Louis, MO) and 0.05% of 3% H2O2 and enhanced with nickel ammonium sulfate (2.5%). It should be noted that sections from each of the groups and ages represented here were processed together in the same staining batch and with the same solutions, reducing the risk for batch to batch variations being responsible for observed effects. Furthermore, penetration of the staining throughout the sections was examined using the stereology microscope with stepwise focusing through the tissue in the z‐plane. Cresyl violet staining was performed on every sixth section throughout the SN to examine the general morphology of the SN.
As every third section throughout the SN was used for TH stereological cell counts, this left only 2 series of every third section, or 4 series of every sixth section for other stains. We therefore utilized the remaining sections for cresyl violet (every sixth), double labeling with TH and dopamine transporter (DAT; every sixth), and double labeling with TH and α‐syn (every sixth), and the last section series was used for Iba1 staining (every sixth). Immunofluorescent labeling was used for co‐localization of TH and DAT antibodies staining on every sixth section passing through the SN, as well as double labeling of TH with α‐syn, using FITC and TRITC, respectively, as fluorescent markers. Free‐floating sections were washed in phosphate buffered saline (PBS, pH 7.4) and incubated for 1 h in a cocktail consisting of 10% normal donkey serum, 0.5% fish skin gelatin and 0.3% Triton X‐100 at room temperature to block non‐specific binding and permeabilize membranes; sections were incubated thereafter in a cocktail of primary antibodies, TH (1:1000; sheep anti‐TH ab‐113, Abcam, Cambridge, MA) and α‐syn (1:250, rabbit anti‐α‐syn, batch no. 2628S, Cell Signaling, Danvers, MA) or DAT (1:500, rabbit anti‐DAT batch no. AB1591P, Millipore, Billerica, MA) in blocking cocktail, for 48 h at 4°C. Sections were washed 3 × 10 minutes in PBS with Tween 20™ (Fisher Scientific, Pittsburgh, PA, USA) (0.05%) and incubated in a cocktail of secondary antibodies, donkey anti‐rabbit Fluorescein isothiocyanate (FITC) (1:200), and donkey anti‐sheep Tetramethyl Rhodamine Iso‐Thiocyanate (TRITC) (1:50) (Jackson ImmuonoResearch Laboratories, West Grove, PA) for 2 h at room temperature. Sections were washed in PBS, mounted on glass slides and protected with ProLong Gold (Invitrogen, Carlsbad, CA, USA) anti‐fading agent in the mounting media. Images were acquired with a Nikon Optiphot microscope™ (Nikon, Melville, NY, USA) using appropriate bandpass filters for FITC and TRITC, and with a confocal microscope (Olympus Fluoview™, Olympus, Lehigh Valley, PA, USA) to obtain confocal images of α‐syn/TH double labeling in the SNpc.
Immunohistochemical image analysis
Quantitation of TH‐immunoreactive (TH‐ir) fiber density was performed on every sixth section through the dorsolateral striatum according to our previously described routine protocol (6). In short, TH‐stained sections of the dorsolateral striatum were digitized in gray scale using a 20× lens. All samples were digitized in the same time frame to ensure a constant light intensity across all groups. The NIH Image Software (Scion Image NIH®) was used to measure the density of TH immunostained fibers. This NIH Image Software measures gray scale values within the range of 0–256, with 0 representing white and 256, black. Subtracting background‐staining values from calculated means provided staining intensities for every sixth section through the dorsolateral striatum area for each animal. Measurements were performed blinded and the values from the images were averaged to obtain one value per animal. The same densitometry system was utilized to measure density of TH staining in the pontine nucleus locus coeruleus (LC), as well as the A10 [ventral tegmental area (VTA)] popoulation of DA neurons (see Supplemental Figure S1), as well as α‐syn immunofluorescent staining in the dorsal striatum, SNpr and SNpc, respectively.
Unbiased stereological cell counts
Quantitative estimates of total numbers of TH immunoreactive neurons in SN were determined by unbiased stereological cell counts from serial sections through the midbrain. We followed the method described by Gundersen (39) and West (110). The optical fractionator system used in the present study was Stereo Investigator stereological software (MicroBrightfield, Colchester, VT) coupled to a Prior H128 computer controlled x‐y‐z motorized stage. Outline contours were drawn at low magnification (10×) and the outlined region measured with a systematic random design of disector‐counting frames. The counting frame area was 2500 µm2 and the sampling grid area was 100 × 100 µm (113). The counting brick was approximately 20 µm thick after excluding the upper and lower guard zones of 2.5 µm each (35). The slides were coded and TH‐ir cells were counted using a 40× objective lens with a 1.4 numerical aperture. The selection of the first section from SN in the rostral position was random; subsequently, every third section was counted, rendering a systematic random design and the total number of neurons was calculated. Outline contours drawn for SN cell counts included areas predominantly containing TH labeled dopaminergic neurons, that is, SNpc and lateralis, and excluded the SNpr area, which contains mostly TH fiber network. The cell area of TH and Iba‐1 immunoreactive neurons and glia, respectively, was measured throughout the SN in all 24 groups on the same (TH) or adjacent (Iba‐1) sections used for stereological assessment, measuring at least 150 cells per brain randomly for each marker across all stereological sections, according to our previously published protocols for SN 6, 36, 114. Complete penetration of antibodies throughout the thickness of the section was assured by focusing stepwise through the sections using the x‐y‐z stage and the stereologer program.
TNFα enzyme‐linked immunosorbent assay (ELISA)
TNFα levels were measured by using the ELISA development kit from PeproTech (Rocky Hill, NJ) according to the manufacturer's instructions and our previous ELISA protocol (103). Tissue samples of the entire striatum were dissected and frozen in pre‐weighed Eppendorf tubes® (Fisher Scientific, Pittsburgh, PA, USA) from one hemisphere of one subject per litter randomly to include 3 and 12 months‐old groups from both treatments and genotypes (different subjects from those treated for immunohistochemistry above). Brain striatal samples were homogenized in lysis buffer [20 mM Tris‐Hcl, pH 7.5 containing 0.25 M sucrose, 2 mM ethylene‐diamine‐tetra‐acetic acid (EDTA), 0.5 mM ethylene glycol Bis (2‐aminoethyl ether) tetraacetic acid (EGTA), 1% Triton X‐100 with protease inhibitor cocktail], centrifuged and 25 µg protein equivalents of each lysate were used for the assay and processed for TNFα, using a murine TNFα ELISA kit from PeproTech (cat #900‐K54). All samples and standards were assayed in duplicate and the results are reported as ng/µg protein.
Statistical analysis
The morphological data were analyzed using a 2(genotype) × 2(treatment) × 3(age) analysis of variance (ANOVA). TNFα concentrations were analyzed using a 2(genotype) × 2(treatment) ANOVA. Pearson correlation coefficient analysis was used to assess possible relationships among the different measures.
RESULTS
LPS effects on body weights and survival rates of litters
As this is the first study, to our knowledge, that assessed the long‐term effects of prenatal LPS exposure on DA systems of mice with growth factor deletions, survival and weight gain were evaluated across the ages examined for the 33 litters used over the 4 years of experiments. Data were analyzed with 2(genotype) × 2(LPS treatment) × 3(age) ANOVAs. To avoid confounding intra‐ vs. inter‐litter variability, the litter was used as the unit of analysis (one pup/litter) by randomly selecting one pup per litter for each age. Body weight increased with age [(1,221); F = 348.113; P < 0.0001] but was not influenced by either genotype or treatment at 0.7, 3 or 12 months of age (data not shown). Body weight data included pups raised by either foster or natural mothers. Maternal LPS did not impact litter size, with 6.2 and 6.7 offspring per litter for saline‐ and LPS‐treated dams, respectively, [(1,33); F = 0.254; P > 0.6], nor did it impact sex distribution within litters, with an average of three pups of each sex; male [(1,33); F = 0.01; P > 0.9], female [(1,33); F = 0.349; P > 0.55], estimated from a total of 33 litters during 4 years of experiments). Further, there were no effects noted of genotype of the female dam upon litter size of the offspring [(1,33); F = 1.586; P > 0.2].
The effects of LPS and age on the DA nigrastriatal system of Gdnf+/− and WT mice: TH immunohistochemistry
TH immunohistochemistry was used to evaluate the impact of the prenatal LPS exposure on DA neurons in dorsal striatum and the SN at the three ages (0.7, 3 and 12 months) for both genotypes. After careful histological examination of tissue from the dorsal striatum and SN of the 12 groups formed by the 2(genotype) × 2(LPS treatment) × 3(age) experimental design, data were quantified to assess TH staining density in the striatum, and the number and average size of TH‐positive neurons in the SN. The results are summarized in Figure 1 for striatal TH‐ir density, in Figure 2 for neurons and neurites in SN, and in Figure 3 for quantification of these measures. This analysis of three ages is followed by a more in‐depth examination of the different treatment and genotype groups at mid‐life (12 months) summarized in Figure 4 for SN TH‐positive neurons and Figure 5 for the presence of neuronal/glial populations in the SN.
Figure 1.
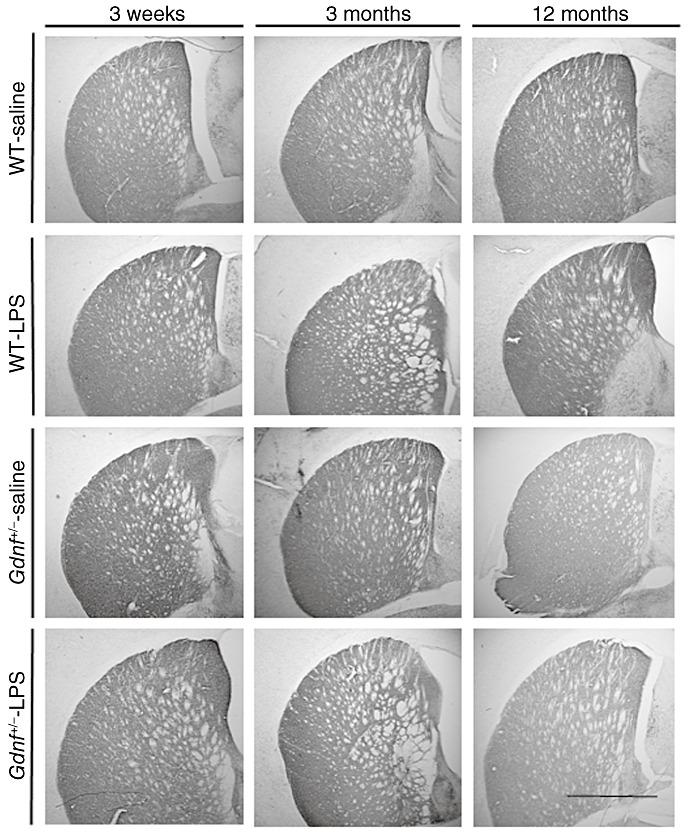
Tyrosine hydroxylase (TH) staining was reduced in the striatum of Gdnf +/− mice with or without lipopolysaccharide (LPS) treatment. TH immunostaining in the striatum of all groups demonstrates that there was a reduction in density in the 12‐month‐old Gdnf +/− mice, regardless of treatment. Prenatal LPS treatment did not alter TH immunostaining observably in wild‐type (WT) mice. Scale bar in lower right corner represents 500 microns.
Figure 2.
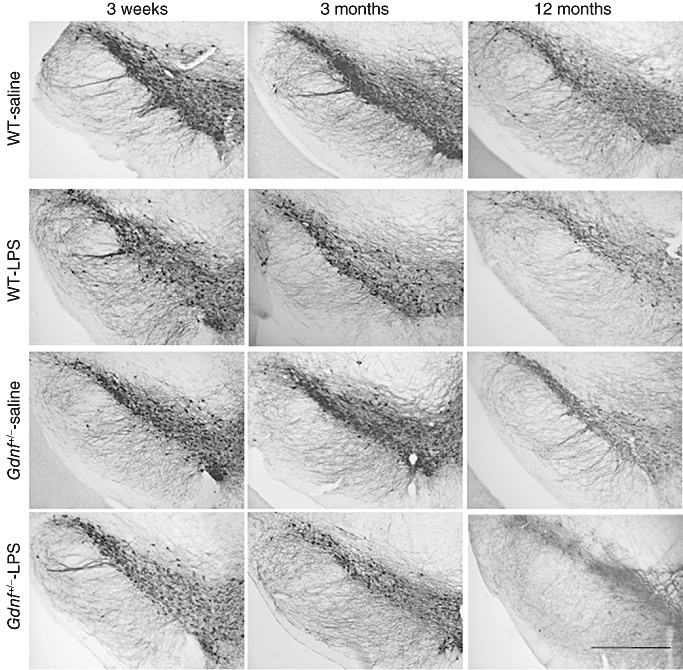
Loss of substantia nigra (SN) tyrosine hydroxylase (TH)‐positive neurons in lipopolysaccharide (LPS)‐treated mice. TH immunohistochemistry of the SN revealed a progressive loss of TH immunoreactive fibers and cell bodies in the SNpc and SNpr in LPS‐treated mice. In particular, it was obvious that LPS lead to lower numbers of TH cell bodies and neurites in the lateral portion of the SN. This appeared to be a progressive loss, as it was observable more in the 12‐month wild‐type (WT) and Gdnf +/− mice, and was found to be more severe in the Gdnf reduced mice than in WT mice. However, a loss of TH cell bodies could be seen already in the 3‐week‐old LPS‐treated Gdnf +/− mice (see panel 1), with fewer neurites and atrophied cell bodies apparent. Scale bar in lower right represents 500 microns.
Figure 3.
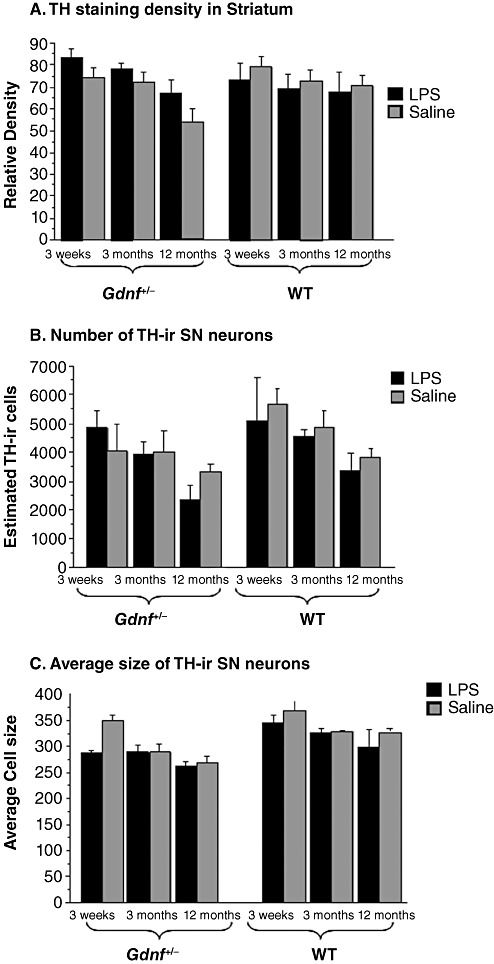
Lower densities of tyrosine hydroxylase (TH) staining in 12‐month‐old Gdnf+/− subjects. (A) TH densitometry in the striatum, (B) TH‐immunoreactive (TH‐ir) cell counts in the substantia nigra (SN) and (C) TH‐ir cell sizes in the SN. Densitometry (A) of striatal TH staining revealed a significant loss of TH‐ir at 12 months of age in the Gdnf +/− mice, regardless of treatment. Lipopolysaccharide (LPS) treatment gave rise to a significant age‐related decrease in TH cell numbers (B) and cell size (C) in Gdnf +/− compared with wild‐type (WT) LPS‐treated mice. There was an overall age effect [(2,48); F = 3.920; P = 0.0265) with a trend toward a genotype–treatment interaction [(1,48); F = 3.388; P = 0.0719], but no interaction between treatment and age.
Figure 4.
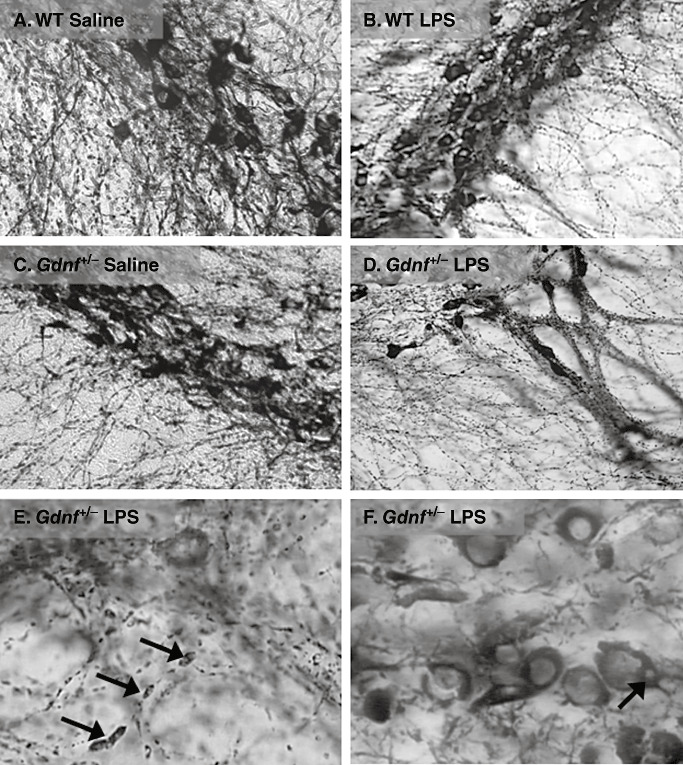
Axonal swellings and cell body inclusions substantia nigra in lipopolysaccharide (LPS)‐treated mice. These close‐up images demonstrate a significant alteration in neurite and cell body morphology as a result of the prenatal LPS treatment in mice of both genotypes. The images are generated from 12‐month mice representing all groups (A–D), followed by two close‐up images (E,F) at high magnification showing the substantia nigra (SN) pc region of LPS‐treated Gdnf +/− mice at 12 months of age, with a high grade of axonal deterioration, pyknotic nuclei and cytoplasmic inclusions (see arrows in E and F, respectively). The tyrosine hydroxylase (TH)‐devoid inclusions were observed in many TH‐immunoreactive (TH‐ir) neurons at this age but only in mice treated with LPS. Abbreviation: WT = wild‐type.
Figure 5.
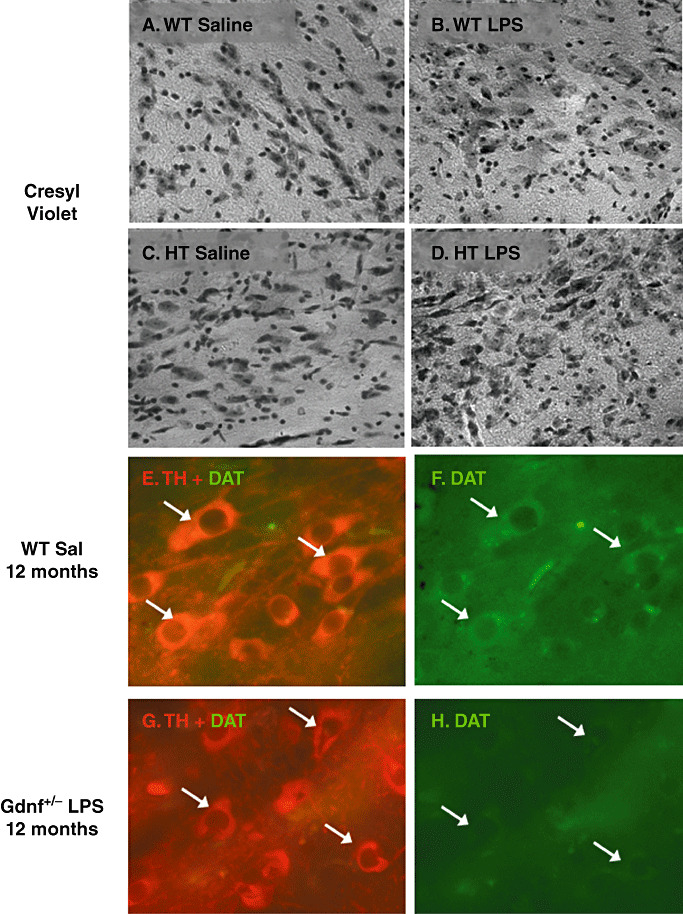
Cresyl violet and dopamine transporter‐immunoreactive (DAT‐ir) staining in substantia nigra (SN) revealed dopamine (DA) neuron degeneration with lipopolysaccharide (LPS) treatment. A–D. Cresyl violet staining of 12‐month‐old LPS‐ and saline‐treated mice. E–H. DAT/tyrosine hydroxylase (TH) double labeling in a wild‐type (WT) saline mouse (E,F) and a Gdnf +/− LPS‐treated mouse (G,H). The routine staining in (A–D) illustrate a general loss of large neuronal‐like cell bodies in the SNpc with the LPS treatment (especially evident in the Gdnf +/− LPS mouse in D), and E–H illustrate that the decline in TH staining neurons in the SN of Gdnf +/− LPS‐treated mice is also coupled with a loss of DAT activity; both of which are consistent with a general cell death and/or phenotypic loss of TH‐ir dopamine neurons as a result of the GDNF/LPS double hit. Abbreviations: HT = heterozygous; Sal = saline.
Dorsal striatum
TH‐immunoreactivity observed in dorsal striatum of Gdnf +/− and WT mice at the three ages after prenatal exposure to LPS or saline injections is depicted in Figure 1. Inspection of these sections indicated differences in TH‐ir staining for only 12 months old Gdnf +/− mice prenatally exposed to LPS or saline injections, both of which exhibited less TH‐ir in the dorsal striatum compared with other treatment groups at any age. TH‐ir density in the dorsal striatum was assessed by NIH Image according to previously published methods (8) and the results graphed in Figure 3A. A genotype × age × LPS treatment ANOVA on these data confirmed an overall age effect [(2,48); F = 3.920; P = 0.0265) with a trend toward a genotype–treatment interaction [(1,48); F = 3.388; P = 0.0719], but no interaction between treatment and age. The results of the tissue stains in Figure 1, and the TH‐ir density quantified in Figure 3A indicate no observable effects of the prenatal LPS exposure on striatal TH‐ir; however, the data confirm previous work from our group indicating lower TH‐ir immunoreactvity in the striatum of both treatment groups of 12 months‐old Gdnf +/− mice (6).
SN
TH immunochemistry of SN tissue sections for the different groups defined by the genotype × LPS × age design are shown in Figure 2. Observation of the panels in Figure 2 indicate that TH‐positive cell bodies in the SNpc and fibers in the SNpr were less numerous for prenatal‐LPS‐exposed than for saline control mice. The frequency and size of axonal swellings were elevated in the prenatal LPS exposed Gdnf +/− mice compared with their WT counterparts, as is particularly evident in the lateral portion of the SN in Figure 2. The extent of this LPS‐induced reduction in TH‐positive immunoreactivity became greater with age. This indicant of cell damage appears to be greater for Gdnf +/− than WT mice, in that it is notable in Gdnf +/− but not WT mice at the time of weaning (3 weeks of age). The increasing prominence and degree of axonal swellings noted for LPS exposed mice at 12 months old compared with the 0.7‐ and 3‐month‐ old Gdnf +/− mice suggests that neuritic degeneration in the SNpr of these mice progressed with age.
Quantification of dopaminergic neurons via unbiased stereological TH‐positive cell counts is summarized in Figure 3B. A three‐way ANOVA (treatment × genotype × age) confirmed significant effects of age [(2,56); F = 10 149; P = 0.0002] and a marginal effect of genotype [(1,56); F = 3.819; P = 0.0557] noted on the graph. Although the 12 months‐old Gdnf +/− LPS group had lower SN TH‐positive cell counts than other groups, the ANOVA indicated no effect of LPS treatment or in the interaction of the three factors (Figure 3B). Examination of Figure 2 demonstrates that the size of TH‐positive cells in the SN indicated neuronal atrophy with the aging process for Gdnf +/− mice, regardless of whether they were prenatally exposed to LPS or not. Quantification of cell size on every third section throughout the SN, as described for stereology cell counts is summarized in Figure 3C. A 2(genotype) × 2(treatment) × 3(age) ANOVA of these data confirmed the reduction of cell size with age [(2,36); F = 9815; P = 0.0004], genotype [(1,36); F = 13 209; P = 0.0009] and LPS treatment [(1,36); F = 7277; P = 0.0106]. There was also a marginally significant interaction of the age × treatment factors [(2,36); F = 3037; P = 0.0604]. Thus, the size of TH‐positive SN neurons was reduced by the genotype, the LPS treatment and age independently, suggesting an additive effect of the three. As can be seen in the bar graph in Figure 3C, the cell size analysis confirmed that Gdnf +/− had smaller SN TH‐positive neurons across age, and this effect was not exacerbated by prenatal LPS treatment but became more evident with age.
Characterization of LPS‐induced damage to DA neurons in the nigrastriatal system of middle aged (12 months) Gdnf+/− and WT mice
To further examine the type of nigrastriatal damage associated with LPS treatment and genetic reduction of GDNF, tissue samples from the older (12 months) Gdnf +/− and WT mice prenatally exposed to the LPS treatment or saline were compared in Figure 4. Examination of Figure 4A–D indicates morphological abnormalities in the cell bodies and neurites of tissue from both Gdnf +/− and WT mice prenatally exposed to LPS. The higher magnification of the SNpc samples from Gdnf +/− mice in Figure 4E–F indicates a high degree of axonal degradation, pyknotic nuclei and cytoplasmic inclusions, which were devoid of TH immunoreactivity. Axonal degeneration was evidenced by the distinctive axonal swellings interspersed along the axonal length identified by the arrows in Figure 4E. Cytoplasmic inclusions, identified by the arrows in Figure 4F, were frequently noted in TH‐positive neuronal cell bodies. These cells often exhibited a pyknotic appearance, with swollen nuclei and peripheral TH‐ir cytoplasm, indicative of ongoing degenerative processes. These inclusion‐like TH‐negative areas were found in LPS‐treated mice of both genotypes, with the greatest frequency in Gdnf +/− mice, and were located distally in the cell bodies. They can be more clearly seen in the TH co‐labeled with α‐syn confocal images displayed in Figure 7 and discussed below.
Figure 7.
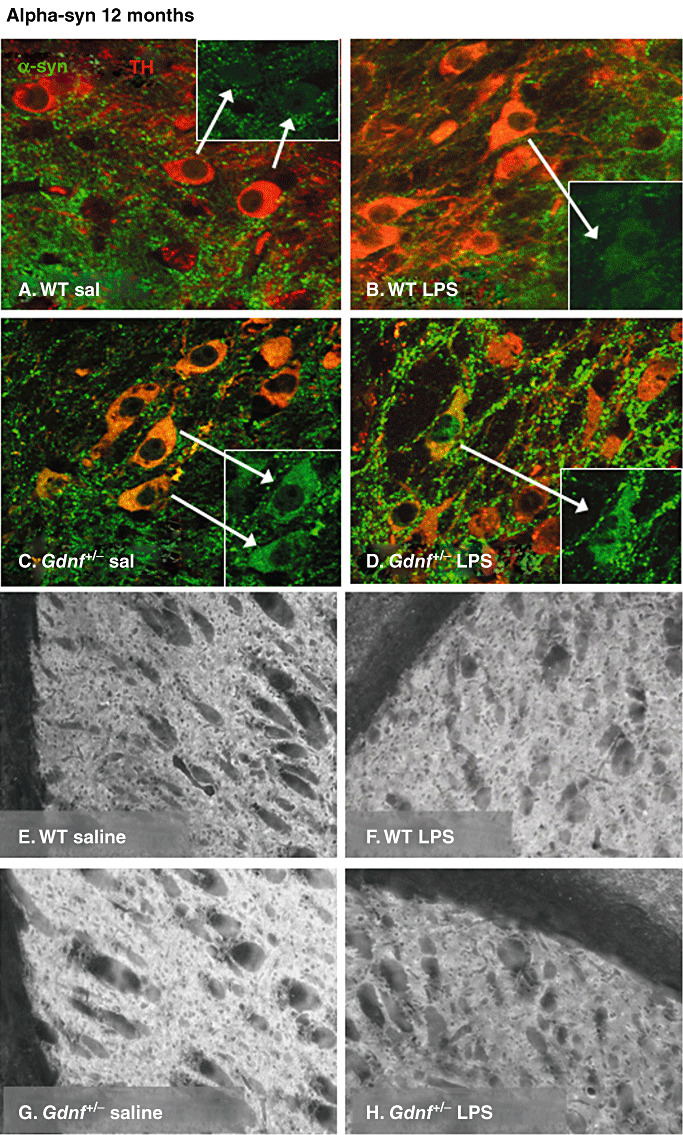
α‐syn‐immunoreactivity in the substantia nigra (SN) and striatum at 12 months of age. (A–D) Confocal micrographs of double labeling of tyrosine hydroxylase (TH; red) and α‐syn (green) in the SN at 12 months of age in all groups and α‐syn immunostaining in the striatum of 12–month‐old mice of all groups (E–H). Both Gdnf +/− groups exhibited vesicular α‐syn labeling adjacent to the TH‐positive cell bodies throughout the pc, while this appearance was only observed in lipopolysaccharide (LPS)‐treated wild‐type (WT) mice. Note that at this age, many cell bodies were double labeled with both markers in all groups except the WT saline group (insets point to α‐syn double‐labeled TH‐immunoreactive (ir) neurons). Further, several accumulations of α‐syn‐ir material were apparent among the cell bodies in LPS‐treated subjects of both genotypes, both inside and outside of TH ir cell bodies, suggesting a widespread distribution of α‐syn even outside of the dopamine (DA) neuronal population. In striatal sections, α‐syn staining was significantly increased in the Gdnf +/− saline‐treated mice compared with all other groups (G), and accumulations of fluorescent material were observed in LPS‐treated Gdnf +/− mice (H).
In order to determine if the apparent loss of TH‐ir neurons in the SN of 12 months‐old Gdnf +/− and WT mice reflected cell death, or merely a loss of the DA phenotype, serial sections throughout the SN (every sixth section for each stain) were stained with cresyl violet (Figure 5A–D) and with DAT/TH double labeling (Figure 5E–H). As can be seen in the cresyl violet stained sections (Figure 5A–D), Gdnf +/− mice had fewer neuron‐like cell bodies, and, observably higher numbers of small, dense nuclei possibly representing an increase in glial‐like cell bodies in the SN, particularly the pc region, as noted especially for the Gdnf +/− LPS group in Figure 5 (see Figure 9 for microglial staining in the SN). TH/DAT‐immunofluorescence also revealed differences between the treatment groups. While WT saline‐treated mice exhibited strong TH‐immunofluorescent profiles in the SNpc, with a medium to strong fluorescent co‐staining with DAT (arrows, Figure 5E–F), Gdnf +/− mice treated with LPS exhibited few if any DAT‐immunofluorescent cells, as shown in Figure 5G–H. Even though the TH‐immunofluorescence was fairly weak and consisted of smaller cell bodies with fewer processes in this group, this was coupled with a reduction in DAT‐immunoflourescence in the SNpc cell bodies. Inspection of the DAT/TH sections suggests that the reduced GDNF, as well as prenatal LPS exposure reduced both TH and DAT immunoreactivity in the SN in middle‐aged (12 months old) mice. This effect coupled with the cresyl violet staining which indicates a much less packed neuronal cell layer in the SNpc, suggests actual cell death in this region caused by the genetic or environmental insult.
Figure 9.
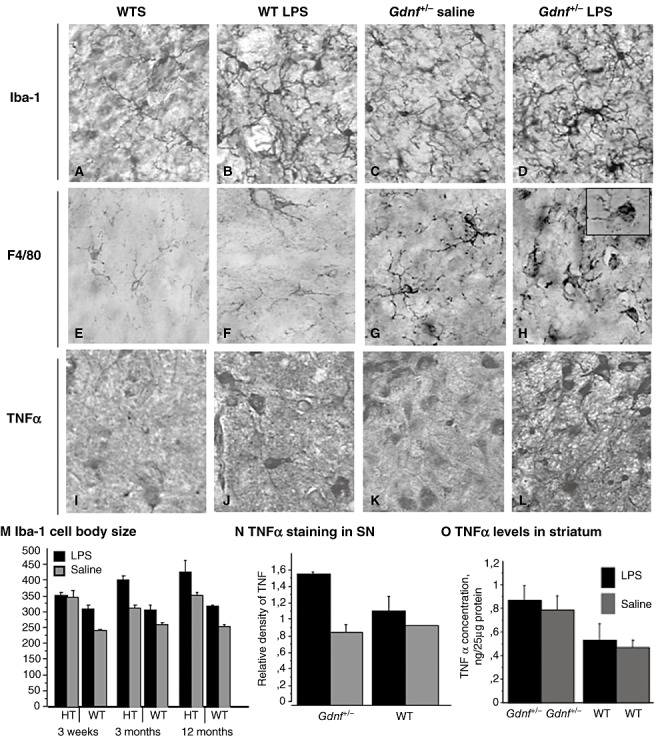
Long‐term activation of microglial cells in the substantia nigra (SN) with prenatal lipopolysaccharide (LPS) treatment. The images are generated from sections of the SN from all four groups at 12 months of age incubated with Iba‐1 (A–D), a marker for activated microglia, F4/80 (E–H), and tumor necrosis factor alpha (TNFα) (I–L). As can be seen here, microglial activation, as seen with the increased cell body size and increased arborizations, and increased staining of F4/80, is enhanced in LPS‐treated mice of both genotypes, but the activation is further increased in mice with a partial Gdnf deletion combined with LPS (D, H, L). M. For Iba‐1 cell body area in the SN there was a significant age × treatment–genotype interaction [(2,40); F = 4.442; P < 0.0181] and individual effects of age [(2,40); F = 4.158; P < 0.05], genotype [(1,40); F = 139.581; P < 0.001] and treatment [(1,40); F = 66.828; P < 0.001]. N. The staining ratio for TNFα staining in SN revealed a significant genotype × treatment effect [F = 647.990; P < 0.0396] and a significant treatment effect [F = 1673.570; P < 0.01], but no effect of genotype (P > 0.15). O. TNFα levels in SN tissue samples revealed a step‐wise increase in levels of this pro‐inflammatory cytokine, and levels were highest in the Gdnf +/−‐ LPS group, as seen in the bar graph. Abbreviations: WT = wild type; WTS = wild type saline.
Effects of prenatal LPS and Gdnf loss on other monoamine populations
As LC Norepinephrine (NE) neurons are known to be especially vulnerable to neurotoxin exposure (104), we explored effects of both treatments (GDNF loss and LPS) upon this pontine neuronal population. We have previously shown that LC NE neurons degenerate in 18 months‐old Gdnf +/− mice (108). Further, we wanted to examine specificity for the SN DA neuron effects by examining VTA DA neurons as well. None of these neuronal populations exhibited observable alterations as a result of GDNF partial deletion or prenatal LPS administration (see Supplemental Figure S1). A three‐way ANOVA (treatment × age × genotype) revealed no interactions for LC or VTA TH‐immunoreactivity staining density (data not shown). Thus, the effects of LPS treatment and partial GDNF gene deletion appeared to be specific for the SN DA neuronal population in the current study. Therefore, the LC NE loss reported previously in Gdnf +/− mice (108) appears to occur sometime between 12 and 18 months of age.
The effects of LPS and age on the DA nigrastriatal system of Gdnf+/− and WT mice: α‐synuclein
As α‐syn appears to play a role in the pathological progression of PD as well as normal DA neurotransmission (53), the distribution of this protein in tissue samples from striatum and SN of the young‐adult (3 months) and middle‐aged (12 months) Gdnf +/− and WT mice prenatally exposed to LPS or saline was examined. TH immunostaining was utilized in order to examine co‐localization of the DA marker with α‐syn. α‐syn stained sections are presented in Figure 6 for SN of 3 months‐old mice and in Figure 7 for SN and striatum of the 12 months‐old mice. Examination of the SN from 3 months‐old brains (Figure 6) revealed differences in α‐syn‐ir patterns for the two genotypes (Figure 6A and B vs. C and D) between the two LPS‐treatment groups (Figure 6A and C vs. B and D). The double labeling with TH revealed α‐syn distributed between the two layers of SN (pc and pr). A dense network of α‐syn‐ir neurites was observed in the pr of all groups but appeared to be more dense in Gdnf +/− than in WT mice. Double labeling with TH revealed that not all α‐syn immunoreactivity was localized in SN DA neurons, but rather distributed to other neuronal populations in the dense fiber network of pr. In addition, immunoreactivity in the pc was much lower in WT saline (Figure 6A) compared with Gdnf +/− mice at 3 months of age whether exposed prenatally to LPS or not (Figure 6C–D).
Figure 6.
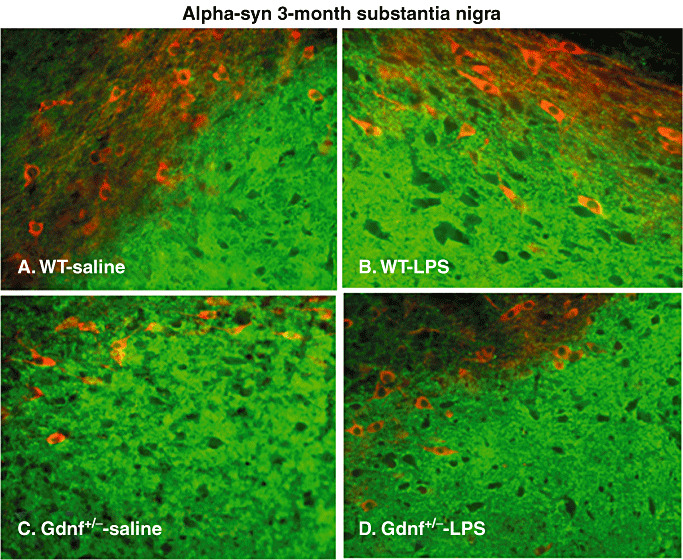
α‐syn staining in substantia nigra (SN) at 3 months of age. Double labeling with α‐syn (green) and tyrosine hydroxylase (TH, red) of the SN region using fluorescence immunohistochemistry (A–D). Note that there were differences in the distribution of this protein between the pc (above) and the pr (below), in that both the Gdnf +/− saline and the wild‐type (WT) lipopolysaccharide (LPS) group had increased α‐syn staining in the pc, compared with the other two groups. At this age, few TH‐ir cell bodies (red staining) were double labeled with α‐syn (green), suggesting that α‐syn is expressed in other neuronal populations in this brain region as well.
Closer inspection of tissue samples from 12 months‐old mice using confocal microscopy (Figure 7), revealed many TH‐ir cell bodies double labeled with α‐syn and TH, as well as α‐syn‐ir punctate staining both within and outside of TH‐ir cell bodies in the SNpc in LPS‐treated mice of both groups, as well as Gdnf +/− saline‐treated mice (see arrows, Figure 7A–D), but not in the WT saline group. The double labeling of TH and α‐syn following LPS was more prominent in Gdnf +/− mice than in WT mice. Thus, both prenatal LPS treatment and partial genetic loss of GDNF lead to accumulation of α‐syn in SN DA neuronal cell bodies, suggesting that these two factors may both participate in neuropathological alterations observed for this protein in PD, and further supporting the present model as a neuropathological model for this disease. The distribution of α‐syn in the striatum was also different between the two genotypes (Figure 7E–H).
Densitometry performed on α‐syn‐ir processed sections are summarized in Figure 8 for SNpr (Figure 8A), SNpc (Figure 8B) and striatum (Figure 8D). Analysis data for SNpr (Figure 8A) indicated a trend toward a significant genotype × age–treatment interaction [(1,23); F = 3.245; P < 0.08], and provided statistical support for the readily apparent reduction in relative density with age [(1,23); F = 44.730; P < 0.001]. Genotype and prenatal LPS exposure had no effect on SNpr α‐syn‐ir within either of the two age groups (Figure 8A), even though there was a significant genotype–treatment interaction [(1,23); F = 4.287; P < 0.0498]. Analysis of data for SNpc (Figure 8B) supported the reduction in α‐syn‐ir relative density for Gdnf +/− compared with WT mice [(1,34); F = 39.726; P < 0.0001] and the genotype–LPS exposure interaction [(1,34); F = 4.465; P < 0.042] indicating different effects of prenatal LPS exposure on the two genotypes. Furthermore, there was a trend toward a genotype–age interaction [(1,34); F = 3.500; P < 0.07] with relative α‐syn‐ir density in the pc tending to increase with age for WT mice but decrease for Gdnf +/− mice (Figure 8B). A follow‐up 2(treatment) × 2(age) ANOVA with WT mice provided statistical support for the increase in α‐syn‐ir with age [(1,15); F = 6.274; P < 0.0243], as well as an increase for mice prenatally exposed to LPS [(1,15); F = 6.319; P < 0.0238], but no interaction, suggesting that LPS increased α‐syn protein expression regardless of age in WT mice.
Figure 8.
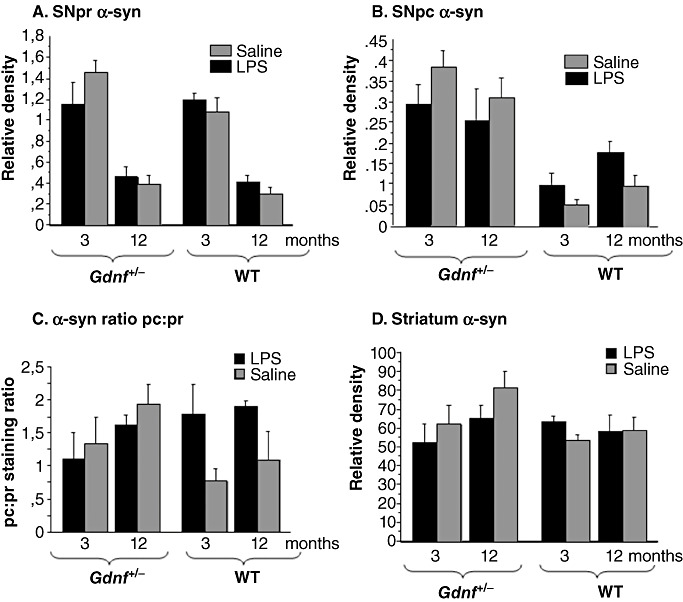
α‐syn densitometry revealed age‐ and genotype differences in staining. Densitometry was performed on α‐syn‐immunoreactive (ir) sections throughout the longitudinal axis of the substantia nigra (SN) and striatum. In pr (A), immunoreactivity to the α‐syn antibodies declined significantly with age in all groups (P < 0.001). B. The lipopolysaccharide (LPS) treatment in the pc of the wild‐type (WT) mice gave rise to a similar significant increase in SNpc α‐syn, compared with WT saline‐treated mice (P < 0.05), and the partial gene deletion of Gdnf lead to increased SNpc α‐syn in both groups (genotype effect: P < 0.001; genotype–treatment interaction P < 0.05), regardless of age. These data present an interesting relationship between LPS treatment and Gdnf deletion, and point to a complicated relationship between expression, transportation or degradation of this protein in our model. C. α‐syn staining ratio between the SNpc and pr. Note that the relative distribution of α‐syn staining is increased gradually with age in Gdnf +/− mice, and in WT mice with prenatal LPS treatment of both ages. There was a significant genotype–treatment interaction [(1,38); F = 5 708; P < 0.0114] of pc:pr in the SN. D. Densitometry of α‐syn immunostaining in the striatum at all ages showed no significant differences, even though a gradual increase in staining density was observed in Gdnf +/− mice across age, similar to what was observed for the ratio in (C).
In order to examine whether there were significant pc: pr distribution differences between groups, we examined the ratio of α‐syn staining between these two regions in 3 and 12 months‐old mice of all groups. The ratio is depicted in Figure 8C. There was a significant genotype–treatment interaction [(1,38); F = 5708; P < 0.0114] of pc:pr in the SN. As we observed large differences between LPS‐ and saline‐treated groups only in WT mice (see Figure 8C), a follow‐up t‐test analysis was performed for WT mice and revealed that LPS gave rise to a significantly increased pc:pr ratio in the SN of WT mice when collapsed over the two ages (3 and 12 months, P < 0.01).
Morphological alterations were observed within the striatum for this marker (Figure 7E–H), with significant “clumping” and extra‐ or intracellular aggregations of the marker observed in LPS‐treated subjects, especially in the dorsolateral striatum. α‐syn relative density measures for the striatum are summarized in Figure 8D. Although mean relative density was greater for the middle‐aged Gdnf +/− mice than for the other groups formed by the 2(genotype) × 2(LPS treatment × 2(age) design, an ANOVA on these data provided no statistical support for differences in α‐syn‐ir relative density in this brain region associated with any of the factors or their interaction.
Correlation analysis of the different measures indicated a positive correlation of cell size and number of DA neurons in SNpc (r = 0.458, P = 0.003) both of which were positively related to striatal TH‐ir density (striatal TH density vs. cell number: r = 0.33, P < 0.05; TH Density vs. cell size: r = 3.35, P < 0.05). Finally, striatal α‐syn‐ir relative density was negatively correlated with SNpr α‐syn‐ir values (r = −0.44, P < 0.05), and the latter measure was negatively related to SN cell size, but not number (r = −0.70, P = 0.001), suggesting that α‐syn levels may be related to shrinkage of TH‐positive cell bodies in the SN. The relationships of the measures in SN and striatum suggest that effects of the different factors are on the entire nigrastriatal system, not just the terminus.
Inflammation as a possible mechanism for degradation of nigrastriatal system function associated with genetic GDNF reduction, prenatal LPS exposure and aging
Prenatal exposure to the LPS endotoxin induces long‐lasting inflammatory responses in the brain. In an effort to examine possible mechanisms for the above‐noted DA neuronal damage, tissue from the two genotypes with and without LPS treatments was further examined using antibodies directed against Iba‐1, F4/80, and TNFα for microglia activation in the SN, and TNFα levels using Western blots in the striatum. F4/80 is a 160 kDa cell surface glycoprotein that is a member of the EGF TM7 family of proteins, upregulated in activated microglial cells and macrophages, and is commonly used as a marker for microglial activation in the mouse brain (100). The inflammatory response assessed with Iba1 antibody on brain sections from the SN of both genotypes LPS treatment groups are summarized in Figure 9A–L, and corresponding image analysis graphs are presented in Figure 9M–O. The Iba‐1 immunostaining revealed presence of resting microglia with small cell bodies and finely branched processes in the mid‐aged WT saline‐treated mice (Figure 9A). A similar pattern was noted for WT + prenatal saline controls at all ages (data not shown). Activated morphological appearance of the microglial cells (larger cell bodies and shorter, thicker processes) was observed for both genotypes prenatally exposed to LPS (Figure 9B and D), with the greatest activation seen in 12 months‐old Gdnf +/− LPS‐treated mice. Gdnf +/− mice not prenatally exposed to LPS (Figure 9C) also exhibited a more activated microglial cell type compared with saline treated WT mice (Figure 9A); also noted for 3 months‐old mice (data not shown). When additional inflammation markers were employed, a significant level of activation of microglia in Gdnf +/− mice was observed using the F4/80 marker (Figure 9G); this was aggravated by prenatal LPS administration (Figure 9H) and the enhanced inflammatory state of the SN cells in LPS‐treated subjects was further corroborated by TNFα immunostaining, as can be seen in Figure 9I–L. Image analysis measurement of Iba‐1 cell body area in the SN of all ages and treatments (Figure 9M) revealed a significant age × treatment–genotype interaction [(2,40); F = 4.442; P < 0.0181] and individual effects of age [(2,40); F = 4.158; P < 0.0229], genotype [(1,40); F = 139.581; P < 0.0001] and treatment [(1,40); F = 66.828; P < 0.0001]. Because of the enhanced effects observed between groups for Iba‐1 staining at the 12‐month age, we also examined additional inflammatory markers at this age. The staining ratio for TNFα staining in SN was examined at 12 months of age and revealed a significant genotype × treatment effect [F = 647.990; P < 0.0396] and a significant treatment effect [F = 1673.570; P < 0.0048], but no effect of genotype (P > 0.15), suggesting that LPS treatment increases TNFα expression in individual cells in the SN (Figure 9N). This was especially evident in the 12‐month‐old Gdnf +/− group treated with LPS.
In order to examine whether a chronic increase in inflammation was occurring also in the striatum of mice treated with prenatal LPS, ELISA measurements of TNFα levels in the striatum of mice from all four groups at 12 months of age were carried out. These data are summarized in Figure 9O. A 2(genotype) × 2(treatment) ANOVA provided statistical support for a higher concentration of TNFα for mid‐aged Gdnf +/− compared with WT controls, with a significant genotype effect [(1,27); F = 7.253; P < 0.0120] but no treatment, or genotype × treatment effects. These findings suggest that maternal LPS injection induced an inflammatory response in the developing fetal brain, which was obvious in both striatum and SN, and was sustained for a long period of time after birth, at least until 12 months of age. Our findings (Figure 9M) suggest that microglial activation increased with age, and that Gdnf partial genetic loss exacerbated these effects. SN has an unusually dense population of microglia compared with other brain regions 55, 60, which might contribute to the fact that this brain region is vulnerable to environmental insults.
DISCUSSION
In this study, we evaluated the effects of an intrinsic factor (GDNF reduction by gene mutation) and an extrinsic insult (maternal LPS injection modeling a bacterial infection) on the developing dopaminergic system, and established that the neuropathology resulting from this “dual hit”, especially pertaining to inflammation, persisted later in life. The nigrostriatal DA pathway of Gdnf +/− and WT control mice prenatally exposed to LPS or saline was examined at 3 weeks, 3 months, and 12 months of age to evaluate the relative and combined effects of maturation/aging, genetic GDNF reduction, and prenatal LPS exposure on the DA system. Both quantification and visual analysis indicated that all three factors impacted the DA system, with the degree of damage depending on the particular measure and brain area. Prenatal LPS exposure reduced DA cell size in the SNpc and tended to interact with the aging effects on this measure. The reduction in cell size of DA neurons in the SN was not observed in the VTA, or for LC pontine NE neurons. Prenatal LPS also tended to interact with genotype and age in its effect on the α‐syn marker of pathology, as well as on inflammation. Age and genotype, however, were the predominant detrimental factors on DA systems. Mice with a single Gdnf gene had reduced DA cell size and number, elevated α‐syn in SNpc, and a trend toward reduced TH‐ir density in the striatum. Aging was associated with the most severe and pervasive DA system changes, with reductions in DA cell size and number in the SNpc, elevated α‐syn in the SNpc and reduced TH‐ir staining density in the striatum.
The age‐related loss of TH‐ir neurons, cell shrinkage and resultant TH‐ir loss in the SN pc and striatum in 12‐month‐old Gdnf +/− mice is consistent with our previous reports for this mouse genotype 6, 7, 114. The reduced effect of the LPS treatment on striatal TH‐ir compared with its effect on SN TH‐ir cell body number may be accounted for by lesion‐induced sprouting in the dorsal striatum, as noted for partial loss of SN neurons in previous rodent studies (26). Close inspection, using cresyl violet routine staining, suggested that the reduction in SN DA neurons for 12‐month‐old mice was due to cell death rather than phenotypic loss. Microglial markers indicated that microglial activation in the SN was elevated to a greater degree in the LPS Gdnf +/− group than Gdnf +/− mice with saline treatment, or the WT LPS groups, suggesting that the double hit produced life‐long microglial activation in the SN. This increase in inflammatory markers was coupled with elevated levels of the pro‐inflammatory cytokine TNFα in the striatum. Finally, results of our studies indicated a redistribution of α‐syn in the SN, with increased cellular localization in TH‐ir cell bodies for mice with LPS treatment and Gdnf loss, and increased accumulation of α‐syn in the SNpc in LPS treated WT mice. These results implicate either increased protein production, failure to transport it to neurites or to metabolize it in the cell bodies.
Prenatal LPS exposure in this study was designed to impact early development of the DA system, a time which pro‐inflammatory cytokines are reported to severely impact developing SN dopaminergic neurons in the rat fetus 48, 64. Since mesencephalic DA neurons are first detected on embryonic day 10 for the mouse (51), LPS was injected into the dam on gestation day 9.5 (E9.5). The prenatal LPS exposure used in our study was at a lower dose and earlier in gestation than reported for models of schizophrenia (1) and cerebral palsy (69). As the developing fetal brain is not uniformly vulnerable to maternal infection throughout gestation (73), the specific time of LPS exposure greatly influences its effect. In our experiments, the relatively low LPS dose (0.01 mg/kg body weight) given on E9.5 produced no gross anatomical abnormalities in the brain. However, this dose of LPS did impact the developing nigrastriatal pathway as reported for rats utilizing a similar dosing paradigm 18, 64. Interestingly, the prenatal LPS strategy utilized here produced no observable effects on either VTA DA neurons or the pontine LC NE neurons (see supplemental Figure S1). Peripheral LPS administration has been reported to impact LC NE neurons, increasing NE turnover (50), and other reports indicate that peripheral LPS injections may differentially affect DA vs. NE neurons in the brain 83, 84. Thus, it is possible that the dose of LPS used here and/or the timing of the LPS injection may have resulted in a lack of LC degeneration in our parameters, and therefore is different from what is observed in patients with PD 4, 9, 29 or in prenatal administration of LPS into rats, as reported previously. The degenerative ability of LPS upon LC NE neurons appears more complicated than effects on the SN DA neurons, as LPS triggers compensatory increase in NE turnover (see above), and NE in itself is involved in the immune response to LPS in the brain (97). Thus, further studies are warranted regarding LPS‐induced LC loss in rat brain following a prenatal exposure. The lack of LPS‐induced effects on VTA DA neurons is consistent with the Liss et al report that this DA population is comparatively protected against neurotoxin exposure compared with SN DA neurons (67). The authors postulate that a difference in distribution and/or function of adenosine triphosphate (ATP)‐sensitive potassium (K‐ATP) channels might account for the differences between these two populations. Future studies will also include additional doses and developmental‐stage times for LPS injections, to examine temporal and regional selectivity of LPS administration effects during development. Current results indicate that relatively low LPS doses relatively early in prenatal development can produce life‐long increases in inflammatory markers in certain vulnerable brain regions. The absence of LPS effects on body weight, litter size, or litter sex distribution minimizes the possibility that reductions in overall well‐being of the fetus and pup might account for its effects on the inflammatory markers and neurite morphology observed in our study. In addition, the absence of LPS effects on body weight or litter distribution in fostered litters indicates that maternal neglect or other maternal factors could account for the observed effects on SN.
The earliest postnatal age examined in the present study, 3 weeks, was after the second apoptotic wave for dopaminergic neurons 14, 18, 64, 81. Hence, whether the prenatal LPS exposure effects existed at birth or became evident during postnatal development is unknown at the present time. However, the focus of our study was on effects of prenatal LPS exposure on DA systems throughout the life span. Although quantitative measures indicated that the LPS exposure paradigm by itself affected only DA cell size, it did tend to interact with age and genotype in α‐syn indicants of DA pathology. Further, morphological assessment of TH‐immunoreactive SN neurons revealed significant degenerative alterations in the “double‐hit group” with more marked effects noted for mice with reduced GDNF plus prenatal LPS exposure (see 2, 4). Obvious signs of neuronal degeneration were present in the 12‐month‐old mice, and morphological abnormalities associated with the LPS treatment were more pronounced in Gdnf +/− than in WT mice, as evidenced by the immunostaining for both α‐syn and TH. Axonal swellings and TH‐devoid cellular inclusions in the cell body were abundant in LPS‐treated dopamine neurons (Figure 4). In an earlier study we demonstrated that mice with a partial Gdnf deletion were more vulnerable to a toxic methamphetamine binge (7) and had elevated microglial activation in the SN. This elevation of microglial activation was by chronic minocycline administration in a subsequent study (8). These combined studies therefore suggest that chronic deletion of Gdnf leads to a heightened inflammatory state, which was further aggravated by prenatal LPS exposure in the current study. Interestingly, previous studies indicate that endogenous production of the pro‐inflammatory cytokine TNFα induces up‐regulation of GDNF production, thereby providing neuroprotection against an impeding inflammatory event (57). Additional reports indicate that a general inflammatory reaction, both in the periphery and the CNS, significantly increases GDNF production by either astrocytes or neurons in the region of inflammation 41, 108. Thus it is reasonable to postulate that the chronically reduced GDNF expression in Gdnf +/− mice might perpetuate the prenatal LPS‐induced damage by preventing the necessary GDNF increase required to protect the developing DA neurons. Work by Hashimoto et al (41) suggests that LPS‐induced increases in GDNF production by inflammatory cells is a necessary component for regeneration after injury. In addition, recent studies by Ghribi et al (30) demonstrated that GDNF injections prevented beta‐amyloid‐induced microglial activation in the brain, providing direct evidence for the inhibiting effects of GDNF upon microglial cell activation. The noted increase in Iba‐1 and F4/80 immunoreactive microglial cells in the SN of Gdnf +/− mice prenatally exposed to LPS certainly suggests that reduced access to this trophic factor exacerbates the inflammatory response of the brain to LPS. This interpretation is supplemented by Gdnf +/− mice with LPS treatment exhibiting the greatest increase in TNFα levels.
Elevated pro‐inflammatory cytokine levels in the nigrostriatal system have been reported for PD patients 5, 74, 75, 111. Moreover, an epidemiological study [Wahner et al (109)] indicated that inflammatory cytokine gene polymorphisms can increase the risk of developing PD. It is also reported that rats prenatally exposed to LPS, according to Ling et al 64, 65, 66, have elevated brain cytokine levels long‐term after exposure, which corroborates our results in mice. In addition, the saline control Gdnf +/− mice, in the present study, had elevated TNFα levels compared with saline control WT mice in keeping with our interpretation of a “basally” activated or “primed” state of microglia serving as a potential source of the cytokine. Prenatal LPS exacerbates this response noted with GDNF reduction. A study on adult mice lacking TNFα receptors (TNF R1/R2–/–) and their WT controls, reported by Qin et al (90) indicated that a single LPS injection elevated TNFα levels and cell body size of Iba‐1 ir microglia in the SN for up to 10 months post‐injection only in WT mice. Furthermore, they observed progressive loss of SN TH‐ir neurons. These findings suggest that pre‐ and postnatal LPS exposure activates microglia to chronically produce pro‐inflammatory cytokines, and also induce progressive loss of dopaminergic neurons in SN, possibly by creating a vicious cycle of pro‐inflammatory cytokine production, neuronal death, and microglial activation, as suggested by Qin et al (90).
The absence of reduced TH‐ir neurons for WT mice in our study suggests that the dose was either too low or given at a different time point in SN DA neuron development relative to those used for the rat studies by Ling et al (64) and also that Gdnf +/− mice exhibit a greater vulnerability to endotoxin exposure. Alternatively, it is possible that the time course for DA neuron development is slightly shifted in Gdnf +/− mice, resulting in them being exposed to LPS at a more vulnerable stage of the DA development; however, our previous work of fetal development in Gdnf complete or partial knockout mice 32, 34, 87 indicates no altered development in terms of birth or development of this neuronal population. It should be noted that these studies did not encompass E9 or E10 of development.
Accompanying the loss of TH‐positive neurons in our study were abnormal morphological features in the SN dopaminergic neurons and fibers of LPS‐treated mice. Moreover, a common feature of 3‐ and 12‐month‐old mice prenatally exposed to LPS were empty spaces in the cytoplasm of SN TH‐ir neurons, indicating cytoplasmic inclusions, as well as numerous axonal swellings. These alterations, coupled with significant TH‐ir cell body atrophy and increased expression of α‐syn‐ir, as discussed below, are consistent with an ongoing and progressive degenerative process of DA neurons in the SN. Interestingly, degenerating neurites or loss of TH‐ir in the striatum were not observed for any of the LPS exposed groups until 12 months of age, at which time prenatal LPS‐exposed or saline control Gdnf +/− mice exhibited significant reductions in TH‐ir striatal density. Although synaptogenesis begins in the neuropil of the SN on or before gestation day 18, most of the striatal synaptogenesis occurs postnatally and continues into adulthood (59), (42). GDNF is expressed at high levels in developing striatum, and is reduced to comparatively low levels in adult rodents 22, 79, 88, 96, 99, 102, 107. Collateral sprouting occurs in the striatum after partial lesions of SNpc, and is proportional to the lesion size (101). Based on the fairly significant TH‐ir cell loss in the SN, a greater effect on TH‐ir in the striatum following the double hit would be expected. However, our results suggest that striatal sprouting of remaining DA neurons has occurred in our model. An interesting extension of the current work therefore would be to examine studies whether this compensatory sprouting of TH‐ir fibers originates solely from SN dopaminergic neurons or also from the neighboring DA neurons in the VTA (A10) which have been documented to sprout into the degenerated striatum following a partial SN lesion 40, 46, 98.
α‐syn is a neuronal protein that is implicated in the control of synaptic vesicle function and in Parkinson's disease (85). The α‐syn inclusion body pathology (Lewy pathology) associated with sporadic PD is distributed throughout the brain, spinal cord and enteric nervous systems (12), indicating its presence throughout the neuraxis, not just DA systems. Recent work by Braak and collaborators (12) indicates that α‐syn‐related pathology is predominantly in motor areas of the brain including the visceromotor, the limbic systems and the somatomotor systems. Mutants of α‐syn, Uch‐L1 and Parkin, indicate that the ubiquitin‐proteosomal system is involved in the progressive pathology observed in PD (52). In the present study, we observed a significant increase of α‐syn‐ir in TH‐ir cell bodies of the SNpc region in both treatment groups of Gdnf +/− mice and in LPS‐treated WT mice, suggesting either that the protein was unable to be transported to terminal regions, or that its production was increased, leading to aggregation of α‐syn‐ir in DA cell bodies. It is possible that this neuropathological finding is similar to the Lewy body‐like inclusions observed in SN DA neurons many months following prenatal LPS exposure in rats (62). However, α‐syn‐ir profiles were also present outside of TH‐ir cell bodies in the pc and pr indicating their presence in other neuronal populations as well. A recent study indicates that α‐syn expression is induced in dopaminergic primary cultures by neurotrophic factors (91). On the other hand, other studies indicate that basal neuronal α‐syn levels are not affected by either cytokines or growth factor expression (94), which suggests that our observations are related more to the re‐distribution than to altered expression of the protein. This interpretation is supported by the absence of significantly elevated α‐syn levels in either pr or the striatum of treated mice in our study. It should be noted, however, that the Satoh and Kuroda (94) experiment utilized neuronal cell lines, and may not generalize to SN DA neurons in situ. α‐syn appears to be involved in DA reuptake mechanisms (52), and we have previously reported alterations in DA reuptake mechanisms in Gdnf +/− mice (7). More detailed studies involving DA transmission mechanisms will be needed to establish the biological mechanisms involved with the LPS‐induced accumulation of α‐syn in the WT mice. GDNF administration has been reported to rescue DA neurons from neurotoxin‐induced damage without affecting α‐syn expression or α‐syn‐induced neuronal damage in in vivo animal models 61, 70, which suggests that GDNF and α‐syn likely function via different, but converging pathways in SN DA neurons. In spite of the lack of clarity about the mechanism, the significant α‐syn elevation in the SNpc of Gdnf +/− mice in our study indicates that a chronic reduction of GDNF does affect α‐syn and/or the processes that this protein controls, perhaps including axonal transport of proteins. Aggregated or modified α‐syn can trigger the activation of microglia, pointing at aggregated and modified forms of α‐syn as a primary cause of inflammation related to PD pathogenesis (92), and therefore suggesting that both GDNF and α‐syn can modify the activation state of microglia. This suggests that a balance between these two proteins is key to modifying the progression of the disease.
CONCLUSION
The present findings demonstrate long‐lasting effects of prenatal LPS exposure as indicated by progressively increasing inflammation in the SN, aggravated by a partial deletion of the Gdnf gene. Studies from the laboratory of Dr. Jau‐Shyong Hong have demonstrated that presence of microglia is essential for LPS‐induced neurotoxicity 27, 89. Our immunohistochemical analysis demonstrated that maternal LPS administration and loss of GDNF induced a progressive loss of TH expression as well as degenerative alterations in TH‐ir SN neurites at 3 and 12 months of age. Gdnf +/− mice exhibited an up‐regulation of TNFα levels in the SN and increased activation of Iba‐1‐ir and F4/80 ir microglia in SN compared with WT mice, suggesting that the loss of this growth factor aggravates LPS induced inflammation. We also found that LPS administration prenatally gave rise to increased accumulation of α‐syn‐ir in SN TH‐ir cell bodies, and that α‐syn expression in SN DA neurons was further elevated by the partial Gdnf deletion. Our studies presented here suggest that an important function for GDNF in the nigrostriatal system may be to modulate inflammatory response to neurotoxic events as well as the normal aging process.
Supporting information
Figure S1. Tyrosine hydroxylase (TH) immunostaining in the pontine nucleus locus coeruleus (LC, A–D) and the ventral tegmental area (VTA, E–H) did not reveal any observable morphological alterations either as a result of the partial Gdnf deletion or the prenatal lipopolysaccharide (LPS) treatment, at least not at 12 months of age. Scale bar in H represents 100 microns.
Supporting info item
ACKNOWLEDGMENTS
The authors would like to thank Mr. A. Moore and Mr. M Nelson for technical assistance. The work was supported by a program project grant from the National Institutes on Aging (AG023630).
REFERENCES
- 1. Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN (2006) The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry 11:47–55. [DOI] [PubMed] [Google Scholar]
- 2. Barlow BK, Cory‐Slechta DA, Richfield EK, Thiruchelvam M (2007) The gestational environment and Parkinson's disease: evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod Toxicol 23:457–470. [DOI] [PubMed] [Google Scholar]
- 3. Bilang‐Bleuel A, Revah F, Colin P, Locquet I, Robert JJ, Mallet J, Horellou P (1997) Intrastriatal injection of an adenoviral vector expressing glial‐cell‐line‐derived neurotrophic factor prevents dopaminergic neuron degeneration and behavioral impairment in a rat model of Parkinson disease. Proc Natl Acad Sci U S A 94:8818–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bing G, Zhang Y, Watanabe Y, McEwen BS, Stone EA (1994) Locus coeruleus lesions potentiate neurotoxic effects of MPTP in dopaminergic neurons of the substantia nigra. Brain Res 668:261–265. [DOI] [PubMed] [Google Scholar]
- 5. Blum‐Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P (1995) Interleukin‐1 beta and interleukin‐6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo Parkinson's disease patients. Neurosci Lett 202:17–20. [DOI] [PubMed] [Google Scholar]
- 6. Boger HA, Middaugh LD, Huang P, Zaman V, Smith AC, Hoffer BJ et al (2006) A partial GDNF depletion leads to earlier age‐related deterioration of motor function and tyrosine hydroxylase expression in the substantia nigra. Exp Neurol 202:336–347. [DOI] [PubMed] [Google Scholar]
- 7. Boger HA, Middaugh LD, Patrick KS, Ramamoorthy S, Denehy ED, Zhu H et al (2007) Long‐term consequences of methamphetamine exposure in young adults are exacerbated in glial cell line‐derived neurotrophic factor heterozygous mice. J Neurosci 27:8816–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boger HA, Middaugh LD, Granholm AC, McGinty JF (2009) Minocycline restores striatal tyrosine hydroxylase in GDNF heterozygous mice but not in methamphetamine‐treated mice. Neurobiol Dis 33:459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bosboom JL, Stoffers D, Wolters E (2004) Cognitive dysfunction and dementia in Parkinson's disease. J Neural Transm 111:1303–1315. [DOI] [PubMed] [Google Scholar]
- 10. Bowenkamp KE, Hoffman AF, Gerhardt GA, Henry MA, Biddle PT, Hoffer BJ, Granholm AC (1995) Glial cell line‐derived neurotrophic factor supports survival of injured midbrain dopaminergic neurons. J Comp Neurol 355:479–489. [DOI] [PubMed] [Google Scholar]
- 11. Bowenkamp KE, David D, Lapchak PL, Henry MA, Granholm AC, Hoffer BJ, Mahalik TJ (1996) 6‐Hydroxydopamine induces the loss of the dopaminergic phenotype in substantia nigra neurons of the rat. A possible mechanism for restoration of the nigrostriatal circuit mediated by glial cell line‐derived neurotrophic factor. Exp Brain Res 111:1–7. [DOI] [PubMed] [Google Scholar]
- 12. Braak H, Del Tredici K (2009) Neuroanatomy and pathology of sporadic Parkinson's disease. Adv Anat Embryol Cell Biol 201:1–119. [PubMed] [Google Scholar]
- 13. Bronstein DM, Perez‐Otano I, Sun V, Mullis Sawin SB, Chan J, Wu GC et al (1995) Glia‐dependent neurotoxicity and neuroprotection in mesencephalic cultures. Brain Res 704:112–116. [DOI] [PubMed] [Google Scholar]
- 14. Burke RE (2004) Ontogenic cell death in the nigrostriatal system. Cell Tissue Res 318:63–72. [DOI] [PubMed] [Google Scholar]
- 15. Burke RE, Antonelli M, Sulzer D (1998) Glial cell line‐derived neurotrophic growth factor inhibits apoptotic death of postnatal substantia nigra dopamine neurons in primary culture. J Neurochem 71:517–525. [DOI] [PubMed] [Google Scholar]
- 16. Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG (2000) Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res 47:64–72. [DOI] [PubMed] [Google Scholar]
- 17. Campbell A (2004) Inflammation, neurodegenerative diseases, and environmental exposures. Ann N Y Acad Sci 1035:117–132. [DOI] [PubMed] [Google Scholar]
- 18. Carvey PM, Chang Q, Lipton JW, Ling Z (2003) Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long‐term losses of dopamine neurons in offspring: a potential, new model of Parkinson's disease. Front Biosci 8:s826–s837. [DOI] [PubMed] [Google Scholar]
- 19. Cass WA (1996) GDNF selectively protects dopamine neurons over serotonin neurons against the neurotoxic effects of methamphetamine. J Neurosci 16:8132–8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castano A, Herrera AJ, Cano J, Machado A (1998) Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem 70:1584–1592. [DOI] [PubMed] [Google Scholar]
- 21. Chauhan NB, Siegel GJ, Lee JM (2001) Depletion of glial cell line‐derived neurotrophic factor in substantia nigra neurons of Parkinson's disease brain. J Chem Neuroanat 21:277–288. [DOI] [PubMed] [Google Scholar]
- 22. Choi‐Lundberg DL, Bohn MC (1995) Ontogeny and distribution of glial cell line‐derived neurotrophic factor (GDNF) mRNA in rat. Brain Res Dev Brain Res 85:80–88. [DOI] [PubMed] [Google Scholar]
- 23. Clarkson ED, Zawada WM, Freed CR (1997) GDNF improves survival and reduces apoptosis in human embryonic dopaminergic neurons in vitro . Cell Tissue Res 289:207–210. [DOI] [PubMed] [Google Scholar]
- 24. Dammann O, Kuban KC, Leviton A (2002) Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment Retard Dev Disabil Res Rev 8:46–50. [DOI] [PubMed] [Google Scholar]
- 25. Emerich DF, Plone M, Francis J, Frydel BR, Winn SR, Lindner MD (1996) Alleviation of behavioral deficits in aged rodents following implantation of encapsulated GDNF‐producing fibroblasts. Brain Res 736:99–110. [PubMed] [Google Scholar]
- 26. Finkelstein DISD, Parish CL, Tomas D, Dickson K, Horne MK (2000) Axonal sprouting following lesions of the rat substantia nigra. Neuroscience 97:99–112. [DOI] [PubMed] [Google Scholar]
- 27. Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B (2002) Microglial activation‐mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem 81:1285–1297. [DOI] [PubMed] [Google Scholar]
- 28. Gash DM, Zhang Z, Cass WA, Ovadia A, Simmerman L, Martin D et al (1995) Morphological and functional effects of intranigrally administered GDNF in normal rhesus monkeys. J Comp Neurol 363:345–358. [DOI] [PubMed] [Google Scholar]
- 29. Gesi M, Soldani P, Giorgi FS, Santinami A, Bonaccorsi I, Fornai F (2000) The role of the locus coeruleus in the development of Parkinson's disease. Neurosci Biobehav Rev 24:655–668. [DOI] [PubMed] [Google Scholar]
- 30. Ghribi O, Herman MM, Pramoonjago P, Spaulding NK, Savory J (2004) GDNF regulates the A beta‐induced endoplasmic reticulum stress response in rabbit hippocampus by inhibiting the activation of gadd 153 and the JNK and ERK kinases. Neurobiol Dis 16:417–427. [DOI] [PubMed] [Google Scholar]
- 31. Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M (2005) Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology 48:903–917. [DOI] [PubMed] [Google Scholar]
- 32. Granholm AC, Mott JL, Bowenkamp K, Eken S, Henry S, Hoffer BJ et al (1997) Glial cell line‐derived neurotrophic factor improves survival of ventral mesencephalic grafts to the 6‐hydroxydopamine lesioned striatum. Exp Brain Res 116:29–38. [DOI] [PubMed] [Google Scholar]
- 33. Granholm AC, Srivastava N, Mott JL, Henry S, Henry M, Westphal H et al (1997) Morphological alterations in the peripheral and central nervous systems of mice lacking glial cell line‐derived neurotrophic factor (GDNF): immunohistochemical studies. J Neurosci 17:1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Granholm AC, Reyland M, Albeck D, Sanders L, Gerhardt G, Hoernig G et al (2000) Glial cell line‐derived neurotrophic factor is essential for postnatal survival of midbrain dopamine neurons. J Neurosci 20:3182–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Granholm AC, Ford KA, Hyde LA, Bimonte HA, Hunter CL, Nelson M et al (2002) Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiol Behav 77:371–385. [DOI] [PubMed] [Google Scholar]
- 36. Granholm AC, Sanders L, Seo H, Lin L, Ford K, Isacson O (2003) Estrogen alters amyloid precursor protein as well as dendritic and cholinergic markers in a mouse model of Down syndrome. Hippocampus 13:905–914. [DOI] [PubMed] [Google Scholar]
- 37. Granholm AC, Boger HA, Emborg ME (2008) Mood, memory, and movement: an age‐related neurodegenerative complex? Current Aging Sci 1:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guan Z, Fang J (2006) Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav Immun 20:64–71. [DOI] [PubMed] [Google Scholar]
- 39. Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K et al (1988) Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS 96:379–394. [DOI] [PubMed] [Google Scholar]
- 40. Hansen JT, Sakai K, Greenamyre JT, Moran S (1995) Sprouting of dopaminergic fibers from spared mesencephalic dopamine neurons in the unilateral partial lesioned rat. Brain Res 670:197–204. [DOI] [PubMed] [Google Scholar]
- 41. Hashimoto M, Nitta A, Fukumitsu H, Nomoto H, Shen L, Furukawa S (2005) Inflammation‐induced GDNF improves locomotor function after spinal cord injury. Neuroreport 16:99–102. [DOI] [PubMed] [Google Scholar]
- 42. Hattori T, McGeer PL (1973) Synaptogenesis in the corpus striatum of infant rat. Exp Neurol 38:70–79. [DOI] [PubMed] [Google Scholar]
- 43. Hawkes CH, Del Tredici K, Braak H (2007) Parkinson's disease: a dual‐hit hypothesis. Neuropathol Appl Neurobiol 33:599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hebert MA, Gerhardt GA (1997) Behavioral and neurochemical effects of intranigral administration of glial cell line‐derived neurotrophic factor on aged Fischer 344 rats. J Pharmacol Exp Ther 282:760–768. [PubMed] [Google Scholar]
- 45. Heindel JJ (2006) Role of exposure to environmental chemicals in the developmental basis of reproductive disease and dysfunction. Semin Reprod Med 24:168–177. [DOI] [PubMed] [Google Scholar]
- 46. Ho A, Blum M (1998) Induction of interleukin‐1 associated with compensatory dopaminergic sprouting in the denervated striatum of young mice: model of aging and neurodegenerative disease. J Neurosci 18:5614–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hudson J, Granholm AC, Gerhardt GA, Henry MA, Hoffman A, Biddle P et al (1995) Glial cell line‐derived neurotrophic factor augments midbrain dopaminergic circuits in vivo . Brain Res Bull 36:425–432. [DOI] [PubMed] [Google Scholar]
- 48. Jarskog LF, Xiao H, Wilkie MB, Lauder JM, Gilmore JH (1997) Cytokine regulation of embryonic rat dopamine and serotonin neuronal survival in vitro . Int J Dev Neurosci 15:711–716. [DOI] [PubMed] [Google Scholar]
- 49. Jonakait GM (2007) The effects of maternal inflammation on neuronal development: possible mechanisms. Int J Dev Neurosci 25:415–425. [DOI] [PubMed] [Google Scholar]
- 50. Kaneko YS, Mori K, Nakashima A, Sawada M, Nagatsu I, Ota A (2005) Peripheral injection of lipopolysaccharide enhances expression of inflammatory cytokines in murine locus coeruleus: possible role of increased norepinephrine turnover. J Neurochem 94:393–404. [DOI] [PubMed] [Google Scholar]
- 51. Kawano H, Ohyama K, Kawamura K, Nagatsu I (1995) Migration of dopaminergic neurons in the embryonic mesencephalon of mice. Brain Res Dev Brain Res 86:101–113. [DOI] [PubMed] [Google Scholar]
- 52. Kazantsev AG (2009) New therapeutic strategies for treatment of neurodegenerative diseases. Curr Pharm Des 15:3917–3918. [DOI] [PubMed] [Google Scholar]
- 53. Kazantsev AG, Kolchinsky AM (2008) Central role of alpha‐synuclein oligomers in neurodegeneration in Parkinson disease. Arch Neurol 65:1577–1581. [DOI] [PubMed] [Google Scholar]
- 54. Kearns CM, Gash DM (1995) GDNF protects nigral dopamine neurons against 6‐hydroxydopamine in vivo . Brain Res 672:104–111. [DOI] [PubMed] [Google Scholar]
- 55. Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS (2000) Regional difference in susceptibility to lipopolysaccharide‐induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20:6309–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kordower JH, Mufson EJ, Granholm AC, Hoffer B, Friden PM (1993) Delivery of trophic factors to the primate brain. Exp Neurol 124:21–30. [DOI] [PubMed] [Google Scholar]
- 57. Kuno R, Yoshida Y, Nitta A, Nabeshima T, Wang J, Sonobe Y et al (2006) The role of TNF‐alpha and its receptors in the production of NGF and GDNF by astrocytes. Brain Res 1116:12–18. [DOI] [PubMed] [Google Scholar]
- 58. Lapchak PA, Miller PJ, Jiao S (1997) Glial cell line‐derived neurotrophic factor induces the dopaminergic and cholinergic phenotype and increases locomotor activity in aged Fischer 344 rats. Neuroscience 77:745–752. [DOI] [PubMed] [Google Scholar]
- 59. Lauder JM, Bloom FE (1975) Ontogeny of monoamine neurons in the locus coeruleus, raphe nuclei and substantia nigra of the rat. II. Synaptogenesis. J Comp Neurol 163:251–264. [DOI] [PubMed] [Google Scholar]
- 60. Lawson LJ, Perry VH, Dri P, Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39:151–170. [DOI] [PubMed] [Google Scholar]
- 61. Li X, Peng C, Li L, Ming M, Yang D, Le W (2007) Glial cell‐derived neurotrophic factor protects against proteasome inhibition‐induced dopamine neuron degeneration by suppression of endoplasmic reticulum stress and caspase‐3 activation. J Gerontol A Biol Sci Med Sci 62:943–950. [DOI] [PubMed] [Google Scholar]
- 62. Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F (1993) GDNF: a glial cell line‐derived neurotrophic factor for midbrain dopaminergic neurons. Science 260:1130–1132. [DOI] [PubMed] [Google Scholar]
- 63. Lindner MD, Winn SR, Baetge EE, Hammang JP, Gentile FT, Doherty E et al (1995) Implantation of encapsulated catecholamine and GDNF‐producing cells in rats with unilateral dopamine depletions and parkinsonian symptoms. Exp Neurol 132:62–76. [DOI] [PubMed] [Google Scholar]
- 64. Ling Z, Gayle DA, Ma SY, Lipton JW, Tong CW, Hong JS, Carvey PM (2002) In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disord 17:116–124. [DOI] [PubMed] [Google Scholar]
- 65. Ling Z, Chang QA, Tong CW, Leurgans SE, Lipton JW, Carvey PM (2004) Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Exp Neurol 190:373–383. [DOI] [PubMed] [Google Scholar]
- 66. Ling ZD, Chang Q, Lipton JW, Tong CW, Landers TM, Carvey PM (2004) Combined toxicity of prenatal bacterial endotoxin exposure and postnatal 6‐hydroxydopamine in the adult rat midbrain. Neuroscience 124:619–628. [DOI] [PubMed] [Google Scholar]
- 67. Liss B, Haeckel O, Wildmann J, Miki T, Seino S, Roeper J (2005) K‐ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat Neurosci 8:1742–1751. [DOI] [PubMed] [Google Scholar]
- 68. Liu YQL, Wilson B, Wu X, Qian L, Granholm AC, Crews FT, Hong JS (2008) Endotoxin induces a delayed loss of TH‐IR neurons in substantia nigra and motor behavioral deficits. Neurotoxicology 29:864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liverman CS, Kaftan HA, Cui L, Hersperger SG, Taboada E, Klein RM, Berman NE (2006) Altered expression of pro‐inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neurosci Lett 399:220–225. [DOI] [PubMed] [Google Scholar]
- 70. Lo Bianco C, Deglon N, Pralong W, Aebischer P (2004) Lentiviral nigral delivery of GDNF does not prevent neurodegeneration in a genetic rat model of Parkinson's disease. Neurobiol Dis 17:283–289. [DOI] [PubMed] [Google Scholar]
- 71. Manning‐Bog AB, Langston JW (2007) Model fusion, the next phase in developing animal models for Parkinson's disease. Neurotox Res 11:219–240. [DOI] [PubMed] [Google Scholar]
- 72. Manning‐Bog AB, Caudle WM, Perez XA, Reaney SH, Paletzki R, Isla MZ et al (2007) Increased vulnerability of nigrostriatal terminals in DJ‐1‐deficient mice is mediated by the dopamine transporter. Neurobiol Dis 27:141–150. [DOI] [PubMed] [Google Scholar]
- 73. Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I et al (2006) The time of prenatal immune challenge determines the specificity of inflammation‐mediated brain and behavioral pathology. J Neurosci 26:4752–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin‐1 beta, interleukin‐6, epidermal growth factor and transforming growth factor‐alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 180:147–150. [DOI] [PubMed] [Google Scholar]
- 75. Muller T, Blum‐Degen D, Przuntek H, Kuhn W (1998) Interleukin‐6 levels in cerebrospinal fluid inversely correlate to severity of Parkinson's disease. Acta Neurol Scand 98:142–144. [DOI] [PubMed] [Google Scholar]
- 76. Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl 60:277–290. [DOI] [PubMed] [Google Scholar]
- 77. Nakamura Y, Si QS, Kataoka K (1999) Lipopolysaccharide‐induced microglial activation in culture: temporal profiles of morphological change and release of cytokines and nitric oxide. Neurosci Res 35:95–100. [DOI] [PubMed] [Google Scholar]
- 78. Nolan Y, Vereker E, Lynch AM, Lynch MA (2003) Evidence that lipopolysaccharide‐induced cell death is mediated by accumulation of reactive oxygen species and activation of p38 in rat cortex and hippocampus. Exp Neurol 184:794–804. [DOI] [PubMed] [Google Scholar]
- 79. Nosrat CA, Tomac A, Lindqvist E, Lindskog S, Humpel C, Stromberg I et al (1996) Cellular expression of GDNF mRNA suggests multiple functions inside and outside the nervous system. Cell Tissue Res 286:191–207. [DOI] [PubMed] [Google Scholar]
- 80. Olanow CW, Tatton WG (1999) Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci 22:123–144. [DOI] [PubMed] [Google Scholar]
- 81. Oo TF, Burke RE (1997) The time course of developmental cell death in phenotypically defined dopaminergic neurons of the substantia nigra. Brain Res Dev Brain Res 98:191–196. [DOI] [PubMed] [Google Scholar]
- 82. Opler MG, Susser ES (2005) Fetal environment and schizophrenia. Environ Health Perspect 113:1239–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ota A, Kaneko YS, Mori K, Nakashima A, Nagatsu I, Nagatsu T (2007) Effect of peripherally administered lipopolysaccharide (LPS) on GTP cyclohydrolase I, tetrahydrobiopterin and norepinephrine in the locus coeruleus in mice. Stress 10:131–136. [DOI] [PubMed] [Google Scholar]
- 84. Ota A, Mori K, Kaneko YS, Nakashima A, Nagatsu I, Nagatsu T (2008) Peripheral lipopolysaccharide administration affects the olfactory dopamine system in mice. Ann N Y Acad Sci 1148:127–135. [DOI] [PubMed] [Google Scholar]
- 85. Parain K, Hapdey C, Rousselet E, Marchand V, Dumery B, Hirsch EC (2003) Cigarette smoke and nicotine protect dopaminergic neurons against the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine Parkinsonian toxin. Brain Res 984:224–232. [DOI] [PubMed] [Google Scholar]
- 86. Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A et al (1996) Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 382:73–76. [DOI] [PubMed] [Google Scholar]
- 87. Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A et al (1996) GDNF is required for kidney development and enteric innervation. Cold Spring Harb Symp Quant Biol 61: 445–457. [PubMed] [Google Scholar]
- 88. Pochon NA, Menoud A, Tseng JL, Zurn AD, Aebischer P (1997) Neuronal GDNF expression in the adult rat nervous system identified by in situ hybridization. Eur J Neurosci 9:463–471. [DOI] [PubMed] [Google Scholar]
- 89. Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B et al (2004) NADPH oxidase mediates lipopolysaccharide‐induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279:1415–1421. [DOI] [PubMed] [Google Scholar]
- 90. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS et al (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rideout HJ, Dietrich P, Savalle M, Dauer WT, Stefanis L (2003) Regulation of alpha‐synuclein by bFGF in cultured ventral midbrain dopaminergic neurons. J Neurochem 84:803–813. [DOI] [PubMed] [Google Scholar]
- 92. Roodveldt C, Christodoulou J, Dobson CM (2008) Immunological features of alpha‐synuclein in Parkinson's disease. J Cell Mol Med 12:1820–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Saliba E, Henrot A (2001) Inflammatory mediators and neonatal brain damage. Biol Neonate 79:224–227. [DOI] [PubMed] [Google Scholar]
- 94. Satoh J, Kuroda Y (2001) A polymorphic variation of serine to tyrosine at codon 18 in the ubiquitin C‐terminal hydrolase‐L1 gene is associated with a reduced risk of sporadic Parkinson's disease in a Japanese population. J Neurol Sci 189:113–117. [DOI] [PubMed] [Google Scholar]
- 95. Sauer H, Rosenblad C, Bjorklund A (1995) Glial cell line‐derived neurotrophic factor but not transforming growth factor beta 3 prevents delayed degeneration of nigral dopaminergic neurons following striatal 6‐hydroxydopamine lesion. Proc Natl Acad Sci U S A 92:8935–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schaar DG, Sieber BA, Dreyfus CF, Black IB (1993) Regional and cell‐specific expression of GDNF in rat brain. Exp Neurol 124:368–371. [DOI] [PubMed] [Google Scholar]
- 97. Schlachetzki JC, Fiebich BL, Haake E, de Oliveira AC, Candelario‐Jalil E, Heneka MT, Hull M (2010) Norepinephrine enhances the LPS‐induced expression of COX‐2 and secretion of PGE2 in primary rat microglia. J Neuroinflammation 7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Song DD, Haber SN (2000) Striatal responses to partial dopaminergic lesion: evidence for compensatory sprouting. J Neurosci 20:5102–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Springer JE, Mu X, Bergmann LW, Trojanowski JQ (1994) Expression of GDNF mRNA in rat and human nervous tissue. Exp Neurol 127:167–170. [DOI] [PubMed] [Google Scholar]
- 100. Sriram K, Miller DB, O'Callaghan JP (2006) Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor‐alpha. J Neurochem 96:706–718. [DOI] [PubMed] [Google Scholar]
- 101. Stanic D, Finkelstein DI, Bourke DW, Drago J, Horne MK (2003) Timecourse of striatal re‐innervation following lesions of dopaminergic SNpc neurons of the rat. Eur J Neurosci 18:1175–1188. [DOI] [PubMed] [Google Scholar]
- 102. Stromberg I, Bjorklund L, Johansson M, Tomac A, Collins F, Olson L et al (1993) Glial cell line‐derived neurotrophic factor is expressed in the developing but not adult striatum and stimulates developing dopamine neurons in vivo . Exp Neurol 124:401–412. [DOI] [PubMed] [Google Scholar]
- 103. Thirumangalakudi L, Prakasam A, Zhang R, Bimonte‐Nelson H, Sambamurti K, Kindy MS, Bhat NR (2008) High cholesterol‐induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem 106:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Thomas B, von Coelln R, Mandir AS, Trinkaus DB, Farah MH, Leong Lim K et al (2007) MPTP and DSP‐4 susceptibility of substantia nigra and locus coeruleus catecholaminergic neurons in mice is independent of parkin activity. Neurobiol Dis 26:312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, Olson L (1995) Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo . Nature 373:335–339. [DOI] [PubMed] [Google Scholar]
- 106. Tomac A, Widenfalk J, Lin LF, Kohno T, Ebendal T, Hoffer BJ, Olson L (1995) Retrograde axonal transport of glial cell line‐derived neurotrophic factor in the adult nigrostriatal system suggests a trophic role in the adult. Proc Natl Acad Sci U S A 92:8274–8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Trupp M, Belluardo N, Funakoshi H, Ibanez CF (1997) Complementary and overlapping expression of glial cell line‐derived neurotrophic factor (GDNF), c‐ret proto‐oncogene, and GDNF receptor‐alpha indicates multiple mechanisms of trophic actions in the adult rat CNS. J Neurosci 17:3554–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. von Boyen GB, Steinkamp M, Geerling I, Reinshagen M, Schafer KH, Adler G, Kirsch J (2006) Proinflammatory cytokines induce neurotrophic factor expression in enteric glia: a key to the regulation of epithelial apoptosis in Crohn's disease. Inflamm Bowel Dis 12:346–354. [DOI] [PubMed] [Google Scholar]
- 109. Wahner AD, Sinsheimer JS, Bronstein JM, Ritz B (2007) Inflammatory cytokine gene polymorphisms and increased risk of Parkinson disease. Arch Neurol 64:836–840. [DOI] [PubMed] [Google Scholar]
- 110. West MJ (1993) New stereological methods for counting neurons. Neurobiol Aging 14:275–285. [DOI] [PubMed] [Google Scholar]
- 111. Whitton PS (2007) Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol 150:963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zaman V, Li Z, Middaugh L, Ramamoorthy S, Rohrer B, Nelson ME et al (2003) The noradrenergic system of aged GDNF heterozygous mice. Cell Transplant 12:291–303. [PubMed] [Google Scholar]
- 113. Zaman V, Nelson ME, Gerhardt GA, Rohrer B (2004) Neurodegenerative alterations in the nigrostriatal system of trkB hypomorphic mice. Exp Neurol 190:337–346. [DOI] [PubMed] [Google Scholar]
- 114. Zaman V, Boger HA, Granholm AC, Rohrer B, Moore A, Buhusi M et al (2008) The nigrostriatal dopamine system of aging GFRalpha‐1 heterozygous mice: neurochemistry, morphology and behavior. Eur J Neurosci 28:1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Tyrosine hydroxylase (TH) immunostaining in the pontine nucleus locus coeruleus (LC, A–D) and the ventral tegmental area (VTA, E–H) did not reveal any observable morphological alterations either as a result of the partial Gdnf deletion or the prenatal lipopolysaccharide (LPS) treatment, at least not at 12 months of age. Scale bar in H represents 100 microns.
Supporting info item