

Abstract
Bone marrow (BM)-derived endothelial progenitor cells (EPC) have a critical role in tumor neovascularization. Vascular endothelial growth inhibitor (VEGI) is a member of the TNF superfamily (TNFSF15). We have shown that recombinant VEGI suppresses tumor angiogenesis by specifically eliminating proliferating endothelial cells (EC). We report here that treatment of tumor bearing mice with recombinant VEGI leads to a significantly decreased population of BM-derived EPC in the tumors. We transplanted whole bone marrow from green fluorescent protein (GFP) transgenic mice into C57BL/6 recipient mice, which were then inoculated with Lewis lung carcinoma (LLC) cells. Intraperitoneal injection of recombinant VEGI led to significant inhibition of tumor growth and decrease of vasculature density compared to vehicle-treated mice. Tumor implantation yielded a decrease of BM-derived EPC in the peripheral blood, while VEGI-treatment resulted in an initial delay of such decrease. Analysis of the whole bone marrow showed a decrease of Lin−-c-Kit+-Sca-1+ hematopoietic stem cell (HSC) population in tumor bearing mice; however, VEGI-treatment caused a significant increase of this cell population. In addition, the number of BM-derived EPC in VEGI-treated tumors was notably less than that in the vehicle-treated group, and most of the apoptotic cells in the VEGI-treated tumors were of bone marrow origin. These findings indicate that VEGI inhibits BM-derived EPC mobilization and prevents their incorporation into LLC tumors by inducing apoptosis specifically of BM-derived cells, resulting in the inhibition of EPC-supported tumor vasculogenesis and tumor growth.
Keywords: Endothelial progenitor cells, Bone marrow-derived, Hematopoietic stem cells, Angiogenesis, Vasculogenesis, Apoptosis
Introduction
Bone marrow (BM)-derived endothelial progenitor cells (EPC) play a critical role in vasculogenesis, the de novo formation of new blood vessels which is essential for organ and tissue growth and wound healing [1–5]. Abnormal neovascularization under disease conditions, such as that in cancer development, also involves postnatal vasculogenesis [6, 7]. Early theories of tumor neovascularization revolved solely around angiogenesis being the source for the tumor’s vascular supply [8, 9]. New evidence shows that EPC migrate from the bone marrow to the tumor site and differentiate into a new endothelium in the tumor bed, providing an alternative method of tumor neovascularization [10–14]. Contributions of EPC to tumor neovasculature are evident from increased EPC markers in tumors of cancer patients [7, 15, 16]. It has also been shown that EPC activate the “angiogenesis switch,” a critical step in the transition of an avascular, dormant tumor to a vascularized, rapidly growing tumor [17].
Vascular endothelial growth inhibitor (VEGI; TNFSF15; TL1A) is a member of the tumor necrosis factor superfamily and an endogenous inhibitor of neovascularization [18]. VEGI is largely produced by vascular endothelial cells (EC) of established blood vessels in a normal tissue, and is a specific inhibitor of EC proliferation, apparently playing a role in the modulation of vascular homeostasis. More specifically, VEGI has the ability to enforce growth arrest of EC in G0 and G1 phases of the cell cycle, while inducing apoptosis of proliferating EC, leading to inhibition of angiogenesis [19, 20]. We have recently demonstrated that recombinant VEGI can prevent BM-derived EPC differentiation into EC in cell cultures [21]. While VEGI is expressed in the vascular EC of normal human adult tissues, it is absent or expressed at low levels in tumor vasculatures in breast cancer [22], prostate cancer [23], urothelial cancer [24], as well as during wound healing [25], supporting the view that VEGI is a negative regulator of neovascularization whose expression needs to be turned down prior to the initiation of physiological or pathological blood vessel growth. Consistently, recombinant VEGI has been shown to be a highly potent inhibitor of EC proliferation in tumors, resulting in specific elimination of EC in a tumor vasculature and inhibition of tumor growth [26–28].
In this paper, we examine whether VEGI can inhibit BM-derived EPC-supported vasculogenesis in a Lewis lung carcinoma (LLC) tumor model. We found that systemically administered recombinant VEGI can inhibit the incorporation of BM-derived EPC into LLC tumors and the differentiation of EPC into EC in these tumors. These findings provide important insights into the mechanism of modulation of EPC-supported vasculogenesis by this unique cytokine and endogenous inhibitor of angiogenesis.
Materials and methods
Antibodies and reagents
Fluorochrome-conjugated anti-mouse Flk-1, CD133, c-Kit, and Sca-1 antibodies were from eBioscience (San Diego, CA). EasySep Mouse Hematopoietic Progenitor Enrichment Cocktail for lineage selection was from Stem Cell Technologies (Vancouver, BC, Canada). Anti-mouse CD31 antibody was from BD Biosciences (San Jose, CA). Cy3 and Cy5 conjugated secondary antibodies were from Jackson ImmunoLabs (West Grove, PA). Terminal Transferase, recombinant, kit for TUNEL staining was from Roche Applied Science (Indianapolis, IN). VEGI isoform VEGI-192 was prepared as described [26]. The endotoxin level in the VEGI preparation is 25 ng/mg.
Cells
Lewis lung carcinoma (LLC) cells were purchased from American Type Culture Collection (Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Lonza, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS, Gemini Bio-Products, West Sacramento, CA).
Mice
Eight week old female C57BL/6 mice (wild-type mice) and C57BL/6 EGFP10sb/J mice (transgenic mice that express GFP in all cell types) were purchased from the Jackson Laboratory (Bar Harbor, ME). All procedures involving experimental animals were performed in accordance with protocols approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Bone marrow transplantation and engraftment analysis
Whole bone marrow from C57BL/6 EGFP10sb/J donor mice was harvested by flushing the femurs and tibias of adult animals. C57BL/6J recipient mice were lethally irradiated with 9 Gray of a cobalt source. After lethal irradiation, the recipient mice received 5 × 106 whole bone marrow cells via lateral tail vein injection. One month after bone marrow transplant (BMT), blood was obtained from the tail veins and engraftment was verified by flow cytometric analysis to confirm stable and complete chimerism, as indicated by >85% of GFP+ cells in peripheral blood.
Tumor inoculation and VEGI administration
One month after BMT, the chimeric mice were injected subcutaneously with 5 × 106 LLC suspended in phosphate-buffered saline (PBS, Lonza). Tumors were allowed to grow for 4 days and measured in a blinded manner with a dial caliper. The tumor volumes were determined using the formula, volume = width × width × length × 0.52. The experimental animals were then randomized and divided into two groups on day 4. The treatment group received one daily i.p. injection of VEGI while the control group received comparable injections of the vehicle (on days 4, 5 and 6 for a total of three treatments). The experimental animals were sacrificed on day 7. Tumor tissues, peripheral blood and bone marrow were collected for pathologic analysis.
Flow cytometry
Cells were harvested (for peripheral blood analysis samples, 100 μl of blood was collected from the tail vein of each experimental mouse, and for bone marrow samples, whole bone marrow was collected from femurs and tibia via bone marrow flush) and lysed with ACK cell lysis buffer (Lonza). Cells were then washed with 2 ml of fluorescence-activated cell sorter (FACS) buffer (1% bovine serum albumin and 0.05% sodium azide in PBS). The cells were collected by centrifugation, resuspended in 100 μl of FACS buffer containing 1 μg of the indicated antibody, dispensed in a minimum of 1 × 105 cells per sample, gently mixed, and incubated at room temperature for 15 min. The cells were washed with FACS buffer, centrifuged, resuspended in 0.5 mL PBS and analyzed within 1 h. Coulter FACS equipment and EXPO analysis software (Beckman Coulter, Fullerton, CA) were used.
Immunohistochemistry
Tumors were fixed with 2% paraformaldehyde in PBS at room temperature for 2 h, then cyroprotected by transferring to a 30% sucrose solution overnight at 4°C. Samples were flash frozen in liquid nitrogen cooled 2-Methylbutane for 30 s, then immersed in liquid nitrogen for an additional 10 s and stored at −80°C until cryostat sectioning. Tumor sections (8 μm thickness) were immunostained by first being blocked for non-specific antibody binding with 2% BSA, followed by incubation with primary antibodies for 60 min at room temperature. After 5 washes with PBS, sections were incubated with secondary antibodies for 60 min at room temperature and then washed again with PBS. After nuclei staining with DAPI, sections were mounted. Immunostained sections from peripheral and central regions of each tumor were imaged using a 60× oil objective on a Fluo-view 1000 confocal microscope (Olympus, Center Valley, PA). A minimum of 10 fields/section and 10 sections/tumor sample were analyzed.
Detection of apoptotic cells
Cryosections were washed 3× with PBS and incubated with TUNEL mixture at 37°C for 30 min, washed with PBS, then streptavidin secondary antibody was added for 30 min at room temperature. DAPI was used to stain for the cell nucleus and slides were mounted before imaging.
Statistical analysis
At least three repeat experiments (n = 3–6 per group) were performed yielding similar results. Results shown are of 1 experiment. Student’s t-test for independent samples was used to compare data between experimental groups. All data presented as the mean ± SE; P < 0.05 was considered statistically significant and is indicated with an asterisk.
Results
VEGI inhibits tumor growth and vascularization
Female mice (C57BL/6) with transplanted bone marrow from green fluorescent protein (GFP) transgenic mice were inoculated on day 0 with LLC cells subcutaneously (5 × 106 cells per injection). On day 4 following the initial inoculation of LLC cells, the experimental animals were treated with recombinant VEGI by intraperitoneal injection (I.P.) (5 mg/kg), with repeats on day 5 and day 6. The control group was treated with vehicle. The size of the tumors was measured daily. We found a significant inhibition of tumor growth rate in the VEGI-treated group compared to that in the control group (Fig. 1a). On day 7, the average tumor volume of the VEGI-treated group was 62% of that of the control group. The tumors were harvested on day 7. Cryostat sections of the tumor samples were analyzed for vascular density using immunohistochemical staining for CD31, a marker of EC. The intensity of CD31 immunostaining was scored on a scale of 1–4. We found that the mean intensity score was significantly lower in the VEGI-treated tumors than in vehicle-treated tumors (2.06 ± 0.07 vs. 3.05 ± 0.06, respectively), indicating less vascular formation (Fig. 1b). These results demonstrate that VEGI treatment leads to retarded growth of the LLC tumors and decreased vascularization in the tumors.
Fig. 1.
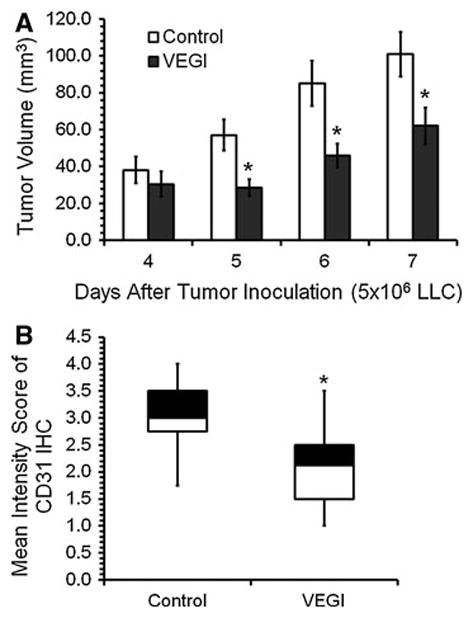
Effect of VEGI treatment on the growth and vascularization of LLC tumors a Average tumor size in mice treated with either vehicle (Control, n = 6) or VEGI (n = 6) given by intraperitoneal injection. b Box plot showing average intensities of CD31 immunostaining scored on Day 7 LLC tumor samples on a scale of 1–4, with 4 being the most
VEGI impact on peripheral blood EPC and bone marrow hematopoiesis
To determine whether VEGI has any impact on BM-derived EPC in the periphery blood as well as in the bone marrow, we first analyzed the number of circulating EPC in the peripheral blood of tumor bearing mice treated with VEGI or vehicle. Blood was collected immediately prior to cancer cell inoculation and analyzed by using flow cytometry to identify BM-derived GFP+ cells that were also positive for both Flk-1 (vascular endothelial growth factor receptor 2), an EC marker, and CD133 (Prominin-1), an indicator of stem cells. Blood was collected again 4 days after tumor inoculation, prior to VEGI treatments, in order to determine any change in the number of BM-derived EPC because of the tumor burden. Comparing to the baseline of day 0, we found an about five-fold decrease in the number of GFP+-Flk-1+-CD133+ cells 4 days after tumor inoculation (Fig. 2a), suggesting active recruitment of periphery blood EPC into the tumor. We also analyzed blood collected on day 5, 6, and 7 for changes in BM-derived EPC resulting from the daily VEGI treatments. Interestingly, 24 h after the initial VEGI treatment, we found a 64% increase in the number of GFP+-Flk-1+-CD133+ cells compared to the vehicle-treated group. However, this difference between the VEGI-treated group and vehicle-treated group disappeared on the following days (Fig. 2a). These findings suggest that VEGI-treatment results in an inhibition of the recruitment of the blood-borne EPC by the tumor, albeit transient under the experimental conditions.
Fig. 2.
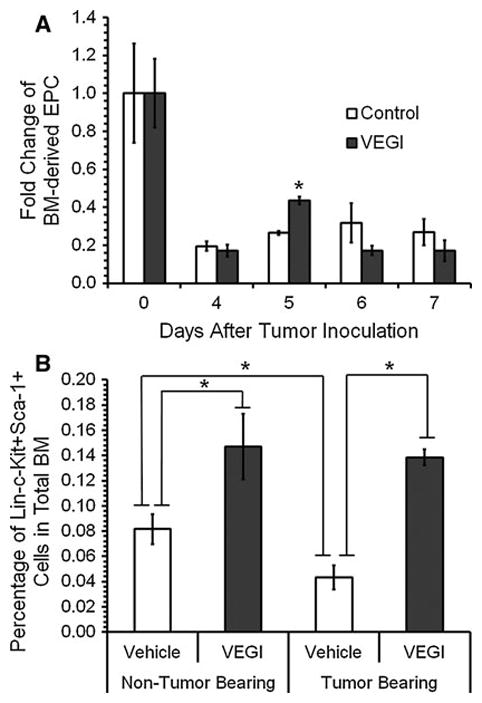
Effect of VEGI treatment on peripheral blood EPC (GFP+-Flk-1+-CD133+) and bone marrow HSC (Lin−-c-Kit+-Sca-1+) a Fold change of circulating EPC. Blood was collected via tail vein on day 0 prior to tumor inoculation and on days 4, 5, and 6 prior to VEGI treatment (n = 3 per group). Day 7 blood samples collected 24 h after the last treatment. b Percentages of HSC in whole bone marrow on Day 7. Samples analyzed by using flow cytometry
We also looked for changes in the hematopoietic stem cell (HSC) population (Lin−-c-Kit+-Sca-1+) in the bone marrow of the VEGI- or vehicle-treated groups by flow cytometry, and compared the results with those of the bone marrow of normal non-tumor bearing mice. We found that there was an about 80% increase in the population of HSC in the bone marrow without tumor burdens because of VEGI-treatment (Fig. 2b). By solely having a tumor burden, the population of purified HSC in the bone marrow exhibited a 53% decrease (Fig. 2b), possibly because of their mobilization induced by vascular trauma such that occurs in tumor neovascularization. VEGI-treatment of the tumor bearing group caused the percentage of Lin−-c-Kit+-Sca-1+ HSC in the bone marrow to increase to a level similar to those found with VEGI-treated, non-tumor bearing mice (Fig. 2b). These findings suggest that VEGI-treatment has an inhibitory effect on the mobilization of BM-derived EPC into the circulation.
VEGI inhibits BM-derived EPC incorporation into LLC tumors
In order to investigate the impact of VEGI-treatment on the incorporation of BM derived-EPC into the LLC tumors, tumor sections were immunostained for EC marker CD31. GFP and CD31 double positive cells were identified by confocal microscopy. In the tumor samples of the vehicle-treated animals, we were able to detect areas highly populated with GFP+-CD31+ cells. In contrast, the tumor samples from the VEGI-treated group consisted of significantly fewer GFP+-CD31+ cells (Fig. 3a). By counting the number of GFP+-CD31+ cells in the tumor sections (Fig. 3b), we found that the number of double positive cells in the tumors of VEGI-treated animals was 56% of that in the control group (Fig. 3c). This finding indicates that VEGI-treatment resulted in a lowered number of EC originating from BM-derived cells in the tumors compared with that of vehicle-treated tumors.
Fig. 3.
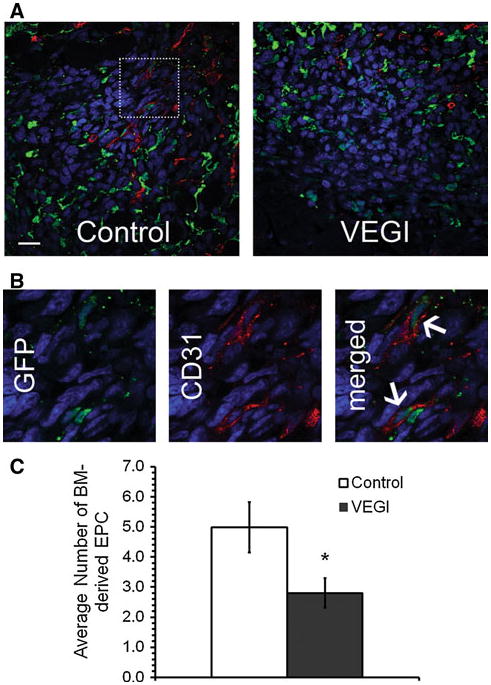
VEGI inhibition of EPC incorporation into LLC tumors a Representative confocal images of immunostaining for BM-derived EPC in LLC tumors collected on Day 7 following cancer cell inoculation and 24-h after the last of three daily VEGI treatments. Green, GFP; Red, CD31; Blue, Dapi; Scale bar 20 μm. b High magnification of the boxed area shown in Panel A. Arrows indicate GFP+-CD31+ cells. c Quantification of the number of GFP+-CD31+ cells from randomly selected fields on each tumor section; a minimum of 10 fields per tumor sample were counted from each animal (n = 4 per group)
VEGI induces apoptosis of BM-derived cells
To investigate the cause of the diminishing population of BM-derived EC in the tumors of VEGI-treated animals, we used TUNEL staining to determine the extent of the apoptosis of BM-derived cells (Fig. 4a). More specifically, we determined overall apoptosis in the tumors as well as apoptosis of GFP+ cells derived from the bone marrow (Fig. 4b). In the VEGI-treated tumors, the total number of apoptotic cells was more than two-times of that in the controls. Remarkably, the number of apoptotic GFP+ cells in the VEGI-treated group was more than three-times of that in the controls. Additionally, while about 40% of all apoptotic cells in the control group were GFP+, nearly 70% of the apoptotic cells in the VEGI-treated group were GFP+ (Fig. 4c). These findings suggest that VEGI treatment gives rise to apoptosis of BM-derived cells that have been recruited into the tumors.
Fig. 4.
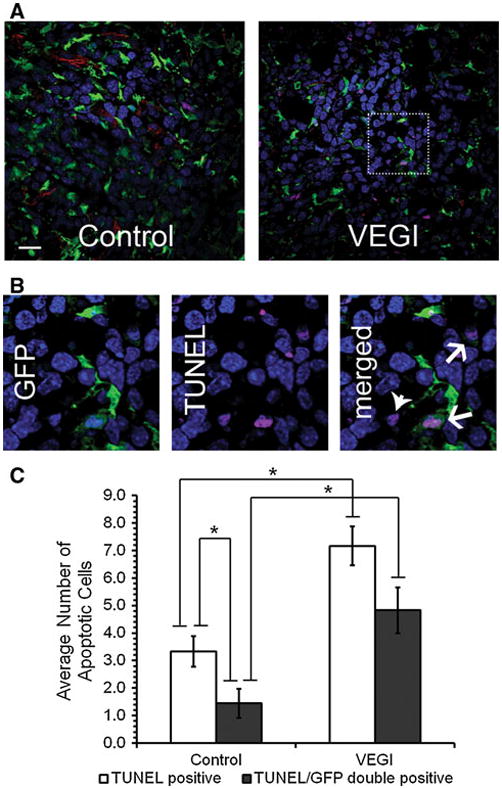
Induction of apoptosis of BM-derived cells by VEGI-treatment a Representative confocal images of Day 7 LLC tumor samples analyzed for apoptosis. Green, GFP; Red, CD31; Magenta, TUNEL; Blue, Dapi; Scale bar 20 μm. b High magnification of the boxed area shown in Panel A. TUNEL positive cells (arrow heads) and GFP+-TUNEL+ cells (arrows) are indicated. c Average number of TUNEL positive cells per field in vehicle-treated control or VEGI-treated tumors. A minimum of 10 fields per tumor sample were counted from each animal (n = 4 per group)
Discussion
Tumor vasculature arises through two processes: angiogenesis supported by EC from existing blood vessels [6], and vasculogenesis supported by an alternative source of EC, known as BM-derived EPC [1–5]. We have previously shown that VEGI inhibits angiogenesis by specifically inhibiting EC proliferation and inducing apoptosis of proliferating EC, resulting in suppression of tumor growth [19, 26]. To demonstrate in animal tumor models whether VEGI, an endogenous cytokine, can inhibit BM-derived EPC-supported vasculogenesis and thereby suppress this alternate method of neovascularization is important to the understanding of the mechanism of tumor neovascularization. The study may also yield pivotal insights into potential anti-cancer therapeutic approaches. The data we show here indicate that VEGI is able to inhibit EPC-supported vasculogenesis in tumors.
The function of EPC in supporting tumor vasculogenesis has been debated extensively in previous studies. In some reports, the role of EPC in tumor vascularization has been shown to be highly significant, not only contributing between 50–100% of the tumor vasculature, but also inducing neovasculogenesis [2, 5, 15, 17, 29–31]. BM-derived EPC are shown to be critical for activating the angiogenic switch, which induces the vascularization of a tumor, as blocking EPC not only inhibits angiogenesis and impairs tumor metastases, but also leads to prolonged survival of tumor bearing mice [17]. Other studies have shown, however, that EPC contribution is negligible, compromising no more than 5% of the EC in the tumor [32–39]. Apparently an important factor attributing to the discrepancies is the timeframe of when EPC incorporation into the tumor vasculature was analyzed. It has been reported that BM-derived EPC participate in early tumor vascularization, forming BM-derived vessels, before their incorporation is diluted by peripheral vessels resulting from angiogenesis [40]. Therefore, we examined the contribution of EPC to tumor vasculogenesis within the timeframe when subcutaneously implanted LLC form palpable tumors, approximately within a few days of cancer inoculation. Treatment of the tumor bearing mice with VEGI resulted in a marked retardation of tumor growth, accompanied by a decrease in vascular density in the tumors. These data are consistent with the view that VEGI is able to negatively regulate EPC-supported nascent neovascularization during the early formation of LLC tumors.
EPC are thought to migrate out of the bone marrow into the periphery and home to a site of vascular trauma, such as occurs during tumor vasculogenesis [10–14]. We examined the peripheral blood of the experimental mice before and after tumor cell inoculation as well as after VEGI treatment to determine if there is any difference in the circulating BM-derived EPC population. Previous studies have indicated that EPC are mobilized from the BM into the circulation, then incorporated into the vascular bed within 48 h after tumor inoculation in animal models [2, 14, 15]. We determined the baseline amount of circulating BM-derived EPC prior to tumor inoculation, and found that 4 days of possessing a tumor burden significantly deprived the amount of BM-derived EPC in the blood. It is likely that the BM-derived EPC population in the blood of the experimental animals diminishes because these cells are incorporated into the tumor. We then treated the tumor bearing mice with VEGI. While no further change was seen in the amount of circulating BM-derived EPC in the vehicle-treated control group, there was an increase of circulating BM-derived EPC in the blood of VEGI-treated mice compared to the control group 1 day after initial VEGI treatment. Interestingly, however, the levels of circulating BM-derived EPC in the VEGI treated group declined to the same level seen for the vehicle-treated control group within the next 24 h. It is plausible that the temporary increase in BM-derived EPC arises from VEGI inhibiting the ability of EPC to adhere to a site of vascular trauma, causing a transient accumulation of BM-derived EPC in the blood. This is supported by our previous findings that EPC in the presence of VEGI is unable to attach to fibronectin-, lamenin- or vitronectin-coated culture surfaces, and that the expression of integrin-α5 or -αV was substantially down-regulated by VEGI [21].
The subsequent decline of circulating EPC in VEGI-treated, tumor bearing animals may be explained in terms of the inhibition of cancer cell-induced HSC mobilization from the bone marrow. We analyzed the whole bone marrow of the experimental mice for the percentage of Lin−-c-Kit+-Sca-1+ HSC, which share a common lineage with EPC [15, 41, 42], after 3 days of VEGI treatment. We compared the percentage of the HSC in the tumor bearing mice to non-tumor bearing mice and found that the tumor burden leads to a marked decrease of the percentage of these cells, while VEGI treatment caused the number of Lin−-c-Kit+-Sca-1+ cells in the bone marrow to increase by about three-fold. A relatively larger percentage of HSC within the bone marrow, but a lower percentage of BM-derived EPC in the circulation, suggests that VEGI inhibits tumor-induced HSC mobilization in the bone marrow. This view is supported by our previous finding that the ability of EPC to migrate markedly declines when the cells were treated with VEGI [21]. Alternatively, the increase of the percentage of Lin−-c-Kit+-Sca-1+ cells in the bone marrow by VEGI-treatment suggests that VEGI promotes HSC production. This possibility will be investigated in future studies.
The significant decrease in the number of EPC incorporated into the tumor site of VEGI-treated mice, compared to that in vehicle-treated mice, may be because of not only VEGI inhibition of the mobilization of HSC in the bone marrow and inhibition of EPC adherence, but also the possibility of VEGI inducing apoptosis of BM-derived EPC [19, 21]. Given the unique ability of VEGI to induce apoptosis of only proliferating EC, we utilized CD31, a widely accepted marker of EC lineage, to visualize BM-derived EPC and their response to VEGI. We found that VEGI treatment not only caused an increase in the number of overall apoptotic cells in the tumor, but specifically induced the apoptosis of cells deriving from the bone marrow, giving rise to a diminished BM-derived EPC population in the tumor. This is also supported by our previous findings with in vitro models that VEGI specifically induces apoptosis of proliferating EC [19] and of late stage EPC [21].
In summary, this study illustrates the role of VEGI as an inhibitor of BM-derived EPC incorporation and differentiation in LLC tumors. Our findings indicate that VEGI inhibits BM-derived EPC-supported tumor vasculogenesis, ultimately inhibiting tumor growth. In addition, we show that tumor burdens mobilize EPC from the bone marrow and recruit circulating EPC, and that VEGI suppresses this process. Further, we show that VEGI directly inhibits BM-derived EPC-supported tumor vasculogenesis by specifically inducing apoptosis in BM-derived cells in the tumors. These findings are consistent with the view that VEGI plays a critical role in the modulation of EPC-supported postnatal vasculogenesis.
Acknowledgments
This work is supported in part by grants to L.Y.L from The National Institute of Health (CA113875), Pennsylvania Department of Health, The Hillman Foundation, and The Ministry of Science and Technology of China (2009CB918901). We thank Dr. Hui Yu, Dr. Richard XuFeng, and Mr. Mark Ross for their skillful technical assistance.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Ethical standards All experiments performed comply with the current laws of the United States of America.
Contributor Information
Paulina H. Liang, Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA. University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA
Fang Tian, Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA. University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA.
Yi Lu, Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA. University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA.
Biyan Duan, College of Pharmacy, Nankai University, Tianjin, China.
Donna B. Stolz, Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Lu-Yuan Li, Email: lil@upmc.edu, Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA. University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA. College of Pharmacy, Nankai University, Tianjin, China.
References
- 1.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 2.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 3.Kalka C, Masuda H, Takahashi T, et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci. 2000;97:3422–3427. doi: 10.1073/pnas.070046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murayama T, Tepper OM, Silver M, et al. Determination of bone marrow-derived endothelial progenitor cell significance in angiogenic growth factor-induced neovascularization in vivo. Exp Hematol. 2002;30:967–972. doi: 10.1016/s0301-472x(02)00867-6. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 7.Davidoff AM, Ng CY, Brown P, et al. Bone marrow-derived cells contribute to tumor neovasculature and, when modified to express an angiogenesis inhibitor, can restrict tumor growth in mice. Clin Cancer Res. 2001;7:2870–2879. [PubMed] [Google Scholar]
- 8.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 9.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 10.Shi Q, Rafii S, Wu MH, et al. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998;92:362–367. [PubMed] [Google Scholar]
- 11.Asahara T, Kawamoto A. Endothelial progenitor cells for postnatal vasculogenesis. Am J Physiol Cell Physiol. 2004;287:C572–C579. doi: 10.1152/ajpcell.00330.2003. [DOI] [PubMed] [Google Scholar]
- 12.Gill M, Dias S, Hattori K, et al. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–174. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- 13.Khakoo AY, Finkel T. Endothelial progenitor cells. Annu Rev Med. 2005;56:79–101. doi: 10.1146/annurev.med.56.090203.104149. [DOI] [PubMed] [Google Scholar]
- 14.Asahara T, Takahashi T, Masuda H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964–3972. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyden D, Hattori K, Dias S, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 16.Hilbe W, Dirnhofer S, Oberwasserlechner F, et al. CD133 positive endothelial progenitor cells contribute to the tumour vasculature in non-small cell lung cancer. J Clin Pathol. 2004;57:965–969. doi: 10.1136/jcp.2004.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 18.Chew LJ, Pan H, Yu J, et al. A novel secreted splice variant of vascular endothelial cell growth inhibitor. FASEB J. 2002;16:742–744. doi: 10.1096/fj.01-0757fje. [DOI] [PubMed] [Google Scholar]
- 19.Yu J, Tian S, Metheny-Barlow L, et al. Modulation of endothelial cell growth arrest and apoptosis by vascular endothelial growth inhibitor. Circ Res. 2001;89:1161–1167. doi: 10.1161/hh2401.101909. [DOI] [PubMed] [Google Scholar]
- 20.Yue TL, Ni J, Romanic AM, et al. TL1, a novel tumor necrosis factor-like cytokine, induces apoptosis in endothelial cells. Involvement of activation of stress protein kinases (stress-activated protein kinase and p38 mitogen-activated protein kinase) and caspase-3-like protease. J Biol Chem. 1999;274:1479–1486. doi: 10.1074/jbc.274.3.1479. [DOI] [PubMed] [Google Scholar]
- 21.Tian F, Liang PH, Li LY. Inhibition of endothelial progenitor cell differentiation by VEGI. Blood. 2009;113:5352–5360. doi: 10.1182/blood-2008-08-173773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parr C, Gan CH, Watkins G, Jiang WG. Reduced vascular endothelial growth inhibitor (VEGI) expression is associated with poor prognosis in breast cancer patients. Angiogenesis. 2006;9:73–81. doi: 10.1007/s10456-006-9033-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhang N, Sanders AJ, Ye L, Kynaston HG, Jiang WG. Vascular endothelial growth inhibitor, expression in human prostate cancer tissue and the impact on adhesion and migration of prostate cancer cells in vitro. Int J Oncol. 2009;35:1473–1480. doi: 10.3892/ijo_00000466. [DOI] [PubMed] [Google Scholar]
- 24.Zhang N, Sanders AJ, Ye L, Kynaston HG, Jiang WG. Expression of vascular endothelial growth inhibitor (VEGI) in human urothelial cancer of the bladder and its effects on the adhesion and migration of bladder cancer cells in vitro. Anti-cancer Res. 2010;30:87–95. [PubMed] [Google Scholar]
- 25.Conway KP, Price P, Harding KG, Jiang WG. The role of vascular endothelial growth inhibitor in wound healing. Int Wound J. 2007;4:55–64. doi: 10.1111/j.1742-481X.2006.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou W, Medynski D, Wu S, Lin X, Li LY. VEGI-192, a new isoform of TNFSF15, specifically eliminates tumor vascular endothelial cells and suppresses tumor growth. Clin Cancer Res. 2005;11:5595–5602. doi: 10.1158/1078-0432.CCR-05-0384. [DOI] [PubMed] [Google Scholar]
- 27.Zhai Y, Ni J, Jiang GW, et al. VEGI, a novel cytokine of the tumor necrosis factor family, is an angiogenesis inhibitor that suppresses the growth of colon carcinomas in vivo. FASEB J. 1999;13:181–189. doi: 10.1096/fasebj.13.1.181. [DOI] [PubMed] [Google Scholar]
- 28.Zhai Y, Yu J, Iruela-Arispe L, et al. Inhibition of angiogenesis and breast cancer xenograft tumor growth by VEGI, a novel cytokine of the TNF superfamily. Int J Cancer. 1999;82:131–136. doi: 10.1002/(sici)1097-0215(19990702)82:1<131::aid-ijc22>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 29.Qin G, Ii M, Silver M, et al. Functional disruption of alpha4 integrin mobilizes bone marrow-derived endothelial progenitors and augments ischemic neovascularization. J Exp Med. 2006;203:153–163. doi: 10.1084/jem.20050459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–1159. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 32.Purhonen S, Palm J, Rossi D, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci USA. 2008;105:6620–6625. doi: 10.1073/pnas.0710516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Palma M, Venneri MA, Roca C, Naldini L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. 2003;9:789–795. doi: 10.1038/nm871. [DOI] [PubMed] [Google Scholar]
- 34.Göthert JR, Gustin SE, van Eekelen JA, et al. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104:1769–1777. doi: 10.1182/blood-2003-11-3952. [DOI] [PubMed] [Google Scholar]
- 35.Peters BA, Diaz LA, Polyak K, et al. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med. 2005;11:261–262. doi: 10.1038/nm1200. [DOI] [PubMed] [Google Scholar]
- 36.Rajantie I, Ilmonen M, Alminaite A, Ozerdem U, Alitalo K, Salven P. Adult bone marrow-derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood. 2004;104:2084–2086. doi: 10.1182/blood-2004-01-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shinde Patil VR, Friedrich EB, Wolley AE, Gerszten RE, Allport JR, Weissleder R. Bone marrow-derived lin(−)c-kit(+) Sca-1+ stem cells do not contribute to vasculogenesis in Lewis lung carcinoma. Neoplasia. 2005;7:234–240. doi: 10.1593/neo.04523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Machein MR, Renninger S, de Lima-Hahn E, Plate KH. Minor contribution of bone marrow-derived endothelial progenitors to the vascularization of murine gliomas. Brain Pathol. 2003;13:582–597. doi: 10.1111/j.1750-3639.2003.tb00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wickersheim A, Kerber M, de Miguel LS, Plate KH, Machein MR. Endothelial progenitor cells do not contribute to tumor endothelium in primary and metastatic tumors. Int J Cancer. 2009;125:1771–1777. doi: 10.1002/ijc.24605. [DOI] [PubMed] [Google Scholar]
- 40.Nolan DJ, Ciarrocchi A, Mellick AS, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21:1546–1558. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–712. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- 42.Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002;2:826–835. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]