

Abstract
Accumulating evidence suggests that reversible protein acetylation may be a major regulatory mechanism that rivals phosphorylation. With the recent cataloging of thousands of acetylation sites on hundreds of proteins comes the challenge of identifying the acetyltransferases and deacetylases that regulate acetylation levels. Sirtuins are a conserved family of NAD+-dependent protein deacetylases that are implicated in genome maintenance, metabolism, cell survival, and lifespan. SIRT3 is the dominant protein deacetylase in mitochondria and emerging evidence suggests that SIRT3 may control major pathways by deacetylation of central metabolic enzymes. Here, to identify potential SIRT3 substrates, we have developed an unbiased screening strategy that involves a novel acetyl-lysine analog (thiotrifluoroacetyl-lysine), SPOT-peptide libraries, machine learning, and kinetic validation. SPOT peptide libraries based on known and potential mitochondrial acetyl-lysine sites were screened for SIRT3 binding, then analyzed using machine learning to establish binding trends. These trends were then applied to the mitochondrial proteome as a whole to predict binding affinity of all lysine sites within human mitochondria. Machine learning prediction of SIRT3 binding correlated with steady-state kinetic kcat/Km values for 24 acetyl-lysine peptides that possessed a broad range of predicted binding. Thus, SPOT peptide-binding screens and machine learning prediction provides an accurate and efficient method to evaluate sirtuin substrate specificity from a relatively small learning set. These analyses suggest potential SIRT3 substrates involved in several metabolic pathways such as the urea cycle, ATP synthesis, and fatty acid oxidation.
Introduction
Accumulating evidence suggests that reversible protein acetylation, which was historically reserved for histone proteins, may be a major regulatory mechanism that controls the functions of non-histone proteins. With the recent cataloging of ~1000 acetylation sites on protein lysine residues (1–3) comes the challenge of assigning functional roles to specific acetylation sites, identifying the acetyltransferases and deacetylases that regulate acetylation levels, and elucidating the physiological cause and effect of specific lysine acetylation sites. Sirtuins (or Sir2-like proteins) are a conserved family of NAD+-dependent protein deacetylases that are implicated in genome maintenance, metabolism, cell survival, and lifespan (4, 5). The NAD+-dependence of the sirtuin reaction suggests that specific protein deacetylation is inextricably linked to metabolism, redox control, and energy status. The observation that the seven mammalian sirtuins (SIRT1-7) display distinct sub-cellular localization further supports the hypothesis that many, if not most, targets of mammalian sirtuins are non-histone proteins.
Recent mass spectrometry studies revealed the widespread occurrence of acetylated proteins within mitochondria, as greater than 20% of all mitochondrial proteins are acetylated on at least one lysine residue (1–3). Mitochondria are central to cellular metabolism, and mitochondrial dysfunction has been linked to neurodegeneration, diabetes, heart disease, and other age-related diseases (6). Understanding the role and function of mitochondrial regulation by reversible protein acetylation could lead to the better understanding of metabolic function and disease etiology. In humans, SIRT3, SIRT4, and SIRT5 are localized to mitochondria. Among these sirtuins, only SIRT3 displays robust deacetylation activity (7). Polymorphisms in human SIRT3 have been linked to survivorship among the elderly (8, 9), suggesting a possible involvement of SIRT3 in age-related phenomenon. The observation that caloric restriction (CR) leads to increases in both transcription and protein levels of SIRT3 (10) suggests a positive effect of SIRT3 activity on parameters that influence aging. CR is the only known environmental intervention that strongly increases maximum life span and retards aging in mammals (11). Hints to the role of SIRT3 in metabolic regulation have come from several recent studies. SIRT3 deacetylates and activates mitochondrial acetyl-CoA synthetase 2 (ACS2) (12, 13). Other reports have suggested that SIRT3 deacetylates and activates complex I of the mitochondrial electron transport chain (14) as well as isocitrate, glutamate, and succinate dehydrogenases (15, 16). SIRT3 deficient mouse embryonic fibroblasts and tissues show lower ATP levels, with basal levels of ATP in the heart, kidney, and liver reduced by >50% (14). SIRT3 also physically interacts with at least one of the known subunits of Complex I, the 39-kDa protein NDUFA9, and functional studies demonstrate that mitochondria from SIRT3 deficient animals display a selective inhibition of Complex I activity (14). SIRT3 may modulate mitochondrial function and metabolism in response to metabolic stress. Consistent with this idea, SIRT3 expression in cardiomyocytes is increased under stress, and plays a protective role by preventing Bax-mediated apoptosis (17). Also, under prolonged fasting, SIRT3 deficient mice display defects in fatty-acid oxidation (18).
To provide an understanding for the molecular basis of SIRT3 function in mitochondria, it is critical that the protein targets and specific lysine deacetylation sites are determined. Unfortunately, only in a few cases have the functional lysines targeted by SIRT3 deacetylation been identified (i.e. acetyl-CoA synthetase 2 (12, 13), cyclophilin D (19), long-chain acyl coenzyme A dehydrogenase (18)). More generally, it will be essential to match functionally important acetylation sites to the enzymes responsible for adding and removing these modifications. Here, to identify potential substrates and understand the pathways controlled by sirtuins, we have developed an unbiased screening strategy that involves a novel acetyl-lysine analog, SPOT-peptide libraries, machine learning, and kinetic validation. We demonstrate the utility of this novel screening and validation approach using the mitochondrial sirtuin SIRT3.
Results and Discussion
General Design of SPOT libraries
To systematically investigate potential sirtuin substrates within a given proteome, we turned to SPOT synthesis (20, 21), which has recently been utilized to determine the substrate specificity of lysine methyltransferases (22–24). The SPOT technique involves the synthesis of peptides covalently attached via their C-terminus to amine-modified cellulose membranes (Figure 1). The hydrophilic nature of cellulose minimizes non-specific binding compared to libraries constructed on hydrophobic polystyrene beads or other surfaces. Because peptides are synthesized in a spatially addressed and parallel manner, SPOT libraries do not require separate sequencing and purification steps to identify queried peptide sequences. The use of SPOT libraries also allows direct quantitation of relative peptide binding affinity.
Figure 1.

Schematic of SIRT3 binding assay to the SPOT library cellulose membranes. Each individual spot within the SPOT libraries is a 9-mer with four randomized residues proximal to a central thiotrifluoroacetyl-lysine residue and C-terminal covalent attachment to amine-modified cellulose membranes.
Here, we utilized SPOT peptide libraries to measure the relative binding affinity of the mitochondrial sirtuin, SIRT3, the major protein deacetylase present in the mitochondria (7). Focusing on the mitochondrial proteome of ~1000 proteins (25) provided a more manageable data set to explore substrate specificity. The overall strategy is as follows: SIRT3 is incubated with the SPOT membrane and binds preferentially to peptides with the highest affinity (Figure 1). The amount of bound SIRT3, which is proportional to the affinity for each peptide, is then determined utilizing a SIRT3 specific antibody. The SIRT3 primary antibody is detected with HRP-conjugated Rabbit IgG secondary antibody. The spots are then visualized and resulting luminescence is quantified with an imaging camera and software (Figure 1), and the peptide binding preferences are analyzed.
Design of SPOT libraries: acetyl-lysine analogs
The SPOT library design required optimization of several parameters including peptide length, evaluated sequences, and affinity of the central lysine residue. To minimize non-specific binding and the amount of SIRT3 required for the binding assays, we developed a novel acetyl-lysine analog that binds to sirtuins tighter than any acetyl-lysine analog previously tested. Previously, we measured the binding of ten acetyl-lysine analog containing peptides to the yeast sirtuin homolog, Hst2, and found that thioacetyl-, trifluoroacetyl-, or propionyl-lysine containing peptides bound significantly tighter than the corresponding acetyl-lysine containing peptide (26). These findings led us to synthesize the thiotrifluoroacetyl- and pentafluoropropionyl-lysine peptides (Scheme S1; Supplemental methods) to discover tighter binding acetyl-lysine analog peptides.
To determine the binding constants of acetyl-lysine analog containing peptides to SIRT3, a fluorescein-labeled thiotrifluoroacetyl-lysine peptide based on the peptide sequence of the known SIRT3 substrate acetyl-CoA synthetase 2 (Fluorescein-LPKTRSGK(CSCF3)VMRR-OH; Fluor-ACS2) was synthesized (12, 13). Fluorescence polarization measurements yielded a Kd value of 1.7 ± 0.3 μM for direct binding of Fluor-ACS2 to SIRT3, which was similar to the Kd value of 0.9 ± 0.2 μM determined by isothermal titration calorimetry (ITC). Subsequently, using Fluor-ACS2 in a competitive fluorescence polarization assay, the Kd values were determined for various acetyl-lysine analog peptides based on the human histone H3 sequence modified at Lys-14 (H2N-KSTGGK(acetyl analog)APRKQ-OH) (Supplemental Figure S1, Table 1). To establish binding trends, the yeast sirtuin Hst2 was also evaluated ((26–28), Table 1 and Supplemental Figure S1).
Table 1.
Binding constants of various acetyl-lysine analog peptides to SIRT3 and Hst2.
| Acetyl-lysine analog | Structure | SIRT3 Kd (μM)1 | Hst2 Kd (μM)2 |
|---|---|---|---|
| acetyl-lysine | 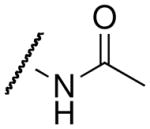 |
77 ± 223 | 21 ± 44 |
| lysine | 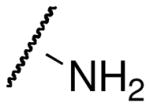 |
>10,000c | >300c |
| thioacetyl-lysine | 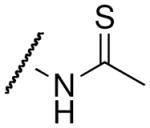 |
8.3 ± 0.8c | 4.7 ± 1.05 |
| trifluoroacetyl-lysine | 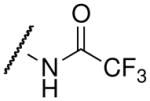 |
45.1 ± 8.2c | 3.3 ± 0.7d |
| propionyl-lysine | 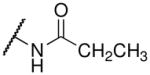 |
49.6 ± 13.8c | 8.6 ± 0.2d |
| pentafluoropropionyl-lysine | 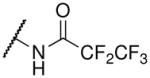 |
29.2 ± 5.6c | 0.95 ± 0.16c |
| thiotrifluoroacetyl-lysine | 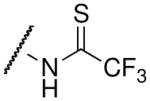 |
5.9 ± 0.3c | 1.0 ± 0.3c |
The thioamide and trifluoromethyl group within the acetyl-lysine analogs provided independent but additive binding effects. The thioacetyl- and trifluoroacetyl-lysine peptide bound SIRT3 9.3-fold and 1.7-fold tighter than the corresponding acetyl-lysine peptide (Table 1 and Supplemental Figure S2). Therefore, we expected the thiotrifluoroacetyl-lysine peptide would bind 15.8-fold (Kd ~ 4.9 μM) tighter to SIRT3 compared to the acetyl-lysine peptide. Indeed, we observed a Kd value of 5.9 μM for thiotrifluoroacetyl-lysine peptide binding to SIRT3. Using the same analysis for Hst2, the thiotrifluoroacetyl-lysine peptide was predicted to bind 28.4-fold tighter (Kd ~ 0.7 μM) to Hst2, and a Kd value of 1.0 μM was observed, suggesting thiotrifluoroacetyl-lysine peptides are general tight-binding sirtuin probes.
Fluorination of the propionyl group to form the pentafluoropropionyl analog also behaved as predicted for both SIRT3 and Hst2 binding. In particular, the propionyl- and trifluoroacetyl-lysine peptides bound SIRT3 1.55-fold and 1.7-fold tighter than the corresponding acetyl-lysine peptide. Therefore, we predicted that the pentafluoropropionyl-lysine peptide would bind 2.65-fold tighter (Kd ~ 29.1 μM) to SIRT3 compared to the acetyl-lysine peptide, and we observed a Kd value of 29.2 μM (Table 1 and Supplemental Figure S2). For Hst2, pentafluoropropionyl-lysine peptide was predicted to bind 15.5-fold (Kd ~ 1.35 μM) tighter to Hst2, and we observed a Kd value of 0.95 μM. Unlike the thiotrifluoroacetyl analog, which exhibited similar fold improvements in SIRT3 and Hst2, the pentafluoropropionyl analog exhibited a significantly greater improvement in Hst2 binding compared to SIRT3 binding. We recently reported that propionyl- and acetyl-lysine peptides exhibited similar overall turnover rates with Hst2; however, significantly slower turnover rates were observed for propionyl- compared to acetyl-lysine peptides with SIRT3 (26). Therefore, the pentafluoropropionyl moiety is likely well accommodated in the Hst2 active-site but not in the SIRT3 active-site, whereas the thiotrifluoroacetyl moiety is well accommodated in both active-sites. As the thiotrifluoroacetyl-lysine analog displayed general tight binding to both SIRT3 and Hst2, this analog was used in subsequent SPOT library screens.
Design of SPOT library: peptide length
Another important consideration in the design of the SPOT libraries was the length of peptides. For maximal effectiveness of the method, the peptides should be short enough to ensure synthetic integrity, but long enough to achieve high binding affinity and discrimination among library members. To this end, we synthesized control SPOT libraries containing thiotrifluoroacetyl-lysine surrounded by peptide sequences based on the general in vitro sirtuin substrate lysine-14 of histone H3 (26, 29), the SIRT1 substrate p53 (30–33), the SIRT2 substrate α-tubulin (32), the SIRT3 substrate acetyl-CoA synthetase 2 (12, 13), and the SIRT6 substrate lysine-9 of histone H3 (34). In these control libraries, peptides longer than nine amino acids did not result in increased SIRT3 binding (Supplemental Figure S3).
We also examined published sirtuin crystal structures in which acetyl-lysine containing peptides were bound in the acetyl-lysine binding pocket (35–43). In these structures, ordered residues were only reported for up to four residues N- or C-terminal to acetyl-lysine despite using peptides of length 8–18 amino acids in the crystallization conditions. This suggests that the majority of specific interactions in the sirtuin acetyl-lysine substrate binding pocket occur within the four residues immediately N- and C-terminal to acetyl-lysine.
A recent bioinformatics study reported acetylation motifs from the computational analysis of existing proteomic acetyl-lysine data sets (44). Although seven N- and C-terminal residues were utilized in training data sets, only the immediate N-terminal residue and four C-terminal residues revealed motifs that were statistically correlated with acetylation.
Finally, the only previous example of machine learning analysis of SPOT libraries, which identified novel antimicrobial peptides, utilized 9-mer peptides (45, 46). For the above four reasons, we utilized 9-mer peptides in our SPOT libraries with four amino acids N- and C-terminal to the central thiotrifluoroacetyl-lysine residue (Figure 1).
Design of SPOT library: peptide sequences
As there are 25.6 billion possible sequences for the 20 natural amino acids at eight randomized positions, screening every possible sequence was impractical. As SIRT3 is localized to the mitochondria (47–50), the sequence space can be narrowed significantly by focusing on the mitochondrial proteome. Even this yields ~22,000 possible 9-mer sequences with a central lysine. Therefore, to provide the greatest coverage of the relevant sequence space, we utilized two distinct libraries. One SPOT library focused on known acetylation sites within the mitochondria and contained the human sequences of acetylated peptides from a recent acetyl proteomics screen (229 peptides) (1). As all acetyl proteomic studies have relied on the ability of anti acetyl-lysine antibodies to enrich for acetylated peptides in tryptic digests, known acetylation sites are potentially biased toward both the specificity of antibodies as well as the length of peptides required for efficient mass spectral sequencing. To minimize any bias, the other SPOT library contained 300 randomly sampled lysine-centered 9-mers from the mitochondrial proteome, independent of evidence for acetylation.
Screening of SPOT libraries for SIRT3 binding
After identification and validation of the proper acetyl-lysine analog, peptide length, and peptide sequences to be utilized, we screened the resulting SPOT libraries for their ability to bind SIRT3. Each library was synthesized in triplicate, screened with SIRT3 and the resulting integrated optical density (IOD) binding scores were averaged to reduce measurement noise and improve the training signal for the machine learning model (see below). A representative SIRT3 library screen of 300 randomly sampled lysine-centered 9-mers from the mitochondrial proteome is shown in Figure 2. These screens exhibited a wide range of binding affinity from high intensity spots representing high affinity peptides to spots with background signal intensity. The identical procedure performed in the absence of SIRT3 revealed no binding above background for both libraries (data not shown), which indicated that the observed SIRT3 binding to the membrane in Figure 2 was not the result of non-directed antibody binding.
Figure 2.

Binding of SIRT3 to a SPOT library of 300 lysine-centered 9-mer peptides randomly selected from the mitochondrial proteome. A representative SPOT library is shown with SIRT3. Performing the same procedure without SIRT3 gave no signal above background (data not shown). SPOT library screens were performed as described under Experimental Procedures.
Machine learning of SIRT3 binding affinity
As mentioned above, testing all 22,000 mitochondrial lysine centered 9-mer sequences was impractical, so we developed a method to model and predict SIRT3 binding for sequences not examined in the SPOT libraries. Machine learning was recently used in the identification of novel antibacterial peptides from SPOT libraries (45, 46). To model binding affinity for peptide sequences, we used a linear regression approach that was trained using the IOD scores from our SPOT library screens, and predicts IOD scores for other peptides not present in our libraries. This method (described below) induces a model by treating a peptide’s IOD score as a linear function of various molecular properties. It performed as well or better than other machine learning algorithms we investigated (e.g. artificial neural networks, Gaussian processes, and decision trees). Linear regression has the added advantage of easily interpretable model parameters (i.e. the coefficients of the learned function).
To represent peptides for the machine learning algorithm, the amino acids at the eight variable positions of each peptide were represented as binary variables. For example, the sequence IINQKRFND has value one for Ile at the −4 position and Arg at the +1 position, and value zero for all other amino acids at those positions, resulting in 8 × 20 = 160 total individual residue features. Additionally, we used ten global chemical properties to describe each peptide. These global properties were hydrophobicity (51), flexibility (52), conformational tendencies for alpha helix, beta strand, beta turn, and coil (53), average area buried on transfer to folded protein (54), and accessibility (55). We desired to separate the conformational preferences for parallel and antiparallel beta strands, so we also included a separate scale for these preferences (56). These scales provide values for individual amino acids, thus we took the average value across all residues in a sequence to produce a real-numbered value for each peptide as a whole. We then normalized the input value for each scale feature to be in the range from zero to one. This simple normalization allowed us to better compare the relative contribution of each scale’s parameter weight in the analysis of our learned model.
To fit the parameters of our model, we used least-squares regression. However, we found that the common approach of computing the coefficients in closed form led to significant “overfitting,” which occurs when a statistical model represents spurious correlations and noise in the training signal rather than an actual underlying relationship between the input and output variables. This sometimes occurs in machine learning when the model has too many degrees of freedom relative to the amount of training data available, which is likely the case in our study with 170 input features and 529 training sequences.
To remedy this, we implemented a gradient-based numerical optimization approach based on the L-BFGS algorithm (57), which finds the least-squares solution iteratively, and we employ “early stopping” to prevent overfitting (58). Specifically, we halt the training process once the reduction in estimated squared error falls below 0.001. One could also use other forms of parameter regularization (59) to maintain predictive accuracy on novel peptides, but we found early stopping to be sufficient. A more detailed discussion of this method, and a comparison to other machine learning approaches we investigated will be discussed elsewhere (Burr Settles, Brian C. Smith, John M. Denu, and Mark W. Craven; manuscript in preparation). We also plan to publicly release a software tool that implements our feature generation and model training for the research community at large.
Parameter weights for the trained machine learning model of SIRT3 substrate specificity are presented in Figure 3. The sequence logo representation in Figure 3a illustrates the relative contribution of residues at specific positions surrounding the central lysine to SIRT3 substrate specificity. Residues above the baseline suggest positive correlation with SIRT3 binding specificity, while residues below the baseline suggest negatively-correlated relationships, and larger characters suggest greater relative impact on binding. We emphasize that these coefficients do not reflect relative frequency of each residue in the libraries (which is the interpretation for some other sequence logos in the literature), but rather how discriminative they are in predicting high or low binding affinity, in terms of IOD score. The bar chart in Figure 3b illustrates the analogous relationship between global parameters and SIRT3 substrate specificity. Again, positive weights suggest positive correlation with measured SIRT3 binding affinity, and negative weights indicate negatively-correlated relationships, and the magnitude of each weight indicates its relative impact on SIRT3 binding.
Figure 3.


(A) Sequence logos illustrating learned linear regression weights for the position-specific residue features. The height of each residue is proportional to the coefficient used by the model in predicting SIRT3 binding specificity. Positively-correlated residue positions are shown above the baseline, while negatively-correlated resides are below. (B) Global feature model parameters. A bar chart illustrating learned linear regression weights for the global peptide scale features. Positive values suggest that the corresponding chemical property (e.g. hydrophobicity) or secondary structure (e.g. antiparallel strand) is positively correlated with SIRT3 binding specificity. Negative values likewise suggest a negatively correlated relationship.
To evaluate the predictive accuracy of our model, we conducted a cross-validation study using the 529 peptide measurements from our SPOT libraries. For each peptide, this involved training a model using the remaining 528 sequences, and predicting a binding score for the single held-out peptide. This procedure is repeated for every sequence in the library, allowing us to fairly estimate how well the model can predict binding on the SPOT membrane for sequences for which it was not trained. The Pearson correlation between predicted scores and the actual IOD measurements is R2 = 0.54, indicating that our model predictions can explain over half of the observed variance in actual IOD measurements from SPOT array screenings.
Machine learning prediction of SIRT3 substrate specificity across the mitochondrial proteome
A significant advantage of our machine learning model is the ability to predict SIRT3 substrate specificity for sequences not present among the original 529 peptides tested in our SPOT libraries. To this end, we used the MitoCarta human inventory of ~1000 mitochondrial proteins (25) to generate a list of ~22,000 lysine-centered 9-mers as potential SIRT3 binding sites. We then generated residue and global scale features for these peptide sequences and predicted their binding affinities using the learned model described above. We have included these predictions in Supplemental Table 2. This table has also been annotated with known acetylation sites from the acetyl-lysine proteomics by Kim et al., Choudhary et al., and Zhao et al. (1–3). Since the acetylation sites discovered in these proteomics screens likely represent a subset of the acetyl-lysine sites present in mitochondria, we have also annotated the spreadsheet with the result of an online acetylation prediction algorithm (phosida.com).
Validation of SPOT library screening and machine learning
Implicit in the machine learning model is the hypothesis that binding correlates with substrate specificity in deacetylation assays. This may not be the case as some peptides may bind outside the active site and potentially uncover other relevant protein:protein interaction sites. However, we have purposely tried to minimize the contribution of binding outside the active-site through engineering a tight binding acetyl-lysine analog (thiotrifluoroacetyl-lysine) that should result in increased binding to only the SIRT3 active-site and not other potential binding sites.
To evaluate the results from the SPOT libraries and the machine learning prediction of SIRT3 substrate specificity, we synthesized the acetyl-lysine versions of sequences with a wide variety of both experimentally-tested (11 peptides) and predicted (12 peptides) binding affinities and measured their steady-state kinetic kcat/Km values with SIRT3 using our previously published continuous sirtuin assay (60). While not the most sensitive sirtuin assay, this assay was an excellent choice to rapidly evaluate the steady-state kcat/Km values among a large diverse group of peptide substrates for SIRT3. The measured kcat/Km values varied ~300-fold over the range of peptides tested. To evaluate the correlation of the results, we plotted our measured IOD scores versus of the log kcat/Km values (61, 62). The IOD scores reflect the relative binding affinity of SIRT3 toward each thiotrifluoracetylated SPOT peptide. The kcat/Km parameter is the apparent second order rate constant between SIRT3 and free, acetylated peptide. This steady-state constant reflects both the affinity for substrate and the efficiency of deacetylation. Therefore, evaluation of the correlation between IOD scores and kcat/Km values was critical as the SPOT libraries may have selected for peptides that bind to the active-site in a conformation that did not allow for efficient deacetylation. This analysis revealed a fairly strong linear correlation (R2 value of 0.37, p = 0.047) of binding affinity of thiotrifluoroacetyl-lysine containing peptides on SPOT membranes and deacetylation efficiency of acetyl-lysine containing peptides in solution for the 11 peptides we analyzed. To further evaluate our machine learning model, we also plotted the predicted IOD scores from the machine learning model versus the log kcat/Km values (Figure 4). This analysis revealed a slightly weaker correlation for the 11 peptides in the experimentally-tested set (R2 value of 0.30, p = 0.079) but an excellent correlation for all 23 of the peptides we analyzed in vitro (R2 value of 0.59, p < 0.001), which suggested that the machine learning model is accurate in predicting deacetylation efficiency for peptides not experimentally tested on the SPOT membranes. Collectively, these analyses provided strong evidence that peptide-binding affinity on membranes revealed in the SPOT library binding screens correlated with deacetylation efficiency in solution.
Figure 4.

Steady state kinetic validation of acetyl-lysine peptides selected from SPOT-library screens of SIRT3. Predicted percentile refers to the relative binding affinity predicted from the learned model trained on the binding intensities from SPOT library screens (Figure 2). Blue circles are sequences tested on the SPOT membrane, and red squares represent sequences that were not tested.
Substrate specificity and mitochondrial sirtuins
The combined approach of SPOT library screening and machine learning analysis revealed several interesting determinants for efficient SIRT3-catalyzed deacetylation. In general, aromatic residues (i.e. Phe, Trp, and Tyr) exhibited tight SIRT3 binding. Another general trend was negatively charged residues (i.e. Asp and Glu) were disfavored across the entire 9-mer peptide (Figure 3a). Likewise, positively charged residues (i.e. Arg and Lys) were correlated with tight SIRT3 binding. This is consistent with the recently published SIRT3 structure (63) that reveals an overall negative charge throughout the peptide binding pocket, particularly at residues C-terminal to acetyl-lysine (Figure 5 and Supplemental Figure S5a).
Figure 5.

Models of acetyl-lysine peptides bound in the SIRT3 active site. Stick models of (a) acetyl-CoA synthetase 2 (ACS2), (b) K88 of ornithine transcarbamoylase (OTC), (c) K230 of the alpha subunit of ATP synthase, and (d) K498 of the flavoprotein subunit of succinate dehydrogenase docked into the SIRT3 active site were based on the structure of a thioalkylamidate bound to SIRT3 resulting from the reaction of a thioacetyl-lysine acetyl-CoA synthetase 2 (ACS2) peptide and NAD+ (PDB code 3GLT) (63). The enzyme surface shown is prior to energy minimization and colored based on electrostatic potential with red, blue, and white surface representing negative, positive, and hydrophobic residues, respectively. Modeling was performed by mutating an acetyl-lysine ACS2 peptide bound to SIRT3 (PDB code 3GLR; (63)) using Pymol (75), and the structure was energy-minimized using CHARMMing (76) using the CHARMM defaults for Shake. Images were generated using Pymol (75).
A previous acetyl-lysine proteomics study revealed that acetyl-lysine sites are enriched in regions with ordered secondary structure, in particular alpha-helical and beta-sheet regions (2). Here, we found that peptides with a high propensity to form alpha-helix or beta-turn structures were disfavored as SIRT3 substrates (Figure 3b). However, peptides with a high propensity to form beta-strand structures were favored overall (Figure 3b). In particular, peptides with a high propensity to form antiparallel-strands were highly favored whereas parallel-strands were slightly favored. This observation is highly consistent with all known sirtuin crystal structures in which acetyl-lysine peptides bind in an extended antiparallel-strand conformation (35–43). Within the SIRT3 active-site, E296, P297, and L398 of SIRT3 form an antiparallel beta strand with the −2 and −1 position and acetyl-lysine of the substrate peptide, and E323, V324, and E325 of SIRT3 form an antiparallel beta strand with the +1, +2, and +3 positions of the substrate peptide (Supplemental Figure S4). In addition, peptide flexibility was less favored towards SIRT3 binding when compared to beta-strand structures. This finding is in contrast to a previous study that reported the yeast sirtuin, Hst2, prefers conformationally flexible substrates within unstructured protein regions (64).
In general, acetyl-lysine residues reside in regions with higher average solvent accessibility than the average of all lysines (1), suggesting that acetyl-lysine residues occur in more exposed environments. However, our machine learning analysis indicated that SIRT3 preferentially binds regions with lower accessibility and higher area-buried scores, reflecting residues that would prefer to be buried given their inherent chemical nature. It is important to note that peptides with low accessibility and high area-buried scores are not necessarily less solvent accessible in specific proteins. As binding to SIRT3 provides a surface to bury these residues, this may provide additional binding energy, analogous to the hydrophobic effect in protein folding (65).
A recent bioinformatics study by Schwartz et al. analyzed known acetyl-lysine sites in mammals and found several motifs that were highly correlated with lysine acetylation (44). Interestingly, several of these motifs were correlated with SIRT3 binding, indicating that peptides predicted to be SIRT3 substrates are predicted to be acetylated. The acetylation motifs K(ac)K, K(ac)R, K(ac)Y, K(ac)XF, and K(ac)XXXK were highly correlated with SIRT3 binding (Figure 3a). The acetylation motifs K(ac)XXK and K(ac)F were less highly correlated with SIRT3 binding (Figure 3a). The only acetylation motif negatively correlated with SIRT3 binding was GK(ac); this motif likely represents nuclear histone acetylation (the GK motif) (2, 66) and would not be expected to correlate with SIRT3 substrate specificity in the mitochondria.
Recall that the 229-peptide library has a potential sample bias as it contains only known acetylated peptides in the mitochondria. To ensure that the observed relationship between the Schwartz et al. motifs and SIRT3 binding was not an artifact of over-representation in our training set, we compared the frequency of each motif in the acetylation library, the randomized library, and the observed frequencies among the full set of ~22,000 lysine-centered 9-mers in the mitochondrion (the background set). Only the K(ac)Y motif appeared at a significantly higher frequency in the acetylated library than in the randomized library or the background set (p < 0.05, using binomial tests with Bonferroni correction, details in Supplemental Table 1). However, the over-representation of the K(ac)Y motif likely does not impact our findings, since (1) the randomized library has virtually the exact same distribution of K(ac)Y as the full set (p = 1.0) which helps alleviate any bias, and (2) other motifs such as K(ac)K and K(ac)R show even stronger correlation with SIRT3 binding while not being over-represented.
To separate acetylation motifs by cellular compartment (mitochondria, nucleus, and cytoplasm), a more recent acetyl-lysine proteomics study analyzed separately the relative abundance of amino acids flanking acetyl-lysine residues (2). Interestingly, Choudhary et al. found that Phe was enriched at the −2 position next to acetyl-lysine residues in the mitochondria, which matches the preference of SIRT3 at this position (Figure 3a). Additionally, Phe and Tyr were enriched at the +2 position for acetyl-lysine residues; SIRT3 has a dual preference for positively charged (i.e. Lys and Arg) in addition to aromatic residues (i.e. Phe, Tyr, Trp) at this position. Similarly, Tyr is enriched at the +1 position next to mitochondrial acetyl-lysine residues, and SIRT3 prefers Lys and Arg at the +1 position but possesses a secondary preference for aromatic residues such as Trp and Tyr. However, Choudhary et al. found that the residues flanking many of the identified mitochondrial acetyl-lysine sites are enriched for negatively charged residues; this is divergent from the preferences of SIRT3 and suggests that this negatively charged subset of mitochondrial acetyl-lysine sites may instead be SIRT4 and/or SIRT5 substrates. Comparing the active-sites of SIRT3 and SIRT4 supports this hypothesis. The peptide binding site in SIRT3 is negatively charged whereas the same site in SIRT4 is positively charged (Supplemental Figure S5). However, the peptide binding site in SIRT5 is mostly hydrophobic suggesting that SIRT3, SIRT4, and SIRT5 possess very distinct protein substrate preferences.
Although we made a concerted effort to construct and utilize the SPOT libraries to provide an efficient method for rapidly screening sirtuin substrate specificity, the ability of this approach to provide insight into cellular targets is limited to sirtuins that discriminate potential substrates based on the amino acid sequence surrounding the acetyl-lysine. To date, the contribution of interacting surfaces outside the active site between sirtuins and their substrate targets is completely unknown. There are no available co-structures with native acetylated proteins. Consequently, it is unclear if there are quaternary-structure contributions to specificity that would not necessarily be revealed in the SPOT library analysis presented here. In addition, it is unknown if the induced backbone conformation of the peptide when present in a whole protein substrate plays a role in substrate specificity. Furthermore, the overall inaccessibility (i.e. buried within tertiary structure) of a particular lysine may restrict its ability to be acetylated by acetyltransferases, and subsequently deacetylated by sirtuins. Therefore, additional information about lysine accessibility within a protein substrate could help eliminate unlikely targets that would otherwise be predicted to be excellent SIRT3 substrates (e.g. lysine sites with high area buried/hydrophobicity and low accessibility scores).
Modeling of SIRT3 with potential substrates
We next sought to rationalize our learned model of SIRT3 peptide substrate specificity in the context of a recently reported SIRT3 crystal structure bound with an acetyl-CoA synthetase 2 (ACS2) peptide (63). The combined total heights of the residues depicted in Figure 3a provide insight into the relative importance of each position to SIRT3 binding. In particular, the −4, −2, +1, +2, and +3 positions provide the greatest contributions towards SIRT3 binding. Therefore, we modeled several peptide sequences into the SIRT3 peptide binding site, and analyzed the −2, +1, and +2 positions of the peptide for interactions with SIRT3. The −4 position was not ordered and the sidechain conformation at the +3 position varied significantly in the SIRT3 structures and therefore we were unable to analyze specific interactions at these positions. We were unable to reliably model the sidechain conformations at the −1 position due to the presence of a glycine at this position in the ACS2 peptide that was used for modeling. However, as mentioned above, our learned model indicates that the −1 position is not as important in dictating substrate specificity as the −4, −2, +1, +2, and +3 positions. To explore structural features at these positions, the ACS2 peptide (Figure 5a) was mutated to three separate peptides, all of which exhibited tight binding in our SPOT libraries: K88 of ornithine transcarbamoylase (OTC), K230 of the alpha subunit of ATP synthase, and K498 of the flavoprotein subunit of succinate dehydrogenase. These modeled peptides were then energy minimized within the SIRT3 structure and examined for salient interactions. It is important to note that these models are necessarily qualitative and strongly suggest the need for further structural work with alternate peptide sequences bound to SIRT3.
For the OTC peptide, the interactions at two residues were particularly striking, Phe at the −2 position and Arg at the +1 position (Figure 5b). Phe was the most highly favored residue at the −2 position in the machine learning analysis, and Phe docked into a hydrophobic cavity on the SIRT3 surface defined by P297, L298, P299, F302, and L303. Arg was the most highly favored residue at the +1 position and this Arg modeled close to the negatively charged E325 on the surface of SIRT3.
The interactions at the +1 and +2 position were of interest for the ATP synthase peptide (Figure 5c). Similar to the OTC peptide, ATP synthase also contains an Arg at the +1 position that may interact with E325 of SIRT3. SIRT3 possesses a hydrophobic hole near the +2 position of the peptide defined by F157, F294, and V324. Phe is the most highly favored residue at the +2 position in the learned model (Figure 4a), and this hole may accommodate hydrophobic residues such as Phe.
The succinate dehydrogenase peptide also contains two interactions of interest (Figure 5d). Similar to the OTC peptide, the succinate dehydrogenase peptide contains a hydrophobic Leu residue at the −2 position (Phe in both OTC and SCHAD) that may dock in the same hydrophobic cavity. The succinate dehydrogenase peptide contains an Arg residue at the +2 position. Arg is the second most highly favored residue at the +2 position (Figure 3a). This Arg may extend beyond the hydrophobic hole at the +2 position to instead interact with the negatively charged E177 or E181. This may explain the dual preference for positively charged and hydrophobic residues at the +2 position in the learned model.
Identifying SIRT3 substrates
One main extension of this study is to accelerate the ability to identify sirtuin substrates. In this section, we will highlight the utility of this method using several examples. A potential SIRT3 substrate identified from our SPOT library screens is ornithine transcarbamoylase (OTC). OTC is a key enzyme in the urea cycle that converts ornithine and carbamoyl phosphate into citrulline. Lysine 88 of OTC ranked in the top 8% of the SPOT library screen of known mitochondrial acetylation sites (Supplemental Table 1). In addition, our machine learning predictions ranked K88 of OTC 1st among the 24 lysines present in the OTC sequence and the top 4% of all mitochondrial peptides (Supplemental Table 2). Interestingly, a recent study revealed that OTC K88 acetylation decreases the affinity for carbamoyl phosphate and the maximum velocity (67). Our SPOT libraries and machine learning analysis strongly suggested that SIRT3 is the enzyme that deacetylates K88 of OTC in vivo. Indeed, in a companion study, we demonstrated in vivo and in vitro that OTC is a bona fide target of SIRT3 and that SIRT3 modulates the urea cycle through direct OTC deacetylation (William C. Hallows, Wei Yu, Brian C. Smith, Mark K. Devries, James J. Ellinger, Shinichi Someya, Michael R. Shortreed, Tomas A. Prolla, John L. Markley, Lloyd M. Smith, Shimin Zhao, Kun-Liang Guan, and John M. Denu, to be submitted).
Other relevant proteins whose corresponding peptides exhibited tight binding to SIRT3 are subunits of ATP synthase and the electron transport chain (NADH dehydrogenase). Indeed, the tightest binding peptide in the SPOT library based on known mitochondrial acetylation sites was from ATP synthase, H1 transporting, mitochondrial F1 complex, alpha subunit 1 (IINQK(ac)RFND) (Supplemental Table 1). Interestingly, both the alpha and beta subunits of ATP synthase were identified as SIRT3 interacting partners in a recent proteomics screen (68). The authors also verified the interaction between the alpha subunit of ATP synthase and SIRT3 by co-immunoprecipitation, but did not examine deacetylation. Previous studies have shown that ATP levels and mitochondrial electron potential are decreased in the sirt3−/− mouse (14). A potential explanation for these lowered ATP levels is that hyperacetylation of ATP synthase upon loss of SIRT3 inhibits ATP synthase and electron transport chain activity.
Several peptides from enzymes involved in fatty acid oxidation exhibited tight binding in our SPOT library screens with SIRT3. Acetylated peptides from short chain L-3-hydroxyacyl-Coenzyme A dehydrogenase (SCHAD) (K202 in top 6% of known mitochondrial acetylation SPOT library and top 3% of all mitochondrial peptides in the learned model), very-long-chain acyl-CoA synthetase (VLACS) (K291 in top 13% of known mitochondrial acetylation SPOT library) and 3-ketoacyl-Coenzyme A thiolase (K137 in top 12% of mitochondrial acetylation SPOT library and top 1% of all mitochondrial peptides in the learned model) scored highly as potential substrates for SIRT3 deacetylation (Supplemental Tables 1 and 2). Consistent with an important role in β-oxidation, sirt3−/− mice exhibit abnormally high levels of a number of acylcarnitines ((18) and WCH and JMD to be submitted). Recently, it was reported that long-chain acyl coenzyme A dehydrogenase (LCAD), is regulated by acetylation and deacetylation by SIRT3 (18).
Identifying SIRT3 substrates is paramount to understanding how reversible acetylation can modulate mitochondrial metabolism, apoptosis, redox, and potentially lifespan extension. This study provides a compelling insight into possible SIRT3 substrates in the mitochondria. We anticipate that this data set will be used to discover novel SIRT3 substrates in one of two ways: 1.) A discovery mode where biologists search among previously cataloged acetylation sites that are functionally uncharacterized and predicted to be SIRT3 substrates by our machine learning analysis. This list of potential SIRT3 substrates can be further narrowed by other factors such as location of the lysine within the protein (e.g. within the active-site of an enzyme or mitochondrial location sequence that is cleaved upon translocation) or location of the protein within the mitochondria (SIRT3, SIRT4, and SIRT5 are present within the mitochondrial matrix). 2.) A validation mode where a biologist has discovered a potential SIRT3 substrate, but the site of deacetylation is unknown. In this case, our learned model would prioritize the lysines as potential sites of SIRT3-catalyzed deacetylation. In this case, a customized SPOT library could be constructed containing only sequences surrounding the lysines within the protein of interest and tested for SIRT3 binding. Overall, this new approach will greatly expand the scope and quicken the pace of sirtuin substrate identification.
Methods
General materials and methods
SIRT3 and Anti Goat-HRP conjugated antibody were purchased from Abcam (Cambridge, MA, USA). Fmoc-protected amino acids were purchased from peptide international (Louisville, KY, USA). All other chemicals used were of the highest purity commercially available and were purchased from Sigma (St. Louis, MO, USA), Aldrich (Milwaukee, WI, USA), or Fisher Scientific (Pittsburgh, PA, USA). Mass spectral analyses were performed at the University of Wisconsin–Madison Biotechnology Center mass spectrometry facility.
Sirtuin expression and purification
Hst2 (69, 70) and SIRT3 (12) was expressed recombinantly in E. coli and purified using Ni-NTA affinity resin as described. The enzyme concentrations were determined using the method of Bradford using BSA as the standard (71).
SIRT3 peptide binding constants by fluorescence anisotropy
SIRT3 (10 μM) was incubated with 20 nM Fluor-ACS2 peptide (see Supplemental Experimental Procedures for synthesis) and various concentrations unlabeled acetyl-lysine analog peptides ranging from 100 nM to 10 mM in 50 mM Tris-HCl (pH 7.5) and 10% (v/v) glycerol in 100 μL total volume. Fluorescence anisotropy (mA) was measured at 25°C using a Panvera Beacon 2000 FP system with 490 nm excitation and 530 nm emission wavelengths. Anisotropy values at each competitor peptide concentration were converted to fraction Fluor-ACS2 bound (FSB) using equation 1.
| (1) |
Where AOBS is the measured anisotropy at the particular competitor peptide concentration, AF is the anisotropy of free Fluor-ACS2, AB is the anisotropy of the fully SIRT3 bound Fluor-ACS2, and Q is the ratio of fluorescence intensities of bound and free Fluor-ACS2. IC50 values were then determined from plots of fraction bound versus competitor peptide concentration using equation 2.
| (2) |
Where FSB0 is the fraction Fluor-ACS2 bound with no added competitor peptide and [peptide] is the concentration of added competitor peptide. Competitor peptide equilibrium dissociation constants were then calculated as previously described (72, 73).
Synthesis of SPOT library membranes
The SPOT peptide libraries were synthesized on an Intavis Respep XL automated peptide synthesis robot. Libraries were synthesized on commercially available amino-functionalized cellulose sheets (INTAVIS; Köln, Germany) according to the manufacturers protocols using standard Fmoc peptide synthesis techniques (for general references of SPOT synthesis see (20, 21)). Double coupling was used for the first seven amino acids, and triple coupling for each subsequent amino acid. Thiotrifluoroacetyl-lysine was added to peptides via use of the Fmoc protected derivative (Fmoc-Lys(CSCF3)-OH; see supplemental methods). Using the Respep XL robot, up to 600 peptides can be synthesized per 10 × 15 cm sheet. After synthesis the side-chain protecting groups were cleaved using 95% v/v TFA, 0.3% v/v Triisopropylsilane, and 4.7% v/v H2O. Membranes were then washed with dichloromethane (4 × 20 mL), DMF (2 × 10 mL), water (2 × 10 mL), and ethanol (2 × 10 mL), and air dried. Successful synthesis was monitored visually by tryptophan fluorescence under a UV lamp at 360 nm.
SIRT3 binding to SPOT libraries
SPOT library membrane was incubated overnight in at 4 °C with 5% w/v BSA in PBST 0.05%. The membrane was incubated with 2 μM of SIRT3 in PBS for 1hr and then with 5% w/v BSA in PBST 0.05% for 30 min. The membrane was then incubated with a primary rabbit antibody specific for SIRT3 for 1 hr in 5% w/v BSA in PBST 0.05%, washed (3 × 5 min with PBST 0.05%), incubated with a goat anti-rabbit IgG-HRP conjugated secondary antibody, washed (3 × 5 min with PBST 0.05%), and developed with Supersignal West Dura from Thermo Scientific. Images of the developed SPOT membranes were captured utilizing Lab Works 4.6 software in a Epi Chem II Darkroom by UVP, and spot intensity was integrated to give integrated optical density (IOD) scores. Machine learning. A linear regression model was trained using a least-squares fit to the IOD measurements obtained via SPOT library screens. In particular, we used a combination of input features that (i) describe the residues flanking the central lysine and (ii) capture global properties of the peptide, such as hydrophobicity and secondary structure information. The induced model provides some insight into the chemical and structural properties that characterize peptides with high SIRT3 binding specificity. The model was then used to predict binding affinity scores for over 22,000 potential SIRT3 binding sites in the mitochondrial proteome. Full details will be described in a subsequent publication (Burr Settles, Brian C. Smith, John M. Denu, and Mark W. Craven; manuscript in preparation).
Solid-phase peptide synthesis of in vitro validation peptides
Acetyl-lysine containing peptides for in vitro validation of our SPOT libraries were synthesized on a 5 μmol scale on an Intavis Respep XL automated peptide synthesis robot according to the manufacturers protocols using standard tBu/Fmoc solid-phase peptide synthesis techniques. Acetyl-lysine was added to peptides via Fmoc-Lys(COCH3)-OH. The protecting groups used were: Boc for lysine and tryptophan, t-butyl for serine and threonine, Pbf for arginine, Trt for cysteine, and tBu for glutamine, asparagine, glutamate, aspartate, and tyrosine. After completion of the synthesis, the resin was rinsed with dichloromethane and dried. The full-length peptide was then deprotected and cleaved from the resin with 95% v/v TFA, 0.3% v/v triisopropylsilane, and 4.7% v/v H2O. The peptide was precipitated with cold ethyl ether and sedimented by centrifuge washing twice with additional cold ethyl ether. The precipitate was dried, redissolved in water, and lyophilized. Masses of the cleaved peptides were confirmed by MALDI-TOF mass spectrometry. Crude peptides were purified by semipreparative RP-HPLC on a C18 small pore column (Grace Vydac, 10 × 250 mm, 10 μm) eluting with a gradient of 0–100% acetonitrile with 0.02% TFA in water with 0.05% TFA. Fractions collected were lyophilized to yield final peptides as dry white powders.
Determination of peptide concentrations
Acetyl-lysine peptide concentrations were determined from the change in NADH absorbance at 340 nm using sirtuin enzyme-coupled assays (60). Typical assay mixtures contained ~100 μM acetyl-lysine peptide, 0.2 mM NAD+, 0.2 mM NADH, 1 mM DTT, 3.3 mM a-ketoglutarate, 1 μM MBP–PncA (nicotinamidase), 3 units of glutamate dehydrogenase from bovine liver, and ~1 μM Hst2 in 20 mM potassium phosphate at pH 7.5. Reactions were run to completion and the peptide concentration calculated from the change in NADH absorbance using an extinction coefficient of 6.22 mM−1 cm−1 and a pathlength of 0.9 cm for 300 μL reactions. Concentrations of peptides containing tyrosine and/or tryptophan were independently determined using calculated extinction coefficients at 280 nm as described previously (74). These two methods routinely gave peptide concentrations within 20% of each other.
Determination of SIRT3 kinetic parameters
The kcat and Km values for different acetyl-lysine containing peptides were determined using a previously described sirtuin enzyme-coupled assay (60). Typical assay mixtures contained 10 to 600 μM acetyl-lysine peptide, 600 μM NAD+, 0.2 mM NADH, 1 mM DTT, 3.3 mM a-ketoglutarate, 1 μM MBP–PncA (nicotinamidase), 3 units of glutamate dehydrogenase from bovine liver, and 0.5 – 1 μM SIRT3 in 20 mM potassium phosphate at pH 7.5. Reactions were carried out in a final volume of 300 μL per well in a clear, flat-bottomed, 96-well plate. All assay components except SIRT3 were preincubated at 25 °C until absorbance at 340 nm stabilized, and the reaction was initiated by the addition of SIRT3. The rates were analyzed continuously by measuring NADH consumption at 340 nm and were determined from the slopes of the initial linear portion of each curve up to 20% conversion using a pathlength of 0.9 cm. The background rates of reactions lacking SIRT3 resulting from the spontaneous formation of nicotinamide or ammonia were subtracted from the initial velocities of the SIRT3-catalyzed reactions. The platereader used for our assay possessed a resolution of 0.001, which corresponds to a detection limit of ~0.2 μM.
Supplementary Material
Acknowledgments
We thank Mark Devries for assistance in SPOT library synthesis and in vitro analysis of SIRT3 substrates and Sam Oliver for help acquiring mass spectra of peptides. This work was supported by a grant from the National Institutes of Health (GM065386). Burr Settles was supported by a grant from the National Human Genome Research Institute (T32HG002760).
References
- 1.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 2.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 3.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 31:212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Lenaz G, Baracca A, Fato R, Genova ML, Solaini G. New insights into structure and function of mitochondria and their role in aging and disease. Antioxid Redox Signal. 2006;8:417–437. doi: 10.1089/ars.2006.8.417. [DOI] [PubMed] [Google Scholar]
- 7.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, BonaFe M, Franceschi C, Tan Q, Boiko S, Yashin AI, De Benedictis G. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol. 2003;38:1065–1070. doi: 10.1016/s0531-5565(03)00209-2. [DOI] [PubMed] [Google Scholar]
- 9.Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, Franceschi C, Passarino G, De Benedictis G. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 11.Weindruch R, Walford RW. The Retardation of aging and disease by Dietary Restriction. C.C. Thomas, Springfield; 1988. [Google Scholar]
- 12.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins sirt3 and sirt5. J Mol Biol. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 16.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49:304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28:6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J Cell Sci. 2010;123:894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 21.Hilpert K, Winkler DF, Hancock RE. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat Protoc. 2007;2:1333–1349. doi: 10.1038/nprot.2007.160. [DOI] [PubMed] [Google Scholar]
- 22.Rathert P, Dhayalan A, Ma H, Jeltsch A. Specificity of protein lysine methyltransferases and methods for detection of lysine methylation of non-histone proteins. Mol Biosyst. 2008;4:1186–1190. doi: 10.1039/b811673c. [DOI] [PubMed] [Google Scholar]
- 23.Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, Jeltsch A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rathert P, Zhang X, Freund C, Cheng X, Jeltsch A. Analysis of the substrate specificity of the Dim-5 histone lysine methyltransferase using peptide arrays. Chem Biol. 2008;15:5–11. doi: 10.1016/j.chembiol.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith BC, Denu JM. Acetyl-lysine Analog Peptides as Mechanistic Probes of Protein Deacetylases. J Biol Chem. 2007;282:37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- 27.Smith BC, Denu JM. Mechanism-based inhibition of sir2 deacetylases by thioacetyl-lysine Peptide. Biochemistry. 2007;46:14478–14486. doi: 10.1021/bi7013294. [DOI] [PubMed] [Google Scholar]
- 28.Smith BC, Denu JM. Sir2 deacetylases exhibit nucleophilic participation of acetyl-lysine in NAD+ cleavage. J Am Chem Soc. 2007;129:5802–5803. doi: 10.1021/ja070162w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 30.Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 32.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 33.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 34.Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, Chang HY, Bohr VA, Ried T, Gozani O, Chua KF. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008 doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell. 2005;17:855–868. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 36.Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell. 2002;10:523–535. doi: 10.1016/s1097-2765(02)00628-7. [DOI] [PubMed] [Google Scholar]
- 37.Cosgrove MS, Bever K, Avalos JL, Muhammad S, Zhang X, Wolberger C. The structural basis of sirtuin substrate affinity. Biochemistry. 2006;45:7511–7521. doi: 10.1021/bi0526332. [DOI] [PubMed] [Google Scholar]
- 38.Hawse WF, Hoff KG, Fatkins DG, Daines A, Zubkova OV, Schramm VL, Zheng W, Wolberger C. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure. 2008;16:1368–1377. doi: 10.1016/j.str.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoff KG, Avalos JL, Sens K, Wolberger C. Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide. Structure. 2006;14:1231–1240. doi: 10.1016/j.str.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Sanders BD, Zhao K, Slama JT, Marmorstein R. Structural basis for nicotinamide inhibition and base exchange in Sir2 enzymes. Mol Cell. 2007;25:463–472. doi: 10.1016/j.molcel.2006.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao K, Chai X, Marmorstein R. Structure of the yeast Hst2 protein deacetylase in ternary complex with 2′-O-acetyl ADP ribose and histone peptide. Structure (Camb) 2003;11:1403–1411. doi: 10.1016/j.str.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 42.Zhao K, Chai X, Marmorstein R. Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli. J Mol Biol. 2004;337:731–741. doi: 10.1016/j.jmb.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 43.Zhao K, Harshaw R, Chai X, Marmorstein R. Structural basis for nicotinamide cleavage and ADP-ribose transfer by NAD(+)-dependent Sir2 histone/protein deacetylases. Proc Natl Acad Sci U S A. 2004;101:8563–8568. doi: 10.1073/pnas.0401057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz D, Chou MF, Church GM. Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol Cell Proteomics. 2009;8:365–379. doi: 10.1074/mcp.M800332-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock RE. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem Biol. 2009;4:65–74. doi: 10.1021/cb800240j. [DOI] [PubMed] [Google Scholar]
- 46.Fjell CD, Jenssen H, Hilpert K, Cheung WA, Pante N, Hancock RE, Cherkasov A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J Med Chem. 2009;52:2006–2015. doi: 10.1021/jm8015365. [DOI] [PubMed] [Google Scholar]
- 47.Bao J, Lu Z, Joseph JJ, Carabenciov D, Dimond CC, Pang L, Samsel L, McCoy JP, Jr, Leclerc J, Nguyen P, Gius D, Sack MN. Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J Cell Biochem. 110:238–247. doi: 10.1002/jcb.22531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper HM, Spelbrink JN. The human SIRT3 protein deacetylase is exclusively mitochondrial. Biochem J. 2008;411:279–285. doi: 10.1042/BJ20071624. [DOI] [PubMed] [Google Scholar]
- 49.Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sweet RM, Eisenberg D. Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. J Mol Biol. 1983;171:479–488. doi: 10.1016/0022-2836(83)90041-4. [DOI] [PubMed] [Google Scholar]
- 52.Bhaskaran R, Ponnuswamy PK. Positional flexibilities of amino acid residues in globular proteins. Int J Pept Protein Res FIELD Full Journal Title:International Journal of Peptide & Protein Research. 1988;32:241–255. [Google Scholar]
- 53.Deleage G, Roux B. An algorithm for protein secondary structure prediction based on class prediction. Protein Eng. 1987;1:289–294. doi: 10.1093/protein/1.4.289. [DOI] [PubMed] [Google Scholar]
- 54.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 55.Janin J. Surface and inside volumes in globular proteins. Nature. 1979;277:491–492. doi: 10.1038/277491a0. [DOI] [PubMed] [Google Scholar]
- 56.Lifson S, Sander C. Antiparallel and parallel beta-strands differ in amino acid residue preferences. Nature. 1979;282:109–111. doi: 10.1038/282109a0. [DOI] [PubMed] [Google Scholar]
- 57.Liu DC, Nocedal J. On the limited memory BFGS method for large scale optimization methods. Mathematical Programming. 1989;45:503–528. [Google Scholar]
- 58.Mitchell TM. Machine Learning. McGraw-Hill; 1997. [Google Scholar]
- 59.Tibshirani R. Regression shrinkage and selection via the lasso. Journal of the Royal Statistical Society: Series B (Methodological) 1996;58:267–288. [Google Scholar]
- 60.Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem. 2009;394:101–109. doi: 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiser AA, Or-Guil M, Tapia V, Leichsenring A, Schuchhardt J, Frommel C, Volkmer-Engert R. SPOT synthesis: reliability of array-based measurement of peptide binding affinity. Anal Biochem. 2005;342:300–311. doi: 10.1016/j.ab.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 62.Tapia V, Bongartz J, Schutkowski M, Bruni N, Weiser A, Ay B, Volkmer R, Or-Guil M. Affinity profiling using the peptide microarray technology: a case study. Anal Biochem. 2007;363:108–118. doi: 10.1016/j.ab.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 63.Jin L, Wei W, Jiang Y, Peng H, Cai J, Mao C, Dai H, Choy W, Bemis JE, Jirousek MR, Milne JC, Westphal CH, Perni RB. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J Biol Chem. 2009;284:24394–24405. doi: 10.1074/jbc.M109.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khan AN, Lewis PN. Unstructured conformations are a substrate requirement for the Sir2 family of NAD-dependent protein deacetylases. J Biol Chem. 2005;280:36073–36078. doi: 10.1074/jbc.M508247200. [DOI] [PubMed] [Google Scholar]
- 65.Lins L, Brasseur R. The hydrophobic effect in protein folding. Faseb J. 1995;9:535–540. doi: 10.1096/fasebj.9.7.7737462. [DOI] [PubMed] [Google Scholar]
- 66.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? Embo J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu W, Lin Y, Yao J, Huang W, Lei Q, Xiong Y, Zhao S, Guan KL. Lysine 88 acetylation negatively regulates ornithine carbamoyltransferase activity in response to nutrient signals. J Biol Chem. 2009;284:13669–13675. doi: 10.1074/jbc.M901921200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Law IK, Liu L, Xu A, Lam KS, Vanhoutte PM, Che CM, Leung PT, Wang Y. Identification and characterization of proteins interacting with SIRT1 and SIRT3: implications in the anti-aging and metabolic effects of sirtuins. Proteomics. 2009;9:2444–2456. doi: 10.1002/pmic.200800738. [DOI] [PubMed] [Google Scholar]
- 69.Jackson MD, Denu JM. Structural identification of 2′-and 3′-O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of beta -NAD+-dependent histone/protein deacetylases. J Biol Chem. 2002;277:18535–18544. doi: 10.1074/jbc.M200671200. [DOI] [PubMed] [Google Scholar]
- 70.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J Biol Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 71.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 72.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 73.Roehrl MH, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein-protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 74.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 75.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 76.Miller BT, Singh RP, Klauda JB, Hodoscek M, Brooks BR, Woodcock HL., 3rd CHARMMing: a new, flexible web portal for CHARMM. J Chem Inf Model. 2008;48:1920–1929. doi: 10.1021/ci800133b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.