

Abstract
Purpose
2ME2 (Panzem®) is a non-estrogenic derivative of estradiol with antiproliferative and antiangiogenic activity. Preclinical data support antitumor activity in prostate cancer. This trial evaluated the efficacy of 2ME2 NCD in patients with taxane-refractory, metastatic CRPC.
Experimental Design
Patients with metastatic CRPC who had progressed on only one prior taxane-based regimen were eligible. All patients received 2ME2 NCD at 1500 mg orally four times daily, repeated in 28 day cycles. The primary endpoint was progression free survival at month 6, with a secondary endpoint of PSA response. An exploratory endpoint was metabolic response on FDG-PET imaging.
Results
A total of 50 pts was planned. The study was terminated after 21 pts when a futility analysis showed the primary endpoint was unlikely to be reached. The median number of cycles on study was 2 (range <1 to 12). Adverse events (AE) of grade ≥3 related to the study drug occurred in 7 unique patients (33%): elevations in liver function tests, fatigue or weakness, gastrointestinal hemorrhage, and hyponatremia. Paired FDG-PET scans were obtained for 11 pts. No metabolic responses were observed.
Conclusions
2ME2 NCD did not appear to have clinically significant activity in this study. 2ME2 NCD was well-tolerated and showed some evidence of biologic activity. Given the aggressive biology in this taxane-refractory population, the potential benefit from a cytostatic agent like 2ME2 might better be realized in the pre-chemotherapy (or rising PSA only) stage of CRPC.
Keywords: Prostate cancer, castrate-resistant, antiangiogenesis, antiproliferative, PET scan, clinical trials
Introduction
In 2009, prostate adenocarcinoma was diagnosed in an estimated 192,280 men and resulted in an estimated 27,370 deaths in the United States [1]. Although androgen deprivation therapy is effective for advanced or recurrent disease, nearly all patients will eventually develop castrate-resistant prostate cancer (CRPC) after a variable duration of response [2–3]. The regimen of docetaxel and prednisone was approved by the United States Food and Drug Administration in 2004 based on a modest improvement (about 2.5 months) in overall survival over mitoxantrone [4] and is the standard of care for first-line treatment of CRPC. However, the duration of response remains short with a median overall survival of approximately 18 months. Currently there is no standard second-line therapy for these men after progression on a taxane-based regimen. Mitoxantrone with corticosteroids was approved for palliative benefit only (i.e. symptom palliation, without proven survival advantage) in 1996 [5–6]. Cabazitaxel with prednisone recently showed a statistically significant improvement in overall survival when compared against mitoxantrone with prednisone in men with CRPC previously treated with docetaxel [7]; however, this regimen is not yet FDA-approved and there are concerns about the toxicity profile of cabazitaxel in this population. While some men with taxane-refractory CRPC may currently go on to receive mitoxantrone, it is clear that better treatments for this population are urgently needed.
As in many solid tumors, angiogenesis appears to have an important role in prostate cancer progression. Increased microvessel density in clinically localized prostate cancer is an independent prognostic factor for progression and survival [8–9] and is associated with higher stage after radical prostatectomy as well as shorter time to recurrence after radiation therapy [10–12]. Plasma levels of vascular endothelial growth factor (VEGF), a potent and specific stimulator of endothelial cell proliferation and angiogenesis [13], are increased in patients with metastatic prostate cancer, both in comparison to patients with localized prostate cancer and normal controls [14]. VEGF level has been demonstrated to be an independent prognostic factor in men with metastatic CRPC [15]. Therefore, inhibition of angiogenesis may be a viable strategy for the treatment of CRPC.
2-methoxyestradiol (2ME2) is a naturally occurring estrogen metabolite with both anti-angiogenic and anti-proliferative activity. 2ME2 binds poorly (0.05% binding) to the estrogen receptor [16] and has not shown estrogenic activity in model systems. Preclinical data support anti-tumor activity of 2ME2 in multiple prostate cancer cell lines, including androgen-independent lines [17]. The antiangiogenic activity of 2ME2 has been demonstrated in vivo in corneal micropocket [18], chick chorioallantoic model systems [19], and Matrigel plug assays [20], as well as by the observation of reduced tumor vasculature in 2ME2-treated mice [21]. 2ME2 appears to inhibit proliferation through induction of apoptosis by activation of p53 [22] and inhibition of hypoxia-inducible factor 1 (HIF-1), an important transcription factor for angiogenesis [23]. In addition, 2ME2 binds to tubulin, inducing mitotic arrest by suppression of microtubule dynamics [24]. The tubulin interaction occurs upstream of the inhibition of HIF-1, providing a mechanistic link between the disruption of the microtubule cytoskeleton and inhibition of angiogenesis [25]. This promising preclinical data supporting antitumor activity of 2ME2 led to further testing in the clinic.
In initial clinical studies, 2ME2 was formulated as a capsule; however, although 2ME2 capsules appeared to be safe, pharmacokinetic results showed that 2ME2 capsules did not achieve sufficient sustained plasma levels of 2ME2 in order to adequately evaluate its therapeutic potential. A phase II study of 2ME2 capsules in chemotherapy-naïve, castrate-resistant prostate cancer (CRPC) patients showed decreases in PSA velocity despite suboptimal exposures of 2ME2 [26]. Therefore, 2ME2 was reformulated as a NanoCrystal® colloidal dispersion (NCD), which demonstrated an improved pharmacokinetic (PK) profile and antitumor activity in preclinical studies. In animal models, anti-cancer activity was enhanced when there was relatively constant plasma exposure to 2ME2 (i.e. when delivered by implanted osmotic pumps or multiple daily oral doses) and a target minimum effective concentration of 3.3 ng/mL was identified [27]. The recommended phase II dose of 2ME2 NCD is 1500 mg by mouth four times daily, as determined in two independent phase Ib studies [28–30]. This dose of 2ME2 NCD was tested in the present study of patients with docetaxel-refractory metastatic CRPC.
Assessing response in metastatic CRPC is difficult due to the disease manifesting primarily as bony metastases, which led to a search for legitimate biomarkers of response. Prostate-specific antigen (PSA) response has been an acceptable surrogate with conventional cytotoxic chemotherapy [31]; however, with novel agents like 2ME2 that are presumably cytostatic, PSA response might not accurately predict clinical benefit. Although paired tumor biopsies (pre- and post-treatment) have provided the primary source for examination of surrogate markers of antiangiogenic therapies, biopsies of osseous metastases in CRPC are invasive and not clinically practical. Fluorodeoxyglucose positron emission tomography (FDG-PET) scanning is a non-invasive, widely available, and technically consistent means to assess the activity of new agents in patients on phase II trials by imaging early tumor-related changes [32]. Although early studies of FDG-PET in prostate cancer suggested a much lower sensitivity for bone metastases than reference bone scans, many of these studies were performed in patients at heterogeneous stages of disease [33]. Concordance of FDG-PET with bone scan findings and PSA changes were much higher, with a greater potential of PET over bone scan in discriminating active lesions from quiescent disease, when studied in more uniform patient populations, such as those with androgen-independent disease [34–35]. A more recent study, in patients with metastatic CRPC undergoing anti-microtubule cytotoxic chemotherapy, revealed a greater than 90% accuracy of FDG-PET in correctly identifying the patients’ response to treatment at 4 and 12 weeks, based on comparison with standard outcomes with PSA measurements, bone scans and computed tomography (CT). The authors concluded that FDG-PET is promising outcome measure in metastatic CRPC for dichotomizing patients as progressors or nonprogressors [36]. For these reasons, FDG-PET was evaluated as a biomarker in the present study.
We conducted a phase II trial of 2ME2 NCD in patients with taxane-refractory, metastatic castrate-resistant prostate cancer. The primary endpoint was progression-free survival (PFS) at 6 months. Secondary endpoints included assessment of PSA response (PCWG1 consensus criteria [31]) and objective response rate by RECIST [37], as well as safety evaluation. An exploratory endpoint was the metabolic response on FDG-PET imaging in a subset of patients with PET-positive metastases. Pharmacokinetics samples were collected to assess the steady-state levels of 2ME2 and its primary metabolite, 2ME1.
Materials and Methods
Study population
To participate in this study, patients were required to have histologically or cytologically confirmed adenocarcinoma of the prostate with evidence of progressive metastatic disease despite prior androgen deprivation therapy (including castration) and a castrate level of testosterone (< 50 ng/dL). Patients were also required to have received only one prior taxane-based regimen for metastatic disease, with evidence of disease progression during treatment or within 6 months of treatment discontinuation. Up to one other prior experimental therapy was permitted. Progression was defined as: new lesions on bone scan (≥1), new/enlarging lesions on computerized tomography (CT) scan by RECIST, or known metastatic disease and rising PSA. Scans were reviewed by independent investigators at each study site for determination of eligibility based on these criteria for progression. Confirmatory scans were not required. In addition, patients with bone metastases only (i.e. lacking soft-tissue disease) were required to have a PSA level of 10 ng/mL or higher. Patients with soft tissue metastases and/or visceral disease were required to have either measurable disease by RECIST or a PSA level of 10 ng/mL or higher. Patients with stable metastatic disease and rising PSA were required to have two consecutive rises in PSA measurement, each separated from the previous by a ≥ two weeks with the most recent PSA value obtained within 1 week prior to registration.
Eligible patients were ≥18 years of age; underwent appropriate antiandrogen withdrawal intervals (≥4 weeks for flutamide, ≥6 weeks for bicalutamide or flutamide) with continued progression confirmed by rising PSA; had no other malignancies within 5 years (excluding non-melanoma skin cancers treated with curative intent); had Eastern Cooperative Oncology Group (ECOG) performance status of two or less; had life expectancy of greater than 12 weeks; and had near normal organ and marrow function within 2 weeks prior to registration (granulocytes > 1500/mm3, platelet count > 100,000/mm3, bilirubin < 1.5 mg/dL, serum glutamate pyruvate transaminase [ALT] < 2 times the institutional upper limit of normal, and creatinine < 1.5 mg/dL or a calculated creatinine clearance > 50 mL/min).
Patients were excluded from the study if they had prior therapy with radioisotopes, active angina pectoris, known heart disease of New York Heart Association Class III to IV, ventricular dysrhythmias, other unstable arrhythmias, known history of carcinomatous meningitis or brain metastases, or major surgery within 21 days of starting treatment. Patients could not have had prior radiotherapy within 4 weeks prior to registration; if palliative radiotherapy had been given, two consecutive rises in PSA values were necessary, each separated from the previous value by at least of two weeks (with baseline value obtained after completion of radiation treatment). No concurrent use of estrogen, or estrogen-like agents (including saw palmetto and other herbal products containing phytoestrogens), or any other hormonal therapy (including megestrol acetate, finasteride, ketoconazole, and systemic corticosteroids) were allowed at any time during the study. Prior use of these agents was discontinued ≥ 4 weeks prior to enrollment followed by confirmation of disease progression as above. Patients with type I insulin-dependent diabetes, poorly-controlled type II insulin-dependent diabetes, or a fasting blood glucose of more than 10 mmol/L (200 mg/dL) were excluded due to difficulty evaluating the tumor metabolic activity using FDG-PET under these conditions. The use of bisphosphonate therapy was allowed provided that the patient had been receiving the therapy for 4 weeks or more with evidence of progressive disease as outlined above; however, patients were not allowed to start bisphosphonate therapy while receiving protocol treatment unless clinically indicated (which required approval of the study principal investigator). Those patients already on a bisphosphonate therapy were allowed to continue to receive the bisphosphonate as previously scheduled.
Before implementation, this study was approved by the Institutional Review Board of the University of Wisconsin-Madison and each of the participating clinical sites. All patients gave written, informed consent before study entry.
Pretreatment evaluation and follow-up studies
Evaluations done at baseline and on day 1 of every 28-day cycle included a physical exam with assessment of weight, vital signs, and ECOG performance status; complete blood count including a differential and platelet count; serum chemistry (albumin, alkaline phosphatase, total bilirubin, bicarbonate, BUN, calcium, chloride, creatinine, glucose, GGT, LDH, potassium, total protein, AST, ALT, sodium, magnesium, and phosphorus, and uric acid), testosterone, and PSA. Coagulation studies and urinalysis were obtained at baseline only. Radiographic evaluation at baseline (within 4 weeks prior to start of therapy) and after every even cycle consisted of a computed tomography scan of the abdomen and pelvis; a chest x-ray or chest CT (if abnormal chest x-ray or known lung metastasis); and a whole body bone scan. Baseline FDG-PET was obtained within 14 days (preferably within 7 days) of the first 2ME2 NCD dose with follow-up scan at 28 days +/− 5 days after treatment started. A subset of 20 patients with matched sets of FDG-PET scans (2 total) was planned; therefore, based on the 65% FDG-PET positivity rate reported in a similar population, it was anticipated at least 30 patients would need to be screened to obtain the 20 matched sets. Plasma levels of 2ME2 and 2ME1 were drawn at baseline and repeated at the beginning of each new cycle of therapy prior to the next scheduled dose of 2ME2 NCD (trough levels).
Study design and treatment schedule
This was a single-arm, phase II efficacy and pharmacodynamic study of 2-methoxyestradiol (2ME2) NanoCrystal® Dispersion (NCD). 2ME2 NCD (Panzem® NCD) was provided by EntreMed, Inc. (Rockville, MD). Patients took 1500 mg of 2ME2 NCD by mouth four times daily in 28 day cycles. Patients were instructed to refrain from eating for 1 hour before and 30 minutes following all 2ME2 doses in order to improve absorption [30]. If patients missed a scheduled dose, they were instructed to take 2ME2 promptly and continue treatment on schedule. 2ME2 was supplied as a 100 mg/mL colloidal dispersion in 8-ounce bottles. Up to a 28-day supply was dispensed to patients. Patients were asked to refrigerate their 28-day supply of drug. Patients who had not undergone bilateral orchiectomy continued their luteinizing hormone-releasing hormone (LHRH) agonist therapy (e.g., leuprolide or goserelin) or LHRH antagonist (e.g. abarelix) while receiving protocol therapy.
Toxicity and dose modifications
Toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. All treated patients were considered evaluable for toxicity.
The first dose reduction for 2ME2 NCD resulted in a 25% decrease compared with the initial dose (i.e. to 4500 mg daily). A second dose reduction mandated a 50% decrease from the initial dose (i.e. to 3000 mg daily). A third dose reduction required a 66% decrease of the initial dose (i.e. to 2000 mg daily). Any patient on the third dose level who would have required further dose reduction according to protocol guidelines was removed from study.
Any grade ≤ 3 diarrhea, constipation, or nausea that resolved to baseline promptly (within 2 days) with best supportive care (i.e. antidiarrheal, stool softener/laxative, or antiemetic medications) did not require a dose reduction. However, recurrent symptoms despite best supportive care required a reduction as outlined above. Subjectively intolerable grade 2 toxicity necessitated holding 2ME2 NCD for up to 2 weeks until recovery to grade 2 or better and restarting at same dose level. Any other grade 3 or higher toxicity or recurrent subjectively unbearable grade 2 toxicity mandated discontinuation of 2ME2 NCD until recovery (up to 2 weeks) followed by resumption of the study drug at the next lower dose level. In general, adverse events that did not resolve to grade 2 or below within 2 weeks of drug discontinuation resulted in patient removal from the study. All dose reductions were permanent for the duration of the study, unless the study sponsor and principal investigator agreed otherwise.
Statistical methods and analysis
The primary efficacy endpoint was progression-free survival (PFS) at 6 months for all patients receiving at least one dose of study medication. PFS was defined as the duration of time from the start of treatment to the time of radiographic or symptomatic progression. A change in PSA by itself was not used to define progressive disease, unless accompanied by either symptomatic deterioration or radiographic progression. RECIST were used to determine radiographic progression. For non-target lesions, appearance of one or more new lesions and/or unequivocal progression of existing non-target lesions defined progression. Any new symptomatic lesion seen on bone scan was considered progressive disease. Scans were reviewed by independent investigators at each study site. Confirmatory scans were required only to confirm partial response (PR) or complete response (CR). Secondary endpoints included objective response rate by RECIST, rate of ≥ 50% decline in the level of PSA (PCWG1 consensus criteria [31]), and evaluation of safety. Exploratory endpoints were determination of 2ME2 and 2ME1 plasma levels and evaluation of metabolic response using FDG-PET.
This study was designed as a single cohort, historically-controlled pilot study. The sample size for the study was based on the PFS at month 6. Historically, at the 3 month time point, patients with metastatic, docetaxel-refractory, castrate-resistant disease had approximately a 50% progression rate, regardless of the exact chemotherapy regimen used [38–39]. Therefore, this population was projected to have a 75% progression rate at month 6 (e.g. PFS of 25%). We hypothesized that 2ME2 NCD would lower this progression rate to 55% (e.g. increase PFS to 45%). The study was designed to enroll up to 50 patients to test the hypothesis that 2ME2 would increase PFS at month 6 by 20%. This fixed sample design tested the null hypothesis that the proportion of PFS at month 6 is at most 25% against the alternative hypothesis that it is at least 45%. Error probabilities of one-sided α = 0.10 and β = 0.05 were used. If 16 or more patients were progression-free at month 6, the regimen was to be considered to warrant further study.
Efficacy analyses of PFS were conducted on an intention-to-treat basis (i.e. all enrolled patients were considered evaluable). Baseline characteristics were summarized using standard descriptive statistics. The PFS curve was calculated using Kaplan-Meier methodology.
Pharmacokinetics
Plasma samples were analyzed for 2ME2 and 2ME1 using validated LC/MS/MS assays with quantifiable limits of 1 ng/mL for both compounds by PPD, Inc. (Madison, WI). The plasma concentrations were used as reported by the bioanalytical laboratory. Standard deviation was not calculated with n <3.
FDG-PET imaging
Participation in the cohort of pharmacodynamic assessment using FDG-PET scanning was required until twenty complete sets of FDG-PET scans were obtained. Based on prior studies [34], the FDG-PET positivity rate was estimated at 70%. Therefore, it was anticipated that approximately 30 patients would need to be screened to obtain 20 complete sets of FDG-PET scans. FDG uptake in primary tumor and metastasis (up to a maximum of three lesions, including the biopsied lesion) were analyzed semi-quantitatively by the maximum Standardized Uptake Value (SUVmax) for body weight calculated according to the following equation:
In cases where there were more than three lesions, the three lesions with maximal uptake were selected from the baseline scan. The location of abnormalities was recorded and the same lesions were followed on subsequent scans. The presence of new lesions was also recorded. FDG-PET metabolic response was assessed based on the European Organization for Research and Treatment of Cancer (EORTC) guidelines [40] and the average tumor SUVmax for the target lesions selected.
Early study closure
The study was terminated after 21 patients had been enrolled. An unplanned futility analysis showed that the primary endpoint was unlikely to be reached, because at that time no patients had remained on study for more than 6 months. Patient accrual was formally discontinued in March of 2008, when the final patient remaining on study had been followed for 6 months. After that time, the one remaining active patient continued to be followed with safety assessments only.
Results
Patient characteristics
Twenty-one patients were enrolled between November 2006 and September 2007. The baseline clinical and biological characteristics are presented in Table 1. The median age of patients was 68 years (range, 49–80) and median Gleason score was 8 (range 6 – 9), although 12 patients (60%) had a Gleason score of 8 or 9. In 3 patients, Gleason scoring was either not performed or not available at baseline. Median PSA concentration at study entry was 140 ng/mL (range, 6.9 – 4784.7 ng/mL). All patients had received docetaxel and none had received paclitaxel. Of these patients, 81% had bone metastases, with the remainder having only soft tissue metastases. Bone-only metastases were present in 33% of patients.
Table 1.
Patient characteristics at baseline. Patients were counted once per category or subcategory of prior therapy and once per metastatic site category.
| Number of Patients | 21 |
| Age (years), median and range | 68 (49 – 80) |
| Median Time from Initial Diagnosis (yrs) | 5 (2 – 14) |
| Gleason Score, median and range (n = 18*) | 8 (6 – 9) |
| PSA Baseline (ng/ml), median and range | 140 (6.9 – 4784.7) |
| Number (%) | |
| ECOG Performance Status (baseline) | |
| 0 | 10 (48%) |
| 1 | 11 (52%) |
| Race | |
| Caucasian | 19 (90%) |
| African-American | 2 (10%) |
| Prior Therapy | |
| Surgery (excluding orchiectomy) | 10 (48%) |
| Hormonal | 21 (100%) |
| Radiotherapy | 14 (67%) |
| Immunotherapy | 1 (5%) |
| Chemotherapy | |
| Docetaxel | 21 (100%) |
| Mitoxantrone | 0 |
| Experimental | 11 (52%) |
| Metastatic Sites | |
| Bone | 17 (81%) |
| Lymph Node | 13 (62%) |
| Lung | 4 (19%) |
| Liver | 1 (5%) |
| Adrenal Gland | 1 (5%) |
Gleason scores were not available for three patients.
Exposure to study medication
The median number of cycles on study was 2 (range, <1 – 12). See table 2 for reasons for study discontinuation. Protocol-mandated dose reductions were necessary for 4 patients.
Table 2.
Reasons for study discontinuation. Patients may be listed in more than one category, as sites were allowed to choose more than one reason for study termination. One patient refused to return for follow-up imaging.
| Reason | Number | % |
|---|---|---|
| Disease Progression | 17 | 81.0 |
| Adverse Event(s) | 7 | 33.3 |
| Clinical or symptomatic disease progression | 2 | 9.5 |
| Noncompliance with follow-up | 1 | 4.8 |
Disease progression
The Kaplan-Meier curve for progression-free survival (PFS) is shown in Figure 1, which includes the 21 patients in the intention-to-treat population. One patient discontinued study treatment at day 329. This patient was alive and without recorded progression of disease at that time and was therefore censored at the discontinuation date. The 6 month PFS rate was only 5.35% (95% confidence interval: 0.795% – 36.0%) and the median PFS was 56 days (95% CI: 53–56 days).
Figure 1.
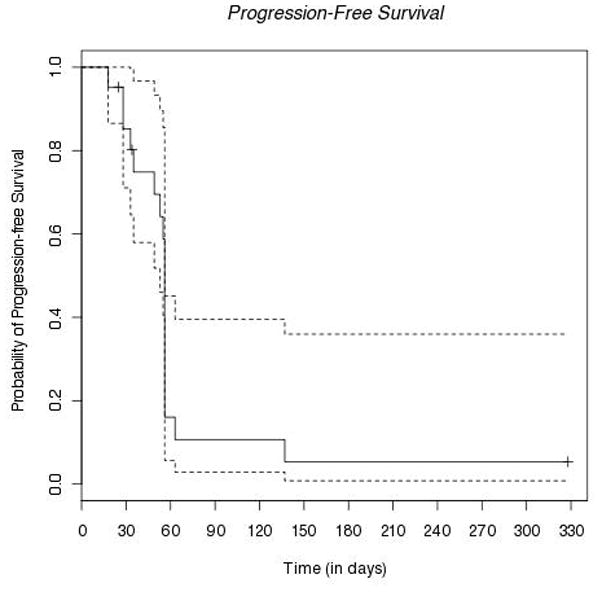
Kaplan-Meier plot of progression-free survival in the intention-to-treat population (solid line). Dashed lines indicate the upper and lower 95% confidence interval bands. One subject terminated at day 329 alive and without recorded progression of disease.
Response to therapy
There were no objective responses by RECIST. One patient had a ≥ 50% decline in PSA after cycle 1 but developed progression by RECIST after 2 cycles of therapy (without PSA progression). Another patient continued on study for 12 cycles of treatment with stable disease by RECIST, after which he developed evidence of progression of disease by bone scan, increasing PSA, and increased fatigue. Figure 2 shows the maximal percent change in PSA at any time point compared with the baseline value (waterfall plot).
Figure 2.

Waterfall plot showing maximal prostate-specific antigen (PSA) change post-therapy (at any time point) compared with baseline (n = 18). Three patients discontinued the study before a post-therapy PSA was drawn. Values are truncated at +100%.
Adverse events
Adverse event (AE) data are available on all 21 patients. 2ME2 NCD was generally well-tolerated. Table 3 lists grade 3 or 4 AEs listed as possibly, probably, or definitely related to the study drug. The least complimentary causality was used. A Grade 3 or 4 AE occurred in 7 unique patients (33%). Elevated liver function tests (alanine transaminase, aspartate transaminase, alkaline phosphatase, gamma glutamyl transpeptidase) occurred in 4 unique patients (19%). Most frequent AE, regardless of grade or attribution, were: pain (55%), nausea (55%), fatigue (45%), anorexia (35%), vomiting (25%) and diarrhea (25%).
Table 3.
Grade 3 or 4 adverse events listed as possibly, probably, or definitely related to the study drug. ALT alanine transamine. AST, aspartate transaminase. ALP, alkaline phosphatase. GGT, gamma glutamyl transpeptidase. GI, gastrointestinal.
| Event (n = 21) | Grade 3 | Grade 4 | Total (%) |
|---|---|---|---|
| Elevated ALT | 2 | 1 | 3 (14%) |
| Elevated AST | 1 | 1 | 2 (10%) |
| Elevated ALP | 1 | 0 | 1 (5%) |
| Elevated GGT | 0 | 1 | 1 (5%) |
| Fatigue or weakness | 2 | 0 | 2 (10%) |
| GI hemorrhage | 1 | 0 | 1 (5%) |
| Hyponatremia | 1 | 0 | 1 (5%) |
| Total | 8 | 3 | 11 |
Pharmacokinetics
Plasma concentrations of 2ME2 and 2ME1 after oral administration of Panzem® NCD four times daily are shown in Figure 3, and were available for 14 patients enrolled in 4 centers. Trough plasma concentrations of 2ME2 and 2ME1 were fairly constant and trough plasma concentrations of 2ME1 were 10 to 20 times higher than plasma concentrations of 2ME2 as expected from previous studies with this agent.
Figure 3.

Pharmacokinetics. Plasma Concentrations of 2ME2 and 2ME2 in Prostate Cancer Patients after Oral Administration of 1500 mg Panzem® NCD Four Times Daily.
2ME2 was not quantifiable in any sample collected before administration of the first dose of Cycle 1. Overall, variations in trough concentrations between patients were small with a coefficient of variance (CV) of 49% (n = 13) before treatment Cycle 2 and a CV of 35% (n = 6) before treatment Cycle 3. The intra-patient variability was less than 100% in patients with more than one plasma concentration determination. Mean trough concentrations of 2ME2 before each treatment cycle ranged between 12.70 and 16.98 ng/mL.
2ME1 was also not quantifiable in any sample collected before administration of the first dose of Cycle 1. Overall, variations in trough concentrations between patients were small with a CV of 63% (n = 13) before treatment Cycle 2 and a CV of 14% (n = 6) before treatment Cycle 3. The intra-patient variability was less than 100% in patients with more than one plasma concentration determination. Mean trough concentrations of 2ME1 before each treatment cycle ranged between 209.00 and 302.54 ng/mL.
FDG-PET imaging
Baseline scans were obtained on 16 patients, of which 11 patients had paired FDG-PET scans and were evaluable for metabolic response. No metabolic responses were observed as defined by the protocol (EORTC guidelines [40]). Mean SUVmax change was +7.73% (range, −27.05% to +20.45%). The patient who had a mean SUVmax change of −27.05% also had a new cervical spine lesion on his follow up PET scan.
Discussion
Patients with metastatic, castrate-resistant prostate cancer (CRPC) who progress on docetaxel currently have no standard options available to them [41] and few treatment options overall. In this poor-prognosis patient population, new treatment strategies are urgently needed. There is strong rationale for targeting angiogenesis in this setting, as angiogenesis appears to play a key role in prostate cancer progression [42]. 2-methoxyestradiol (2ME2) is a non-estrogenic derivative of estradiol with both antiangiogenic and antiproliferative activity that has been re-formulated as NanoCrystal® colloidal dispersion (NCD), with improved pharamacokinetics and antitumor activity compared with the original capsule form.
This phase II, single-arm, open-label study tested the hypothesis that 2ME2 NCD would improve the progression-free survival (PFS) at 6 months over historical controls, from 25% to 45%. Unfortunately, the observed PFS at 6 months was only 5%. With the NCD formulation, plasma concentrations of 2ME2 necessary for antitumor effect as predicted by preclinical data (e.g. ≥3.3 ng/mL) were achieved. Regrettably, 2ME2 NCD did not appear to have clinically significant activity in this study and cannot be recommended for use as monotherapy in men with taxane-refractory CRPC.
The time on-study was short, at a median of 2 cycles. 2ME2 NCD was relatively well-tolerated, with the main drug-related grade 3 or 4 adverse events being elevated liver function tests. There was some evidence of biologic activity, with one PSA response (PCWG1 consensus criteria [31]) and one other patient who remained on-study for 12 cycles with stable disease. However, given the aggressive biology in this taxane-refractory population, the potential benefit from a cytostatic agent like 2ME2 NCD might better be realized in the pre-chemotherapy (or rising PSA-only) stage of CRPC. A 2ME2 analog with increased bioavailability and anti-tumor activity has completed Phase I clinical testing [43].
One strength of our study is the design, which incorporated a biomarker (FDG-PET imaging) as a secondary endpoint into a standard, historically-controlled single-arm study with PFS as the primary endpoint. A weakness in the study design is that FDG-PET appears to be useful mainly for dichotomizing patients with CRPC treated with antimicrotubule therapy into progressors and nonprogressors [36], so FDG-PET should have been evaluated as a biomarker for progression (instead of response). The use of randomized phase II clinical trials in oncology has increased with the advent of molecularly targeted agents [44]. Experts have made disparate recommendations as to how often randomized phase II design should be employed, from recommending use in “select circumstances” by an international task force [45] to recommending use as a “standard approach” by Ratain et al. [46]. However, historically-controlled studies are more statistically efficient, requiring significantly fewer patients, oftentimes fewer than half the number required for a randomized trial. With the large number of anti-cancer agents currently in development, maximizing patient exposure to effective versus ineffective agents will be crucial. The incorporation of functional and/or molecular imaging has the potential to make clinical trials more efficient by predicting early responders to treatment and thereby shortening trials of ineffective agents [47]. Molecular imaging also represents a possible intermediary between treatment exposure and survival as an outcome. However, molecular imaging and other prospective biomarkers (e.g. enumeration of circulating tumor cells [CTC]) will need to undergo rigorous surrogate evaluation before they can be used in larger clinical trials as primary endpoints.
The results of several phase II trials of antiangiogenic agents as monotherapy have been reported in the docetaxel-refractory CRPC population, including with the vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitors (TKI) sunitinib and sorafenib. It should be noted that sunitinib and sorafenib both have some antiproliferative activity as well. Two small studies tested sunitinib on the 4 weeks on, 2 weeks off schedule. In the first study by Periman et al. in 19 patients, the primary objective of PFS at 12 weeks was 75.8% and PSA decline ≥ 50% was documented in 4 patients (12.1%) [48–49]. A second study by Michaelson et al. in 17 patients (Group B, docetaxel-refractory) observed only one ≥ 50% PSA decline and seven men with stable PSA at 12 weeks [50]. The authors conclude that since assessments of radiographic disease status were often discordant with changes in PSA, alternate end points are important in future trials. In a study by Dahut et al., in which 55% of the 22 enrolled patients had received prior docetaxel, sorafenib provided some evidence of benefit by clinical criteria (two patients with improved bony metastatic lesions), but not PSA criteria or RECIST [51]. Interestingly, in this study there were also discordant PSA and radiographic responses; therefore, PSA increase was removed as a criterion of disease progression. These studies highlight the overall disappointing results for anti-angiogenic agents as monotherapy in the setting of docetaxel-refractory CRPC and the challenges of using PSA as an endpoint in trials of novel biologic agents.
In summary, 2ME2 (Panzem®) NCD did not have clinically significant activity in this population of men with docetaxel-refractory CRPC. However, interest remains in the clinical evaluation of anti-angiogenic agents for the treatment of prostate cancer. It will be crucial to identify the subset of patients who may best benefit from these agents. Because of the aggressive biology of docetaxel-refractory CRPC, conventional wisdom held that anti-angiogenic agents might best be combined with cytotoxic chemotherapy in this disease stage. However, preliminary reports indicate that CALGB 90401, a randomized phase III study evaluating docetaxel chemotherapy and prednisone with or without bevacizumab in men with chemotherapy-naïve CRPC, failed to meet its primary endpoint of extending overall survival [52]. This data has been submitted for presentation at the American Society of Clinical Oncology (ASCO) annual meeting in June 2010. Clearly, much work remains to determine how to measure clinical benefit most expeditiously in early-phase trials and to find the combination schedule and dosing that optimize the efficacy to toxicity ratio of antiangiogenic therapies in CRPC.
Acknowledgments
We thank our patients and their families for their participation in this study. Special thanks to the investigators and staff of the Genitourinary Program at the University of Wisconsin Carbone Cancer Center, Indiana University Simon Cancer Center, Johns Hopkins Kimmel Comprehensive Cancer Center, and Dana-Farber Cancer Institute.
Grant Support: EntreMed, Inc. (sponsor/funding); Department of Defense Prostate Cancer Clinical Trial Consortium Award (P30 CA14520); Prostate Cancer Foundation Therapy Consortium.
Footnotes
ClinicalTrials.gov Identifier: NCT00394810
Prior Presentations: ASCO 2008 Annual Meeting, abstract 5173.
This clinical research article is original work and has not been published or submitted elsewhere.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics, 2009. CA Cancer J Clin. 2009 doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Crawford ED, Eisenberger MA, McLeod DG, Spaulding JT, Benson R, Dorr FA, et al. A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. N Engl J Med. 1989;321:419–24. doi: 10.1056/NEJM198908173210702. [DOI] [PubMed] [Google Scholar]
- 3.Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 4.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 5.Tannock IF, Osoba D, Stockler MR, Ernst DS, Neville AJ, Moore MJ, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol. 1996;14:1756–64. doi: 10.1200/JCO.1996.14.6.1756. [DOI] [PubMed] [Google Scholar]
- 6.Kantoff PW, Halabi S, Conaway M, Picus J, Kirshner J, Hars V, et al. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the cancer and leukemia group B 9182 study. J Clin Oncol. 1999;17:2506–13. doi: 10.1200/JCO.1999.17.8.2506. [DOI] [PubMed] [Google Scholar]
- 7.Sartor AO, Oudard S, Ozguroglu M, Hansen S, Machiels JH, Shen L, et al. Cabazitaxel or mitoxantrone with prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC) previously treated with docetaxel: Final results of a multinational phase III trial (TROPIC). ASCO Genitourinary Cancers Symposium 2010; San Francisco, CA. 2010. [Google Scholar]
- 8.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143:401–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Borre M, Offersen BV, Nerstrom B, Overgaard J. Microvessel density predicts survival in prostate cancer patients subjected to watchful waiting. Br J Cancer. 1998;78:940–4. doi: 10.1038/bjc.1998.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettencourt MC, Bauer JJ, Sesterhenn IA, Connelly RR, Moul JW. CD34 immunohistochemical assessment of angiogenesis as a prognostic marker for prostate cancer recurrence after radical prostatectomy. J Urol. 1998;160:459–65. [PubMed] [Google Scholar]
- 11.Brawer MK, Deering RE, Brown M, Preston SD, Bigler SA. Predictors of pathologic stage in prostatic carcinoma. The role of neovascularity. Cancer. 1994;73:678–87. doi: 10.1002/1097-0142(19940201)73:3<678::aid-cncr2820730329>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 12.Hall MC, Troncoso P, Pollack A, Zhau HY, Zagars GK, Chung LW, et al. Significance of tumor angiogenesis in clinically localized prostate carcinoma treated with external beam radiotherapy. Urology. 1994;44:869–75. doi: 10.1016/s0090-4295(94)80173-8. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara N. The role of vascular endothelial growth factor in pathological angiogenesis. Breast Cancer Res Treat. 1995;36:127–37. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 14.Duque JL, Loughlin KR, Adam RM, Kantoff PW, Zurakowski D, Freeman MR. Plasma levels of vascular endothelial growth factor are increased in patients with metastatic prostate cancer. Urology. 1999;54:523–7. doi: 10.1016/s0090-4295(99)00167-3. [DOI] [PubMed] [Google Scholar]
- 15.George DJ, Halabi S, Shepard TF, Vogelzang NJ, Hayes DF, Small EJ, et al. Prognostic significance of plasma vascular endothelial growth factor levels in patients with hormone-refractory prostate cancer treated on Cancer and Leukemia Group B 9480. Clin Cancer Res. 2001;7:1932–6. [PubMed] [Google Scholar]
- 16.Martucci C, Fishman J. Uterine estrogen receptor binding of catecholestrogens and of estetrol (1,3,5(10)-estratriene-3,15alpha,16alpha,17beta-tetrol) Steroids. 1976;27:325–33. doi: 10.1016/0039-128x(76)90054-4. [DOI] [PubMed] [Google Scholar]
- 17.Qadan LR, Perez-Stable CM, Anderson C, D'Ippolito G, Herron A, Howard GA, et al. 2-Methoxyestradiol induces G2/M arrest and apoptosis in prostate cancer. Biochem Biophys Res Commun. 2001;285:1259–66. doi: 10.1006/bbrc.2001.5320. [DOI] [PubMed] [Google Scholar]
- 18.Klauber N, Parangi S, Flynn E, Hamel E, D'Amato RJ. Inhibition of angiogenesis and breast cancer in mice by the microtubule inhibitors 2-methoxyestradiol and taxol. Cancer Res. 1997;57:81–6. [PubMed] [Google Scholar]
- 19.Yue TL, Wang X, Louden CS, Gupta S, Pillarisetti K, Gu JL, et al. 2-Methoxyestradiol, an endogenous estrogen metabolite, induces apoptosis in endothelial cells and inhibits angiogenesis: possible role for stress-activated protein kinase signaling pathway and Fas expression. Mol Pharmacol. 1997;51:951–62. doi: 10.1124/mol.51.6.951. [DOI] [PubMed] [Google Scholar]
- 20.Sweeney CJ, Miller KD, Sissons SE, Nozaki S, Heilman DK, Shen J, et al. The antiangiogenic property of docetaxel is synergistic with a recombinant humanized monoclonal antibody against vascular endothelial growth factor or 2-methoxyestradiol but antagonized by endothelial growth factors. Cancer Res. 2001;61:3369–72. [PubMed] [Google Scholar]
- 21.Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, et al. The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature. 1994;368:237–9. doi: 10.1038/368237a0. [DOI] [PubMed] [Google Scholar]
- 22.LaVallee TM, Zhan XH, Johnson MS, Herbstritt CJ, Swartz G, Williams MS, et al. 2-methoxyestradiol up-regulates death receptor 5 and induces apoptosis through activation of the extrinsic pathway. Cancer Res. 2003;63:468–75. [PubMed] [Google Scholar]
- 23.Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Updat. 2003;6:355–61. doi: 10.1016/j.drup.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Attalla H, Makela TP, Adlercreutz H, Andersson LC. 2-Methoxyestradiol arrests cells in mitosis without depolymerizing tubulin. Biochem Biophys Res Commun. 1996;228:467–73. doi: 10.1006/bbrc.1996.1683. [DOI] [PubMed] [Google Scholar]
- 25.Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–75. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 26.Sweeney C, Liu G, Yiannoutsos C, Kolesar J, Horvath D, Staab MJ, et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin Cancer Res. 2005;11:6625–33. doi: 10.1158/1078-0432.CCR-05-0440. [DOI] [PubMed] [Google Scholar]
- 27.Folger W, Volker K, Swartz G, et al. The antitumor activity of 2ME2 is maximized by maintaining a threshold concentration within a 24-hour dosing period. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; 2006; Washington, D.C.. 2006. [Google Scholar]
- 28.Liu G, Quon C, Sidor C, Feierabend C, Eun J, Alberti D, et al. Phase I trial of 2-methoxyestradiol (2ME2), administered orally as a Nanocrystal® Colloidal Dispersion (NCD), in patients with advanced cancer. Clin Can Res. 2005;11:B14. [Google Scholar]
- 29.Sweeney C, Porter J, Selbe K, Treston A, Quon C, Sidor C. A single-center, safety and pharmacokinetic study of 2-methoxyestradiol Nanocrystal® Colloidal Dispersion (Panzem® NCD), administered orally to patients with advanced cancer. Clin Can Res. 2005;11:B121. [Google Scholar]
- 30.Tevaarwerk AJ, Holen KD, Alberti DB, Sidor C, Arnott J, Quon C, et al. Phase I trial of 2-methoxyestradiol NanoCrystal dispersion in advanced solid malignancies. Clin Cancer Res. 2009;15:1460–5. doi: 10.1158/1078-0432.CCR-08-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bubley GJ, Carducci M, Dahut W, Dawson N, Daliani D, Eisenberger M, et al. Eligibility and response guidelines for phase II clinical trials in androgen-independent prostate cancer: recommendations from the Prostate-Specific Antigen Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 32.Weber WA. Positron emission tomography as an imaging biomarker. J Clin Oncol. 2006;24:3282–92. doi: 10.1200/JCO.2006.06.6068. [DOI] [PubMed] [Google Scholar]
- 33.Fogelman I, Cook G, Israel O, Van der Wall H. Positron emission tomography and bone metastases. Semin Nucl Med. 2005;35:135–42. doi: 10.1053/j.semnuclmed.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Morris MJ, Akhurst T, Osman I, Nunez R, Macapinlac H, Siedlecki K, et al. Fluorinated deoxyglucose positron emission tomography imaging in progressive metastatic prostate cancer. Urology. 2002;59:913–8. doi: 10.1016/s0090-4295(02)01509-1. [DOI] [PubMed] [Google Scholar]
- 35.Kurdziel KA, Figg WD, Carrasquillo JA, Huebsch S, Whatley M, Sellers D, et al. Using positron emission tomography 2-deoxy-2-[18F]fluoro-D-glucose, 11CO, and 15O-water for monitoring androgen independent prostate cancer. Mol Imaging Biol. 2003;5:86–93. doi: 10.1016/s1536-1632(03)00039-8. [DOI] [PubMed] [Google Scholar]
- 36.Morris MJ, Akhurst T, Larson SM, Ditullio M, Chu E, Siedlecki K, et al. Fluorodeoxyglucose positron emission tomography as an outcome measure for castrate metastatic prostate cancer treated with antimicrotubule chemotherapy. Clin Cancer Res. 2005;11:3210–6. doi: 10.1158/1078-0432.CCR-04-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 38.Michels J, Montemurro T, Murray N, Kollmannsberger C, Nguyen Chi K. First- and second-line chemotherapy with docetaxel or mitoxantrone in patients with hormone-refractory prostate cancer: does sequence matter? Cancer. 2006;106:1041–6. doi: 10.1002/cncr.21695. [DOI] [PubMed] [Google Scholar]
- 39.Ross RW, Beer TM, Jacobus S, Bubley GJ, Taplin ME, Ryan CW, et al. A phase 2 study of carboplatin plus docetaxel in men with metastatic hormone-refractory prostate cancer who are refractory to docetaxel. Cancer. 2008;112:521–6. doi: 10.1002/cncr.23195. [DOI] [PubMed] [Google Scholar]
- 40.Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA, et al. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35:1773–82. doi: 10.1016/s0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- 41.Berthold DR, Sternberg CN, Tannock IF. Management of advanced prostate cancer after first-line chemotherapy. J Clin Oncol. 2005;23:8247–52. doi: 10.1200/JCO.2005.03.1435. [DOI] [PubMed] [Google Scholar]
- 42.Armstrong AJ, George DJ. New drug development in metastatic prostate cancer. Urol Oncol. 2008;26:430–7. doi: 10.1016/j.urolonc.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Q, Gustafson D, Nallapareddy S, Diab S, Leong S, Lewis K, et al. A phase I dose-escalation, safety and pharmacokinetic study of the 2-methoxyestradiol analog ENMD-1198 administered orally to patients with advanced cancer. Invest New Drugs. 2010 doi: 10.1007/s10637-009-9383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubinstein L, Crowley J, Ivy P, Leblanc M, Sargent D. Randomized phase II designs. Clin Cancer Res. 2009;15:1883–90. doi: 10.1158/1078-0432.CCR-08-2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Booth CM, Calvert AH, Giaccone G, Lobbezoo MW, Eisenhauer EA, Seymour LK. Design and conduct of phase II studies of targeted anticancer therapy: recommendations from the task force on methodology for the development of innovative cancer therapies (MDICT) Eur J Cancer. 2008;44:25–9. doi: 10.1016/j.ejca.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 46.Ratain MJ, Humphrey RW, Gordon GB, Fyfe G, Adamson PC, Fleming TR, et al. Recommended changes to oncology clinical trial design: revolution or evolution? Eur J Cancer. 2008;44:8–11. doi: 10.1016/j.ejca.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stephen RM, Gillies RJ. Promise and progress for functional and molecular imaging of response to targeted therapies. Pharm Res. 2007;24:1172–85. doi: 10.1007/s11095-007-9250-3. [DOI] [PubMed] [Google Scholar]
- 48.Periman PO, Sonpavde G, Bernold DM, Weckstein DJ, Williams A, Zhan F, et al. Sunitinib malate for metastatic castration resistant prostate cancer following docetaxel-based chemotherapy. J Clin Oncol. 2008;26:a5157. doi: 10.1093/annonc/mdp323. [DOI] [PubMed] [Google Scholar]
- 49.Sonpavde G, Periman PO, Bernold D, Weckstein D, Fleming MT, Galsky MD, et al. Sunitinib malate for metastatic castration-resistant prostate cancer following docetaxel-based chemotherapy. Ann Oncol. 2009 doi: 10.1093/annonc/mdp323. [DOI] [PubMed] [Google Scholar]
- 50.Dror Michaelson M, Regan MM, Oh WK, Kaufman DS, Olivier K, Michaelson SZ, et al. Phase II study of sunitinib in men with advanced prostate cancer. Ann Oncol. 2009;20:913–20. doi: 10.1093/annonc/mdp111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahut WL, Scripture C, Posadas E, Jain L, Gulley JL, Arlen PM, et al. A phase II clinical trial of sorafenib in androgen-independent prostate cancer. Clin Cancer Res. 2008;14:209–14. doi: 10.1158/1078-0432.CCR-07-1355. [DOI] [PubMed] [Google Scholar]
- 52.Roche provides update on phase III study of Avastin in men with late stage prostate cancer. [cited 2010 April 5]; Available from: http://www.roche.com/media/media_releases/med-cor-2010-03-12.htm.