

Abstract
Blacklegged ticks (Ixodes scapularis) transmit the causative agent of Lyme disease in the Northeastern and upper Midwestern US. Current research focuses on elucidating biochemical pathways which may be disrupted to prevent pathogen transmission, thereby preventing disease. A genome screening process reported transcripts coding for two putative sulfotransferases in salivary glands of adult Ixodes scapularis and in whole tick extracts of the nymphal and larval stages. Sulfotransferases are known to sulfonate phenolic and alcoholic receptor agonists such as 17β-estradiol, thereby inactivating the receptor ligands. We used bioinformatic approaches to predict substrates for these two sulfotransferases (designated Ixosc Sult 1 and Ixosc Sult 2), and tested the predictions with biochemical assays. Amino acid sequence analysis showed that Ixosc Sult 1 and 2 are members of the cytosolic sulfotransferase superfamily. Homology models of 3D-protein structure were prepared for each tick sulfotransferase based on sulfotransferase X-ray crystal structures. Visualization of the electrostatic surface of the ligand binding cavities showed regions of negative electrostatic charge. Molecular docking identified potential substrates including dopamine, R-octopamine and S-octopamine, which docked into Ixosc Sult 1 with favorable binding affinity and correct conformation for sulfonation. Dopamine, but not R- or S-octopamine, also docked into Ixosc Sult 2 in a catalytic binding mode. Other molecules including 17β-estradiol, pregnenolone and serotonin, did not dock in catalytically active position to either protein. The predictions were tested and confirmed using cytosolic fractions of extracts of whole ticks. Dopamine was found to be a good substrate (Km 0.1 – 0.4 μM) for the native Ixodes scapularis sulfotransferases from larval and nymphal stages regardless of their fed/unfed status. Octopamine–sulfonation was only detected after feeding when gene expression data suggests that only Ixosc Sult 1 is present. These results agree with the docking predictions that octopamine is sulfonated only by Ixosc Sult 1, whereas dopamine is sulfonated by both Ixosc Sult 1 and Sult 2. Because dopamine is known to stimulate salivation in ticks through receptor stimulation, these results imply that the function/s of Ixosc Sult 1 or Sult 2 in the Ixodid tick may include inactivation of the salivation signal via sulfonation of dopamine and/or octopamine.
Keywords: dopamine, octopamine, cytosolic sulfotransferase, tick, Lyme disease, salivation, blacklegged tick, Autodock 4
Blacklegged ticks (Ixodes scapularis) transmit the causative agent of Lyme disease in the United States. The disease is most prevalent in the North East and Great Lakes regions, with 12 to 92 confirmed cases per 100,000 population in 2008 in these states (CDC, Division of Vector-Borne Infectious Diseases, http://www.cdc.gov/ncidod/dvbid/lyme/ld_rptdLymeCasesbyState.htm, accessed 2/15/2010). This tick species serves as the vector for the pathogen, Borrelia burgdorferi, which is a spirochete bacterium responsible for Lyme disease in humans. Small animals such as wild mice, chipmunks and other wild rodents provide the reservoir and seem not to be adversely affected by presence of the bacterium. The spirochete can be passed to humans after a tick, which has previously fed on an infected small animal, subsequently feeds on an individual. Blacklegged ticks (also known as deer ticks) have three life stages (larval, nymphal, adult) and feed once at each life stage. A single infected tick can transmit the pathogen, but only after feeding for greater than about 24 hours (1, 2). Thus, strategies to control incidence of Lyme disease include methods to discourage or shorten feeding duration.
Successful feeding requires an alternating cycle of host blood drawn into the tick and tick saliva secreted into the host through a single buccal canal. The saliva of Ixodid ticks contains many pharmacological substances which overcome host-derived defenses including substances that prevent coagulation of blood at the site, mask pain and itch signals from the site, and suppress the host immune system (3). These substances are secreted into the tick saliva and subsequently are transmitted into the host for action against the host defenses. In addition, successful feeding requires endogenous signaling molecules within the tick to turn on and off salivation and control release of substances into the saliva. Current research is focused on elucidating biochemical pathways which may be disrupted to prevent transmission of pathogen to the host, thereby preventing disease.
As part of a genome screening process, Ribeiro et al (4) identified transcripts coding for two putative sulfotransferases located in the salivary glands of adult Ixodes scapularis and in the whole tick extracts of the nymphal and larval stages. Relative expression levels of the two sulfotransferase proteins (named Ixosc Sult 1 and Ixosc Sult 2) in the tissue was found to be altered upon feeding of larval and nymphal stage ticks (Pichu et al, unpublished results), which suggests the function of the Ixosc Sult 1 and Sult 2 may be important within the process of tick feeding. Based on current knowledge of sulfotransferase function in other species (5, 6, 7), these enzymes could function to control (i) blood clotting in the host, (ii) protein secretion in the tick, or (iii) steroid, neurotransmitter, or thyroid activity in the host or tick. Because any of these activities may be relevant to interruption of the tick feeding process or to interruption in the pathogen transfer process, we sought to determine substrates for Ixosc Sult 1 and Ixosc Sult 2 using bioinformatics and biochemical approaches. All known sulfotransferases catalyze the transfer of an −SO3− moiety from the physiological donor, 3′-phosphoadenosine-5′-phosphosulfate (PAPS), to an acceptor nucleophile such as −OH. This results in formation of a negatively charged sulfate from a neutral −OH. Based on current knowledge of sulfotransferase function (5, 6, 7), there are three possible substrate structure classifications: (a) macromolecules including sugar residues of carbohydrates and proteoglycans, (b) tyrosine residues of proteins and peptides, and (c) small molecule phenols and alcohols. The sulfotransferases catalyzing these activities in other species include two major divisions based on their subcellular locations: membrane-bound sulfotransferases utilize a- and b-class substrates, while cytosolic sulfotransferases utilize c-class substrates.
This paper describes comparative analyses of structure and sequence amongst diverse sulfotransferases with preparation of 3D-models of Ixosc Sult 1 and 2 in order to predict ligand binding and the interaction between the ligand and Ixosc Sult 1 and 2. Our approaches include global and local amino acid sequence similarity, 3D structure conservation among sulfotransferases, and protein structure modeling with ligand docking. The predictions were then tested and confirmed using native enzyme tissue homogenates. Dopamine was found to be a good substrate for both Ixosc Sult 1 and Sult 2, while (R, S)-octopamine was sulfonated only by Ixosc Sult 1.
RESULTS AND DISCUSSION
Global sequence similarity
Our first approach to bioinformatic characterization of the black-legged tick sulfotransferases was to conduct multiple protein sequence comparisons for global sequence similarity of Ixosc Sult 1 and Sult 2 amongst over 500 distinct members of the Sulfotransfer-1 family of Pfam from diverse species. As shown in Figure 1a, the sulfotransferases clearly diverge according to their subcellular location and function. Membrane bound-sulfotransferases group in clads shown on the left side of this circular dendrogram; for instance, the tyrosine protein sulfotransferases (TPST), and the glycosaminoglycan sulfotransferases (OST, NDST, chondroitin ST, CHST). On the right half of the circular dendrogram group the clads containing the cytosolic sulfotransferases referred to most commonly by the abbreviations ST or SULT. The SULT 1, 2 and 3 families (mostly mammalian), are well-characterized and have known substrates including endogenous and exogenous phenols, alcohols. The SULT 4, 5 and 6 families are also mostly mammalian, but do not yet have known substrates. Our results show that Ixosc Sult 1 and Sult 2 globally align within the cytosolic sulfotransferase superfamily, and thus are likely to utilize small molecule alcohols or phenols as substrates. This global sequence similarity analysis also indicates that Ixosc Sult 1 and 2 are not closely related to the membrane-bound sulfotransferases and, thus, are unlikely to control blood clotting in the host or protein secretion in the tick.
Figure 1.

Phylogenetic relationship of Ixosc Sult 1 and Sult 2 to other sulfotransferases. (A) Full distance tree of protein family Sulfotransfer-1 ClustalW+ multiple alignment displayed in circular form. Abbreviations are defined in the text. (B) Phylogenetic best scoring maximum likelihood tree showing relation to representative cytosolic sulfotransferases. Numbers indicate bootstrap values from 100 trees. All proteins are identified by their UniProt codes. Proteins discussed in the text are noted with their common abbreviated names and example substrates.
Phylogenetic analysis
The global amino acid sequence analysis also allowed evaluation of the phylogenetic relationship of Ixosc Sult 1 and Sult 2 within the cytosolic sulfotransferase superfamily (Figure 1b). Specifically, the maximum likelihood tree analysis shows that Ixosc Sult 1 and Sult 2 amino acid sequences are most closely related to the cytosolic sulfotransferases from C. elegans (ceST1, Uniprot q9u2z2_caeel) and C. Brassica (Uniprot a8xgq0_caebr). The bootstrap values for these relationships are 84 and 91, reflecting good confidence. ceST1 has been shown to catalyze sulfonation of a variety of phenols, but no endogenous substrate has yet been identified (8).
We found it interesting that Ixosc Sult 1 and Sult 2 were more closely related phylogenetically to the mammalian sulfotransferases (SULT1, SULT2, and SULT3 families) than to sulfotransferases from other arthropods and non-mammalian species. In the phylogenetic analysis many non-mammalian cytosolic sulfotransferases located to a separate clad with bootstrap value of 100 (Figure 1b). A few of these non-mammalian sulfotransferases have been characterized including bmST (Bombyx mori, silkworm, Uniprot a0pcf9_bommo), dmST1-4 (Drosophila melanogaster, fruitfly), and that from Spodoptera frugiperda (fall army worm, Uniprot q26490_spofr). The sulfotransferase isolated from Bombyx mori was found to catalyze sulfonation of 4-nitrocatechol although no endogenous substrate has yet been identified (9). The Spodoptera frugiperda enzyme sulfonates retinol (10). The cytosolic sulfotransferases from D. melanogaster (dmST1-4) catalyze sulfonation of vanillin, 1-naphthol, 4-nitrophenol, 4-hydroxyecdysone, and dopamine (11).
We also compared overall sequence identity and homology of Ixosc Sult 1 and Sult 2 versus individual proteins from other species after pairwise alignment. As observed previously for sulfotransferases from diverse species, the sequence identity was in the range of 25-35%, and sequence homology in the range of 40-55% over the circa 300 residue sulfotransferase domain.
Local sequence similarity and motif regions
After observing that the global sequence analysis showed the Ixosc Sult 1 and Sult 2 to be cytosolic sulfotransferases, local amino acid sequence comparisons were made. Because all sulfotransferases bind PAPS, the residues folding into the PAPS binding pocket are highly conserved. We found six conserved motifs spaced throughout the ~300 amino acid sulfotransferase domain of the cytosolic sulfotransferases (Figure 2c). Motifs 1, 2, 3 and 6 are the classic cytosolic sulfotransferase motif regions (7, 12) previously known to make up a portion of the binding cavity for the cofactor PAPS, and containing the catalytic histidine. Motif 1 contains PKxGTTW which has been referred to as signature sequence Region I (Weinshilboum 1997) and the 5′PSB motif (7, 12). Motif 2 contains the absolutely conserved catalytic histidine. Motif 3 contains RNPKD××VS which has been referred to as the 3′PB motif (7, 12). Motif 6 contains RKG××GDWK which has been referred to as signature sequence Region IV (13). Phylo-mLogo detected two additional motif regions that 3D-structure analysis found also to locate in the PAPS binding region (Motifs 4 and 5). All six local sequence motif regions were highly conserved within Ixosc Sult 1 and Sult 2 (Figure 2a).
Figure 2.
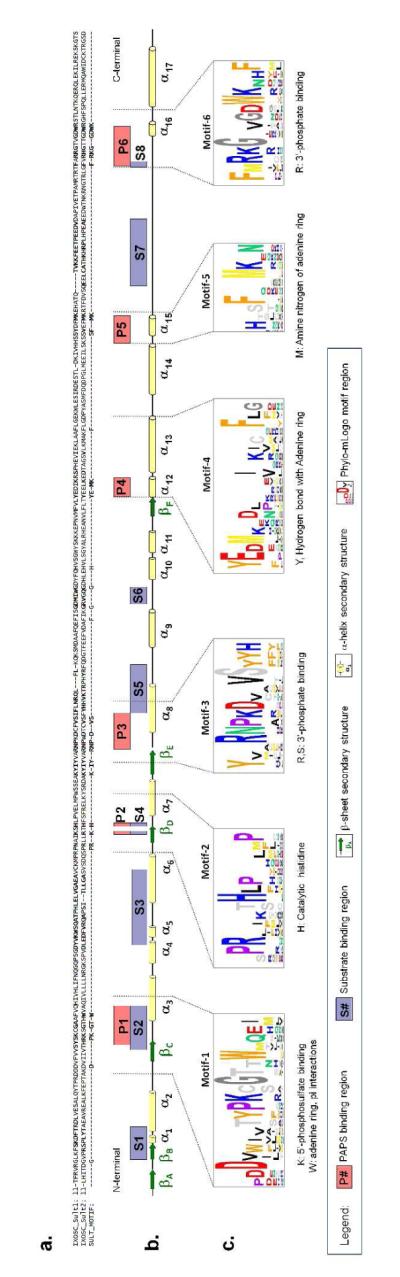
Mapping of cytosolic sulfotransferase primary and secondary structure to sequence motif and ligand binding regions. (A) Ixosc Sult 1 and Sult 2 primary sequence aligned with conserved sulfotransferase motif residues. (B) Conserved sulfotransferase secondary protein structure is noted by cylinders (α--helix), arrows (β--strands), and lines (loop/turn). Sequence regions folding to form the PAPS binding pocket are labeled P1-P6 (in red highlight). Sequence regions folding to form the substrate binding pocket are labeled S1-S9 (in blue highlight). (C) Phylo-mLogo motif regions are shown in detail; the one amino acid letter height corresponds to the relative conservation of the particular amino acid. α--Helix and β-strand numbering is consistent with Alalli-Hassani et al (6).
Sulfotransferase structure conservation and integrated analysis
Integrated 3D-structural and protein sequence analysis of the diverse set of crystallized sulfotransferases was used to further predict function of the Ixosc Sult 1 and Sult 2 by comparison (Figure 2). The sequence regions folding into the PAPS binding pocket were previously identified (7, 12). Thus, we specifically sought to identify regions of sequence which fold into the substrate binding pocket, and compare these residues in Ixosc Sult 1 and Sult 2 sequence with characterized sulfotransferases. Substrate binding pocket residues are difficult to identify by primary sequence comparisons because they are minimally conserved across sulfotransferase families. We found that superimposition of 31 published cytosolic sulfotransferase structures and integrated analysis with the aligned sequences identified succinct aligned regions which consistently fold into the substrate binding pockets of the crystallized sulfotransferase structures (Figure 2b, S1 – S8). From this information, we predicted that the residues listed in Table 1 would contain the residues folding into the substrate-binding pocket for Ixosc Sult 1 and Ixosc Sult 2.
Table 1.
Residues of Ixosc Sult 1 and 2 predicted to fold into the substrate binding pocket.
| Protein | S1a | S2 | S3 | S4 | S5 | S6 | S7 |
|---|---|---|---|---|---|---|---|
| Ixosc Sult 1 | GLKFSKDFT | SYSKCGAAFVQ | D–YVKWSQATPHLELVGAEA– | K–H | VSIFLNRQLFL | GDMIWG | FEETPEEDVDA |
| Ixosc Sult 2 | GVPRSPLYT | THRKSGTHWVA | D–LEDFVRQAPSITLLGASVS | R–H | YHHVKTRPHYR | GRVGQG | HRPLHPEAEED |
| SULT1A3 | GVPLIKYF– | TYPKSGTTWVS | YVRVPFLEVNDPGEPSGLET– | K–H | YYHFHRMEKAH | GEVSYG | YTTVPQELMDH |
S#, refers to substrate-binding region number, as indicated on Figure 2b.
The residues underlined provide charge interaction for dopamine and octopamine ligands.
Protein modeling and binding pocket characterization
As described in the methods section, multiple test homology 3D-structure models were created and evaluated (~500 of Ixosc Sult 1, ~400 of Ixosc Sult 2). After evaluation of all model preparation criteria, one model each for Ixosc Sult 1 (#470) and Sult 2 (#780) were judged the highest quality models and were chosen for further studies. These models were based on the X-ray crystal structure coordinates of 1LS6 (human SULT1A1). Ligand accessible cavities of both models (Ixosc Sult 1 and 2) were calculated in the absence of any ligands. The resulting pocket demonstrated the characteristic shape (14) for cytosolic sulfotransferases which is composed of a ligand binding region and a cofactor binding region connected by a channel through which −SO3 transfer occurs (Figure 3a, 3b).
Figure 3.

Protein structure modeling for Ixodes scapularis Sult 1 and Sult 2. Homology model of Ixosc Sult 1 (A) and Ixosc Sult 2 (B): yellow closed surface indicates water-accessible cavity available for ligand binding. Electrostatic charge surface display of exposed binding cavity of Ixosc Sult 1 (C) and Ixosc Sult 2 (D). Interior of binding pockets were exposed by dissection. Red color indicates partial negative charge, blue color indicates partial positive charge, white color indicates neutral charge. Substrate binding cavity region of Ixosc Sult 1 (C) contains distinct red color indicating negatively charged binding surface available to the substrate. Molecules in stick form show results from docking experiments (PAPS, dopamine).
Electrostatic surface calculation facilitated identification of key features of the defined cavities. Visualization of the electrostatic charge surface of the residues surrounding the substrate binding cavity of Ixosc Sult 1 and Sult 2 homology models revealed a region with substantial negative charge; this region may provide attractive binding interactions for a positive charge region in a ligand (Figure 3c, 3d). In the case of Ixosc Sult 1, Aspartate 229 provided the negatively charged side-chain; whereas in Ixosc Sult 2, Glutamate 245 provided the negatively charged side-chain. The negatively charged region located on the edge of the binding pocket, ~10 Å away from the sulfate group of PAPS. This suggests that molecules with a basic aliphatic amine functional group ‘tail’, such as the monoamine neurotransmitters, may be preferred ligands for the Ixosc sulfotransferases. Specific dopamine sulfotransferases are known to exist in humans (SULT1A3) and in mice (Sult1d1). The 31 superimposed crystal structure coordinate sets (see above) contained two structures of SULT1A3 (pdb: 1cjm, pdb: 2a3r, highest quality model). Comparison of the Ixosc Sult 1 and Sult 2 models with the crystal structure coordinates of human SULT1A3 (2a3r) showed that the critical amine-interacting residue in SULT1A3 (Glu146) (15) nearly superimposed with Asp229 and Glu245 identified visually for Ixosc Sult 1 and Sult 2, respectively. Surprisingly, the Glu146 of SULT1A3 was located in substrate-binding-region number 5 (S5) of the primary sequence, while the Asp229 and Glu245 of Ixosc Sult 1 and Sult 2 located in substrate-binding-region number 7 (S7) (Table 1). In the 3D structure these two regions folded just adjacent to one another. Thus, primary sequence alone seems to provide very limited utility for prediction of substrate selectivity in distant sulfotransferases.
Docking of PAPS and potential substrates
Molecular docking studies were used to quantitatively explore ligand binding preferences on the homology model for each sulfotransferase using Autodock 4 and MGL AutodockTools. First, the cofactor PAPS was docked into each model. The resulting binding mode of PAPS was consistent with the binding mode in the published sulfotransferase crystal structures, and placed the 5′-phosphosulfate in catalytically competent position relative to the conserved histidine. Subsequently, potential substrate molecules were docked into the PAPS/enzyme complex. Dopamine, octopamine and serotonin were chosen for docking based on the results of the binding pocket characterization indicating presence of negatively-charged binding region in the substrate binding pocket. 17β-Estradiol and pregnenolone were chosen for docking because of their neutral molecular charge and characteristic of being common sulfotransferase substrates. We used a docking strategy which repeatedly searches for low energy binding conformations and calculates the frequency of each low energy binding mode (16). High frequency denotes greater confidence. Catalytically competent binding modes were defined as those in which the docked small molecule (dopamine, octopamine, serotonin, estradiol, or pregnenolone) oriented with the −OH within 3-4 Å of the NZ of catalytic histidine and with the oxygen within 3-4 Å of the PAPS sulfur atom simultaneously. Dopamine consistently docked in catalytic mode with reasonable affinity to both Ixosc Sult 1 and Sult 2, indicating that it would be a substrate for either sulfotransferase (Figure 4a, 4d; Table 2). (R)-Octopamine and (S)-octopamine consistently docked as a substrate to Ixosc Sult 1, but not to Ixosc Sult 2, indicating some level of substrate selectivity between the two sulfotransferases (Figure 4b, 4c; Table 2). Serotonin docked to both sulfotransferases, but only in binding modes that positioned the −OH too distant (5-6 Å) from the catalytic atoms for catalysis to occur. Neither 17β-estradiol nor pregnenolone docked in a catalytically competent mode.
Figure 4.

Binding interactions between Ixosc Sult 1/PAPS complex and docked (A) dopamine, (B) (R)-octopamine, and (C) (S)-octopamine, respectively. Similarly, interactions between (D) Ixosc Sult 2 complex and docked dopamine. PAPS, ligands, and catalytic residues (Ser-Lys-His) are displayed in stick and other interacting residues are displayed in line form with text labels. Interactions were calculated using Ligand Explorer.
Table 2.
Computational prediction of Ixodes scapularis sulfotransferase substrate selectivity by molecular docking.
| Docked Ligand |
Ixosc Sult 1 model Predicted Kd (μM)a |
Ixosc Sult 2 model Predicted Kd (μM)a |
|---|---|---|
| Dopamine | 40 | 67 |
| (S)-Octopamine | 71 | ndb |
| (R)-Octopamine | 76 | nd |
| Serotonin | nd2 | nd |
| 17β-Estradiol | nd | nd |
| Pregnenolone | nd | nd |
Predicted Kd values determined as described in Methods section.
Not dock, Indicates ligand did not consistently dock in a catalytically competent binding mode.
Sulfotransferase activity in whole tick extracts
In order to test the computational predictions, the cytosolic (100,000×g supernatant) fractions of native enzyme preparations were incubated with each potential substrate under sulfotransferase conditions. We utilized whole tick extracts from two life stages of the Ixodes scapularis ticks: larval and nymphal. The larval stage is significant because larval ticks feeding on infected animals become infected with the Lyme disease spirochete bacterium. The nymphal stage is significant because nymphal ticks most often transfer the spirochete to human hosts. We utilized ticks which had been fed (allowed access to laboratory animal host under controlled conditions), and ticks which had not been allowed to feed. The rationale for comparing fed and unfed ticks comes from our work which will be published separately (Pichu, Mather, and King, unpublished) that showed that expression levels of Ixosc Sult 1 and Sult 2 was altered by feeding status. Dopamine was found to be a good substrate (Km 0.1 – 0.4 μM) for the native Ixodes scapularis sulfotransferases at larval and nymphal stage regardless of their fed/unfed status (Table 3). Octopamine–sulfonation only was detected after feeding when the gene expression data suggests that only Ixosc Sult 1 is present (Pichu et al, unpublished). These results agree with the docking predictions that octopamine should be sulfonated only by Ixosc Sult 1, whereas dopamine should be sulfonated by both Ixosc Sult 1 and Sult 2. In agreement with the docking predictions, β-estradiol, pregnenolone, and serotonin were not substrates (at 1 μM or 20 μM) for the native tick sulfotransferases.
Table 3.
Sulfotransferase activity in cytosolic fractions of larval and nymphal stage Ixodes scapularis whole tick extracts.
| Dopamine | Octopamine | ||||
|---|---|---|---|---|---|
| Tissue | Vmaxa pmol min−1 mg−1 |
Km dopamine a μM |
Km PAPS a μM |
μM | ν b pmol min−1 mg−1 |
| Nymph un-fed | 25 ± 5 | 0.4 ± 0.5 | 3.2 ± 1.8 | 1, 10 | None detected |
| Nymph fed | 48 ± 7 | 0.2 ± 0.4 | 1.1 ± 0.9 | 1 | 6.4 |
| Larvae un-fed | ν = 0.24 pmol min−1 mg−1 at 10 μM dopamine b | Not done | |||
| Larvae fed | 30 ± 6 | 0.1 ± 0.5 | 2.5 ± 1.7 | 1 | 1.8 |
Vmax, Km dopamine, and Km PAPS were calculated after analysis of sulfonation including a range of dopamine (1.48 – 3.7 μM) and PAPS (2 – 10 μM) concentrations. Data plots are available in supplementary materials, Figure 5.
ν was calculated after analysis of sulfonation under single PAPS concentration (4 μM) and limited number of substrate concentrations (1, 10 μM).
The significance of these findings relate to the known role of dopamine in stimulation of the salivation signal in ticks. Dopamine binds to a dopamine D1 receptor and stimulates saliva secretion in ticks through the activation of adenylyl cyclase and formation of intracellular cyclic AMP causing salivary secretion (17). Octopamine also exhibits neuromodulatory functions in arthropods (18), but it is not known if it functions by the same mechanism as dopamine. It is generally accepted that sulfated ligands do not bind to their receptors because of the replacement of neutral hydrogen-bonding −OH with the negatively-charged sulfate (5). To directly test this assumption, we used a system to test for agonist activity of dopamine-sulfate at the D2 dopamine receptor by expressing D2R with KIR3 in Xenopus oocytes (19, 20). However, even with high levels of D2R expression, dopamine-sulfate (4 nM) did not elicit detectable activation of D2R in any of the oocytes injected with both D2R and KIR3 cRNA. In contrast and in the same oocytes, subsequent perfusion of 4 nM dopamine produced robust activation of D2R as measured by an increase in KIR3 conductance (Supplementary Figure 6). This data supports the conclusion that dopamine-sulfonation in Ixodes scapularis ticks may result in inactivation of salivary secretion. Saliva secretion is an alternating cycle, where ticks draw in host blood and salivate out excess fluid and pharmacologically active molecules. Interruption of this cycle may interfere with successful feeding and pathogen transmission into host. Taken together, these results imply that the function/s of Ixosc Sult 1 and Sult 2 in the Ixodid tick salivary gland may include inactivation of the salivation signal via sulfonation of dopamine and/or octopamine.
METHODS
Global sequence similarity
The Ixosc Sult 1 and Sult 2 sequences were compared with a robust set of 768 highly diverse sulfotransferases which were identified via the Protein Family database using the Sulfotransfer-1 family (21) (http://www.sanger.ac.uk/Software/Pfam/, PF00685, accessed July 27, 2007, downloaded with all gaps removed). The sulfotransferase domain contains circa 300 amino acids, therefore fragments (defined as <200 residues) were discarded; duplicate sequences were also discarded. Multiple amino acid sequence comparisons of Ixosc Sult 1 and Sult 2 with the remaining 586 sulfotransferase sequences were conducted using Accelrys SeqLab (GCG version 11.1). The guide (distance) tree for the multiple sequence alignment was prepared after slow/accurate pairwise alignments with ClustalW+ (1.0 gap opening penalty, 0.1 gap extension penalty, GONNET scoring matrix). The multiple sequence alignment was subsequently conducted in ClustalW+ using the BLOSUM scoring matrix (1.0 gap opening penalty, 0.1 gap extension penalty, 4 residue gap separation minimum). The distance tree showing the relationship of the set of 586 sulfotransferases was displayed in circular form using the Interactive Tree of Life online tool (22) (http://itol.embl.de/).
Phylogenetic analysis
A set of 50 sulfotransferase amino acid sequences was analyzed for phylogenetic relationships; the set included representatives from clads containing SULT families 1-6, and the clad marked arthropod in Figure 1A). Because different alignment methods and parameters will produce different results, we used MCoffee (23) (server version: http://www.tcoffee.org/) to compute and then combine the output of several multiple sequence alignment packages (PCMA, POA, MAFFT, Muscle, TCoffee, ClustalW, ProbCons, DialignTX). The resulting multiple alignment was then further refined by including X-ray crystal structure information via Expresso (3DCoffee) (available at http://www.tcoffee.org/) (24, 25). This server based software combines sequence-sequence (ClustalW), structure-structure (SAP), and sequence-structure (Fugue) information to assemble a progressive alignment (26). Expresso was used under the advanced mode with user control of input structures (the coordinates of the superimposed crystal structures, described in Sulfotransferase structure conservation section). The MCoffee and Expresso (3DCoffee) methods also estimate the local consistency between the final alignment and the individual alignments by measuring the fraction of individual alignments that are consistent for each residue.
Subsequently, the refined alignment was used to prepare a phylogenetic maximum likelihood tree with bootstrap support values. The tree was prepared using RAxML (Randomized Axelerated Maximum Likelihood) rapid bootstrapping and subsequent ML search (27) accessed through the MSA hub on the web server of the Vital-IT unit at the Swiss Institute of Bioinformatics (http://myhits.vital-it.ch). The best scoring maximum likelihood tree (prepared from 100 bootstrapped trees) was displayed using the Interactive Tree of Life online tool (22). Several flavanoid sulfotransferases from plants were used as the out-group (FSTL arath, F3ST flach, F4ST flach).
Local sequence similarity and sequence motifs
A set of 174 sulfotransferase amino acid sequences (clads containing all members of cytosolic SULT families 1-6, and clad marked arthropod in Figure 1A) was analyzed for local sequence motifs, and the conserved motif residues of cytosolic sulfotransferases were determined with Phylo-mLogo (28) (http://cg1.iis.sinica.edu.tw/new/phylomlogo/). Phylo-mLogo considers the frequency of every amino acid in each column and calculates the variable and homogenous regions of multiple sequence alignments by base frequencies and entropies. The resulting sequence logo diagrams represent informative patterns in a multiple sequence alignment.
Sulfotransferase structure conservation and integrated analysis
Structural 3D coordinate sets were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB, www.rcsb.org, 36 structures downloaded July 2006) using the keyword search “sulfotransferase” (29). The keyword search collected one additional protein, not considered a sulfotransferase (1SUR, PAPS reductase), which was discarded from the analysis. UCSF Chimera software (www.cgl.ucsf.edu/chimera, 6/21/2007 version, win32 platform) was used for integrated 3D structural and protein sequence analyses (30, 31). Using Chimera, each structure was superimposed onto the coordinates of 1AQU using the MatchMaker feature (alpha-carbon of each aligned residue overlaid, iterated by pruning long atom pairs). 1AQU, the first published cytosolic sulfotransferase structure, was chosen as the reference. The transposed coordinates of each of the 36 superimposed structures, including crystal ligands, were saved and used for all further analyses. The advantage of using Chimera MatchMaker feature for the superimposition step was the pruning feature–that is, improving the superimposition in an iterative fashion starting with all atoms matched, and each iteration disregarding the most mismatched atoms and refining the superimposition based on the remaining atom pairs. The structural coordinates of the crystal ligands of the superimposed structures were used to identify the ligand binding regions across all of the sulfotransferases. Specifically, residues contributing to substrate binding were identified by highlighting all protein atoms within 5 Å of any crystal ligand atom (substrate or inhibitor).
Protein modeling and characterization
Homology model preparation and evaluation
Because the success of homology modeling is dependent on the accuracy of the amino acid alignment of template and model sequence, the highly refined alignment prepared for the phylogenetic analysis was used as the starting point for homology modeling. All homology models were created using the commercial software Accelrys Discovery Studio (DS) v 2.1, Build Homology Models protocol, which is based on the MODELER algorithm (32). Parameter settings were generally as follows: cut overhangs, true; disulfide bridges, false; cis-prolines, false; additional restraints, false; copy ligands, false; optimization level, none; refine loops, false. Multiple models (circa 900) were created to test the effect of template choice and variables such as side-chain refinement (CHI-ROTOR) (33), loop refinement (LOOPER) (34), and CHARMM minimization. In separate experiments, low, medium and high optimization levels were also tested.
Each model was evaluated based on a number of parameters. The DS Protein Health tool provided quantitation of the validity of main-chain conformations (Ramachandran), side-chain conformations (rotamer library), peptide planarity, cis-peptides, alpha-carbon distances, bond lengths, and bond angles. In addition, each model was superimposed on its template and judged for replication of the general sulfotransferase fold. Based on these criteria, one model each for Ixosc Sult 1 (#470) and Sult 2 (#780) were chosen for further use. For simplicity, these will be referred to as Ixosc Sult 1 model and Ixosc Sult 2 model.
Binding pocket characterization
The binding sites of the Ixosc Sult 1 and Sult 2 models were defined with DS based on the cavities inside the proteins. These solvent accessible cavities were calculated with an opening site of 10 Å and grid size of 0.7 Å. The analyzed grid points were converted to hydrogen atoms and visualized in soft, closed surface form by DS.
The electrostatic charge calculation for atoms in the Ixosc Sult 1 and Sult 2 models was performed in DS using Delphi with default parameters. Delphi allows electrostatic potential determination within the whole protein’s electrostatic field, resulting in more physically realistic computational experiments. Because the catalytic region is located in the core of the folded protein, each protein was visualized as dissected into half in order to visualize the electrostatic surface of the residues surrounding the ligand cavity.
Homology model optimization
The homology model for Ixosc Sult 1 was optimized for ligand docking via limited side-chain refinement and minimization in the presence of cofactor PAPS. The side-chain refinement module of DS 2.1 optimizes the protein side-chain conformation based on systematic searching of side-chain conformation and CHARMm energy minimization using the ChiRotor algorithm (33). Based upon the binding pocket characterization results, the following residues of the Ixosc Sult 1 model were selected for the side-chain refinement: Asp9, Phe10, Asp13, Ala39, Leu118, Asn119, Phe123, Ser207, Tyr208, Arg248, Gly249, and Thr250. Subsequently, PAPS was docked to the side-chain refined structure, and the structure was minimized in the presence of docked PAPS as follows. A CHARMm forcefield was applied to the receptor and PAPS complex, and minimization was carried out with following settings: Algorithm, Smart Minimizer; Max Steps, 200; RMS Gradient, 0.1; Energy Change, 0.0; Implicit Solvent Model, None; Nonbond List Radius, 14.0; Nonbond Higher Cutoff distance, 12.0; Nonbond Lower Cutoff distance, 10.0; Electrostatics: Spherical Cutoff (Kappa, 0.34, Order, 4); Minimization Constraints, None; Apply SHAKE Constraint, False.
Based upon the binding pocket characterization results, the Ixosc Sult 2 homology model was used without side-chain refinement. Thus, PAPS was docked directly to the Ixosc Sult 2 model and the structure was minimized in the presence of docked PAPS as described for Ixosc Sult 1.
Docking of PAPS and potential substrates
Because current docking protocols can only dock a single ligand at a time, PAPS was first docked into the empty Ixosc Sult 1 and 2 model receptors. Then dopamine, (R)-octopamine, (S)-octopamine, 17β-estradiol, pregnenolone, and serotonin were individually docked as ligands into each receptor-PAPS complex. Autodock 4 (35, 36, 37) with Autodock Tools 1.4.6 (38, 16) on a win64 platform was used for all docking experiments. General docking method was as described previously (14) including Lamarckian genetic algorithm search method with 2500000 evaluations, 27000 generations, and 300 population size. We used the option in Autodock 4 for a fully flexible ligand with a non-flexible receptor for docking. The grid box dimensions, based on the identified binding cavities from DS, were large enough to cover all possible rotations of the ligand. Eight separate docking experiments (of 100 runs each) starting from different random conformations were conducted. The 800 resulting docked conformations were clustered, and those within 2.0 Å rmsd were considered a single reproducible result. Docked clusters were visualized, binding interactions were assessed, and atomic distances calculated using Autodock Tools and Discovery Studio. Clusters were considered to be in catalytic position when all of the following conditions were met: OH of ligand within 3-4 Å of the nitrogen atom of the catalytic histidine, OH of ligand within 3-4 Å of sulfur atom of the 5′-phosphosulfate of PAPS. Binding affinity (kcal/mol) was calculated in Autodock 4 as the difference in Gibbs free energy between the bound state and unbound state of the ligand, incorporating van der Waals interactions, hydrogen bonding interactions, desolvation energy, electrostatic interactions, total free energy, and torsional free energy. The dissociation constant (Kd) was calculated by the equation, ΔG = R*T*ln(Kd), where R is the gas constant, and T is 298.15 Kelvin.
Sulfotransferase activity in native tissue
Tissue preparation
All animal studies were approved by the Institutional Animal Care and Use Committee (protocol number AN01-12-014) and were performed in accordance with all national and local guidelines and regulations. Laboratory-derived Ixodes scapularis larvae and nymphs were used for the study. The fed nymphs and larvae were allowed to feed on mice for 48 hr (39), while un-fed nymphs and larvae from the same lot were kept in control conditions for the same time period. The un-fed larval tissue was derived from a pool of 200 whole un-fed larvae which were homogenized in 0.1 mL ice-cold 20 mM potassium phosphate pH 7.0, 1 mM dithiothreitol, 1 mM PMSF. The un-fed nymphal tissue was derived from a pool of 180 whole un-fed nymphs which were homogenized in 0.1 mL buffer. Whole fed larval (200/0.1 mL) and whole fed nymphal (50/0.1 mL) tissues were prepared similarly. The tissue was homogenized by sonication followed by centrifugal separation (1 hr) and collection of the cytosolic fraction (100,000 × g supernatant). All manipulations were at 4°C.
Sulfotransferase assay
Reaction mixtures contained radiolabeled sulfonyl donor [35S]-3′-phosphoadenosine-5′-phosphosulfate (PAP35S), substrate in 20 mM potassium phosphate/1 mM EDTA (pH 7.0), tissue cytosol, and 20 mM potassium phosphate with 1 mM EDTA (pH 7.0) (40). After timed incubation at 37°C, reactions were stopped by heating in boiling water for 30 sec. After centrifugation at 10,000×g for 1 min to pellet the protein, an aliquot of the reaction mix was injected onto a Hypersil Duet C18/SAX column (150 × 4.6 mm, 4 micron). Components were separated and eluted using a linear salt gradient of 10-50 mM ammonium bicarbonate pH 8.0 in 10% acetonitrile. Radiolabel was detected and quantified on a flow scintillation analyzer (Packard Bioscience, 500TR series) with Perkin Elmer Ultima Flo-M scintillation cocktail. PAP35S was eluted at 1.9 min, dopamine-sulfate at 2.9 min, and octopamine-sulfate at 3.0 min.
Assay conditions were adjusted to ensure initial-rate conditions for the kinetic experiments (linear with time, linear with protein concentration, less than 20% substrates utilized). Accordingly, the kinetic experiments utilized 1.48 – 3.7 μM dopamine, 2 – 10 μM PAP35S, 8 mg/mL un-fed larval cytosol, 0.4 mg/mL fed larval cytosol, 0.85 mg/mL un-fed nymphal cytosol, or 0.2 mg/mL fed nymphal cytosol, and were incubated for 30 min at 37°C. Product formation was quantified by flow scintillation counting, and rates of sulfonation were calculated as pmol product formed per minute per mg cytosolic protein. Kinetic constants (Km dopamine, Km PAPS, Vmax) were determined using non-linear regression fitting to the Michaelis-Menten two-substrate equation by the Enzyme Kinetic (Pharmacology) Module of Sigma Plot 11.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by the University of Rhode Island Council for Research (RSK), and by the University of Rhode Island Foundation (RSK, fund #5920). E. B. Yalcin was partially supported by National Institute of Health grant RO1 AI 37230 (TNM). Accelrys Discovery Studio software was made available through the RI-INBRE Bioinformatics Core Facility (NIH P20 RR16457).
Abbreviations
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
The authors report no conflicts of interest.
REFERENCES
- 1.Piesman J, Mather TN, Sinsky RJ, Spielman A. Duration of tick attachment and Borrelia burgdorferi transmission. J. Clin. Microbiol. 1987;25:557–558. doi: 10.1128/jcm.25.3.557-558.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piesman J, Eisen L. Prevention of tick-borne diseases. Annu. Rev. Entomol. 2008;53:323–343. doi: 10.1146/annurev.ento.53.103106.093429. [DOI] [PubMed] [Google Scholar]
- 3.Wikel SK. Tick modulation of host immunity: an important factor in pathogen transmission. Int. J. Parasitol. 1999;29:851–859. doi: 10.1016/s0020-7519(99)00042-9. [DOI] [PubMed] [Google Scholar]
- 4.Ribeiro JM, Alarcon-Chaidez F, Francischetti IM, Mans BJ, Mather TN, Valenzuela JG, Wikel SK. An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochem, Mol. Biol. 2006;36:111–129. doi: 10.1016/j.ibmb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Chapman E, Best MD, Hanson SR, Wong CH. Sulfotransferases: Structure, mechanism, biological activity, inhibition, and synthetic utility. Angew. Chem. Int. Ed. Engl. 2004;43:3526–3548. doi: 10.1002/anie.200300631. [DOI] [PubMed] [Google Scholar]
- 6.Allali-Hassani A, Pan PW, Dombrovski L, Najmanovich R, Tempel W, Dong A, Loppnau P, Martin F, Thornton J, Edwards AM, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biol. 2007;5:e97, 1063–1078. doi: 10.1371/journal.pbio.0050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Negishi M, Pedersen LG, Petrotchenko E, Shevtsov S, Gorokhov A, Kakuta Y, Pedersen LC. Structure and function of sulfotransferases. Arch. Biochem. Biophys. 2001;390:149–157. doi: 10.1006/abbi.2001.2368. [DOI] [PubMed] [Google Scholar]
- 8.Hattori K, Inoue M, Inoue T, Arai H, Tamura HO. A novel sulfotransferase abundantly expressed in the dauer larvae of Caenorhabditis elegans. J. Biochem. 2006;139:355–362. doi: 10.1093/jb/mvj041. [DOI] [PubMed] [Google Scholar]
- 9.Hattori K, Hirayama M, Suzuki H, Hamamoto H, Sekimizu K, Tamura HO. Cloning and expression of a novel sulfotransferase with unique substrate specificity from Bombyx mori. Biosci. Biotechnol. Biochem. 2007;71:1044–1051. doi: 10.1271/bbb.60703. [DOI] [PubMed] [Google Scholar]
- 10.Grun F, Noy N, Hammerling U, Buck J. Purification, cloning, and bacterial expression of retinol dehydratase from Spodoptera frugiperda. J. Biol. Chem. 1996;271:16135–16138. doi: 10.1074/jbc.271.27.16135. [DOI] [PubMed] [Google Scholar]
- 11.Hattori K, Motohashi N, Kobayashi I, Tohya T, Oikawa M, Tamura HO. Cloning, expression, and characterization of cytosolic sulfotransferase isozymes from Drosophila melanogaster. Biosci. Biotechnol. Biochem. 2008;72:540–547. doi: 10.1271/bbb.70647. [DOI] [PubMed] [Google Scholar]
- 12.Kakuta Y, Pedersen LG, Pedersen LC, Negishi M. Conserved structural motifs in the sulfotransferase family. Trends Biochem. Sci. 1998;23:129–130. doi: 10.1016/s0968-0004(98)01182-7. [DOI] [PubMed] [Google Scholar]
- 13.Weinshilboum RM, Otterness DM, Aksoy IA, Wood TC, Her C, Raftogianis RB. Sulfotransferase molecular biology: cDNAs and genes. FASEB J. 1997;11:3–14. [PubMed] [Google Scholar]
- 14.Yalcin EB, Struzik SM, King RS. Allosteric modulation of SULT2A1 by celecoxib and nimesulide: Computational analyses. Drug Metab. Lett. 2008;2:198–204. doi: 10.2174/187231208785425755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu JH, Li HT, Liu MC, Zhang JP, Li M, An XM, Chang WR. Crystal structure of human sulfotransferase SULT1A3 in complex with dopamine and 3′-phosphoadenosine 5′-phosphosulfate. Biochem. Biophys. Res. Commun. 2005;335:417–423. doi: 10.1016/j.bbrc.2005.07.091. [DOI] [PubMed] [Google Scholar]
- 16.Huey R, Morris GM. Using AutoDock 4 with AutoDockTools: A Tutorial. version 1.4.5b The Scripps Research Institute; La Jolla, California: 2007. [Google Scholar]
- 17.Sauer JR, Essenberg RC, Bowman AS. Salivary glands in ixodid ticks: control and mechanism of secretion. J. Insect Physiol. 2000;46:1069–1078. doi: 10.1016/s0022-1910(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 18.Pannabecker T, Needham GR. Effects of octopamine on fluid secretion by isolated salivary glands of a feeding ixodid ticks. Arch. Insect Biochem. Physiol. 1984;2:217–226. [Google Scholar]
- 19.Kovoor A, Celver JP, Wu A, Chavkin C. Agonist induced homologous desensitization of mu-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol. 1998;54:704–711. [PubMed] [Google Scholar]
- 20.Celver J, Sharma M, Kovoor A. RGS9-2 mediates specific inhibition of agonist-induced internalization of D2-dopamine receptors. J. Neurochem. 2010;114:739–749. doi: 10.1111/j.1471-4159.2010.06805.x. [DOI] [PubMed] [Google Scholar]
- 21.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:281–288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Letunic I, Bork P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 23.Moretti S, Armougom F, Wallace IM, Higgins DG, Jongeneel CV, Notredame C. The M-Coffee web server: a meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res. 2007;35:W645–648. doi: 10.1093/nar/gkm333. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J. Mol. Biol. 2004;340:385–395. doi: 10.1016/j.jmb.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 25.Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, Notredame C. Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006;34:604–608. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poirot O, Suhre K, Abergel C, O’Toole E, Notredame C. 3DCoffee@igs: a web server for combining sequences and structures into a multiple sequence alignment. Nucleic Acids Res. 2004;32:W37–40. doi: 10.1093/nar/gkh382. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxMLWeb servers. Syst. Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 28.Shih AC, Lee DT, Peng CL, Wu YW. Phylo-mLogo: an interactive and hierarchical multiple-logo visualization tool for alignment of many sequences. BMC Bioinformatics. 2007;8:63. doi: 10.1186/1471-2105-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics. 2006;7:339. doi: 10.1186/1471-2105-7-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 33.Spassov VZ, Yan L, Flook PK. The dominant role of side-chain backbone interactions in structural realization of amino acid code. ChiRotor: a side-chain prediction algorithm based on side-chain backbone interactions. Protein Sci. 2007;16:494–506. doi: 10.1110/ps.062447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spassov VZ, Flook PK, Yan L. LOOPER: a molecular mechanics-based algorithm for protein loop prediction. Protein Eng. Des. Sel. 2008;21:91–100. doi: 10.1093/protein/gzm083. [DOI] [PubMed] [Google Scholar]
- 35.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olsen AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 37.Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 38.Sanner MF. Python: A Programming Language for Software Integration and Development. J. Mol. Graphics Mod. 1999;17:57–61. [PubMed] [Google Scholar]
- 39.Mather TN, Mather ME. Intrinsic competence of three ixodid ticks (Acari) as vectors of the Lyme disease spirochete. J. Med. Entomol. 1990;27:646–650. doi: 10.1093/jmedent/27.4.646. [DOI] [PubMed] [Google Scholar]
- 40.King RS, Ghosh AA, Wu J. Inhibition of human phenol and estrogen sulfotransferase by certain non-steroidal anti-inflammatory agents. Curr. Drug Metab. 2006;7:745–753. doi: 10.2174/138920006778520615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.