

Abstract
Evidence suggests that lung structure and function is partly maintained by a balance between the competing arginine-metabolizing enzymes arginase and nitric oxide synthase. Asymmetric dimethylarginine is an endogenous inhibitor of nitric oxide synthase. It is metabolized by dimethylarginine dimethylaminohydrolase-2, which is oxidant-sensitive. The mechanism that induces excess lung collagen deposition in burned patients has not yet been explored. Our objective is to investigate the role of asymmetric dimethylarginine and the arginase pathway in acute lung injury. An ovine model for burn and smoke inhalation injury was used to assess excess lung collagen deposition. Sheep were deeply anesthetized during the injury, mechanically ventilated, resuscitated with fluid, and sacrificed after either 2 or 3 weeks. Lungs were assessed histologically and biochemically for collagen content, arginase activity, lipid peroxidation product and antioxidant concentration, and protein concentrations. Plasma was assessed for amino acid and nitrate/nitrite concentrations. Burn and inhalation injury resulted in significantly reduced pulmonary function and increased lung collagen deposition. These physiological changes were associated with significantly increased lung arginase activity, collagen synthesis precursor ornithine aminotransferase, and ornithine decarboxylase, which is associated with cell proliferation. Significant decreases in plasma nitrate/nitrite after injury were associated with increased lung asymmetric dimethylarginine concentrations and decreased dimethylarginine dimethylaminohydrolase-2 expression. The decreased dimethylarginine dimethylaminohydrolase-2 expression was associated with significantly increased lipid peroxidation product and decreased antioxidant content in the lung. This data supports that excess lung collagen deposition and reduced pulmonary function in acute lung injury following burn and inhalation injury is mediated through the arginase pathway.
Keywords: fibrosis, nitric oxide synthase, oxidative stress, arginine metabolism
INTRODUCTION
In the United States there are 1.25 million victims of thermal injury, resulting in 5,000 deaths annually (1). Approximately 23,000 of these casualties suffer concomitant injury from the inhalation of smoke. Inhalation injury is a significant contributor to mortality and morbidity in burn injury (2). Severe inhalation injury commonly results in acute lung injury (ALI), which frequently progresses to acute respiratory distress syndrome (ARDS) in patients with concomitant burn injury (3).
Mlcak, et al., investigated late residual pulmonary impairment in 17 patients with a 67 ± 32% total body surface area (TBSA) burn injury 8 years post-injury (2). Thirteen of the 17 children were diagnosed by bronchoscopy with inhalation injury, and 9 patients had obstructive and restrictive diseases (2). A characteristic of restrictive lung disease is the excessive lung collagen deposition, contributing to fibrosis (4).
Nitric oxide (NO) is formed from L-arginine by nitric oxide synthase (NOS) (5). Arginine depletion is linked to ALI, and the supplementation of arginine attenuates inflammation (6). NOS and arginase compete for arginine. Enhanced arginase activity results in less arginine substrate for NOS and reduced NO production. NOS converts arginine into citrulline and NO. Arginase converts arginine to urea and ornithine. Ornithine is converted by ornithine aminotransferase (OAT) to proline, which is an essential component of hydroxyproline and collagen. Ornithine decarboxylase (ODC) also metabolizes ornithine to putrescine, which forms the polyamines spermine and spermidine that increase cell proliferation (7). Increased arginase activity may contribute to lung airway remodeling by increasing collagen deposition and cell proliferation (8).
Asymmetric dimethylarginine (ADMA) is an analog of arginine and an endogenous inhibitor of all three isoforms of NOS (9, 10). It is synthesized by hydrolysis of proteins with methylated arginine residues (11). ADMA is excreted through urine (12) and is metabolized by an isoform of dimethylarginine dimethylaminohydrolase (DDAH), either DDAH-1 or DDAH-2. DDAH-2 is sensitive to oxidation (13). In the presence of increased ADMA, NOS can become uncoupled to produce superoxide (O2−) (14, 15), and because ADMA is a competitive inhibitor of NOS, the reduction of NO in the presence of O2− can result in peroxynitrite production (16). Thus, in the presence of ADMA, NOS can produce reactive oxygen species (ROS) and reactive nitrogen species (RNS) in addition to NO, which may contribute to inflammation.
The mechanisms that induce excess lung collagen deposition in burned patients are unknown. To date, there are no studies that report excess lung collagen deposition in large animals after burn and inhalation injury. The well-established ovine model is clinically relevant because of similarities between human and ovine pulmonary physiology. We hypothesize that excess lung collagen deposition and reduced pulmonary function following burn and inhalation injury is mediated through the arginase pathway in the ovine model.
MATERIALS AND METHODS
Animal Care and Use
This study was approved by the Animal Care and Use Committee of the University of Texas Medical Branch and conducted in compliance with the guidelines of the National Institutes of Health and of the American Physiology Society for the care and the use of laboratory animals. The studies were completed at UTMB’s Investigative Intensive Care Unit, which is a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC). All animals in the two study groups survived throughout the experimental time period.
Animal Model
The acute, 24–96 hr model of this burn and inhalation injury has previously been described in detail (17). The animals in the current 3-week study were prepared in a similar manner. Briefly, 18 adult female Merino sheep (body weight, 30–40 kg) were surgically prepared under isoflurane anesthesia with a right femoral artery catheter (Intracath, 16GA, 24IN, Becton Dickinson Vascular Access, Sandy, UT), a thermodilution catheter (Model 131F7, Edwards Lifesciences LLC, Irvine, CA), a left atrial catheter (0.062 in. ID, 0.125 in. OD; Dow Corning, Midland, MI), and a lymphatic catheter (0.025 in. ID, 0.047 in. OD; Dow Corning, Midland, MI). After a 7-day recovery period, sheep were randomly divided into three groups: sham (noninjured, nontreated; n = 6), 2-week, and 3-week injured animals. Then, 2- and 3-week injured animals were anesthetized with isoflurane and given flame burn (20% TBSA, third degree) and inhalation injury (36 breaths of cotton smoke, <40°C). The injury was intentionally smaller than the injury used previously by our lab in order to have 100% survival during the study. Sham animals were anesthetized with isoflurane anesthesia and treated in the same manner as the injured sheep but were insufflated with air rather than smoke and were not given a burn injury. After the burn and smoke inhalation injury or the sham procedure, all sheep were awakened and placed on a ventilator with positive end expiratory pressure set to 5 cm H2O and tidal volume maintained at 15 mL/kg. The sheep were ventilated with 100% oxygen for the first 3 hrs after injury for rapid clearance of CO to reduce their carboxyhemoglobin. Following this procedure the fraction of inspired oxygen (FiO2) was adjusted according to blood gas analysis to maintain PaO2 above 80 mm Hg. Respiratory rate was initially set at 30 breaths/minute and thereafter adjusted to keep PaCO2 between 25–35 mm Hg. All sheep were resuscitated with Ringer’s solution using the formula 4 mL/kg/% burned body surface for 24 hrs. The experiment continued for either 2 or 3 weeks to study long-term collagen deposition and to allow time for the burn wound to heal.
Early Excision and Skin Autografting
It is standard clinical practice to surgically remove the dead burned skin (eschar) and cover the wound with a meshed autograph obtained from the contralateral side. At 24 hrs post-injury, early excision was performed to the muscular fascia to the burned skin under isoflurane anesthesia. The skin was autografted using split-thickness skin (20/1000 inch) that was harvested from the flank of the non-burned skin on the contralaterial side using an electric dermatome (Padgett Electro-Dermatome, Padgett Instruments Inc., KC, USA) and meshed 4:1 using a mesh dermatome (Padgett Mesh-Dermatome, Padgett Instruments Inc., KC, USA). The wound was covered using non-adhering dressing (ADAPTIC, Johnson & Johnson, Skipton, UK) in the graft area and non-adhesive hydrocellular polyurethane dressing (ALLEVYN, Smith & Nephew Medical Limited, Hull, UK) in the donor site. The tie-over dressing was attached using rubber bands and removed 4–6 days after the operation. Operating time was limited to 2 hrs, and ambient temperature was maintained at 30°C to prevent hypothermia. The sham group was exposed to anesthesia with isoflourane for 100 minutes in the operation room for comparison.
Masson Trichrome Collagen Stain
The middle, right lobe of the ovine lungs were fixed with 3% paraformaldehyde, processed, embedded in paraffin blocks, serially sectioned at 4 μM, and mounted on Superfrost Plus Slides (VWR, West Chester, PA; ). Sections of lung were stained with the Masson Trichrome Stain Kit (Sigma, Catalog No. HT15) to qualitatively assess the degree of collagen deposition.
Lung Hydroxyproline Content
The protocol to determine hydroxyproline content in lung tissue supernatant has previously been described in detail (18). Briefly, lung supernatant was incubated with trichloracetic acid, centrifuged, and resuspended in HCl. The pellets were dried and reconstituted with water. The samples were incubated at room temperature with chloramine-T solution, combined with Ehrlich’s solution at 65°C, and optical density was measured at 550 nm.
Plasma Arginine, Ornithine, Citrulline, and Hydroxyproline Analysis
Ovine plasma was precipitated to remove proteins by mixing equal volumes of 7.5% SSA in 0.02N HCl containing internal standard with plasma sample. The sample was centrifuged for 15 min at 10,000 x g. The supernatant was filtered through a 0.22 μm filter and sample was injected on the Hitachi L8800 and analyzed using standard physiological analysis methods as provided by the manufacture.
Arginase Activity
The protocol to determine arginase activity in lung tissue homogenate has previously been described in detail (18). It is a colorimetric assay that uses α-isonitrosopropiophenone to assess urea formation. Protein levels for each sample were determined using the Protein Quantification Kit (Dojindo Molecular Technologies, Rockville, MD) the according to manufacturer’s instructions. All results are expressed as μMol urea/μg protein.
Western Blot Analysis
DDAH-2, ODC, and OAT protein expression in lung tissue was determined using anti-DDAH-2 (C-19) (sc-26071), anti-ODC (D-15) (sc-34181), and anti-OAT (G-20) (sc-55732) antibodies with donkey anti-goat IgG HRP secondary antibody (R&D Systems, Minneapolis, MN, Catalog No. HAF109), as described previously (19). Blots were completed using 20 μg of protein and were quantified by National Institutes of Health IMAGE J (Image and Processing and Analysis in Java) scanning densitometry, and normalized to total actin (I-19) (sc-1616) expression.
Plasma Nitrite/Nitrate (NOx)
The plasma NO levels were evaluated by measuring the intermediate and end products, which are nitrates and nitrites (NOx). Concentrations were determined at 0, 6, 12, 18, 24, 36, 48, 96 hrs and at 1, 2, and 3 weeks using a commercially available assay (Nitrate/Nitrite Colorimetric Assay Kit, Cayman Chemicals, Ann Arbor, MI, Catalog No. 780001).
Lung Homogenate Asymmetric Dimethylarginine Concentration
ADMA concentration was evaluated in lung homogenate supernatant by the liquid chromatography mass spectrometry technique (LC-MS). Briefly, 25 μL of [2, 3, 3, 4, 4, 5, 5-2H7]-ADMA (25 nmol/ml) was added to 100 μL of homogenized lung. Selective ion monitoring of ADMA was conducted using positive electrospray ionization. The samples were analyzed by LC-MS performed with the Agilent Series 1100 LC-MSD System (Agilent Technologies, Livermore, CA) and the analytical Atlantis T3 column (Waters Corporation, Milford, MA), and ADMA was measured at (IS) m/z 210.1. A linear equation weighted by 1/x was constructed to establish the relationship between concentrations and peak area ratio using the SPSS 11.0 statistical software. ADMA concentration was calculated by interpolation of the linear equation.
Antioxidant Determinant
The protocol to determine malondialdehyde, in addition to gamma- and alpha-tocopherol in lung tissue homogenate has previously been described in detail (20). All results were expressed as nMol/g.
Lipid Peroxidation Product
Lung tissue malondialdehyde was measured by a modified version Lykkesfeldt’s method (21). Briefly, lung tissue (~50 mg) was added to 1.5 mL KCl (1%, w:v) and homogenized. A 0.5 mL aliquot of the homogenate was diluted with an equal volume of water, 0.1 mL BHT (0.06% w:v), and 0.2 mL SDS (8% w:v). To this mixture, 3 mL TBA reagent (8 g/L diluted 1:1 with 200 mL/L acetic acid and adjusted to pH 3.5 with NaOH) was added and the reaction mixture was heated at 95°C for 60 minutes. The MDA(TBA)2 adduct was then partitioned out of the mixture with 3 mL of n-butanol. Following vortexing and centrifugation, an aliquot of the supernatant was dried under nitrogen, resuspended in methanol and 50 mL injected onto a Shimadzu HPLC reverse phase system including an autosampler and a Beckman 5μ ODS, 4.6 × 250 mm column. The MDA(TBA)2 adduct was eluted using an isocratic mobile phase consisting of 50% methanol and 50% 25 mM phosphate buffer at pH 6.5 at a flow rate of 1.5 mL/minute. Malondialdehyde was detected by fluorescence at excitation 532 nm and emission 533 nm. Quantitation was done using an external standard of 1, 1, 3, 3-tetraethoxypropane (Sigma, St Louis, MO, USA) prepared using the same method as the samples.
Statistical Analysis
All values are expressed as mean ± SEM using GraphPad Prism Software (GraphPad Software, La Jolla, CA, USA). Results were compared between groups using repeated measures (analysis of variance) and the Newman-Keuls multiple comparison tests. A value of p < 0.05 was accepted as statistically significant.
RESULTS
Burn and Inhalation Injury Caused Reduced Pulmonary Function and Increased Collagen Deposition
To establish a clinically relevant model of long-term 3-week pathophysiology of lung injury, we performed studies in sheep with combined burn and smoke inhalation. A 20% TBSA burn was administered to the sheep in addition to 36 breaths of cotton smoke under heavy anesthesia. Pulmonary function, measured by the ratio of the partial pressure of arterial oxygen (PaO2) to the fractional concentration of oxygen in inspired air (FiO2), is significantly decreased over time compared to baseline values after the burn and inhalation injury (p < 0.05; Figure 1A). Direct staining of collagen of lung slices using the Masson trichrome stain revealed increased deposition of collagen in sheep injured after 2 and 3 weeks (Figure 1C–D) compared to uninjured sheep (Figure 1B). The intense blue stain for collagen is increased after 2 and 3 weeks. Lung hydroxyproline content was quantified in lung tissue to measure lung collagen deposition. Lung hydroxyproline significantly increased after 3 weeks compared to uninjured sheep (1.12 ± 0.08 in uninjured animals versus 1.57 ± 0.16 in 3-week injured animals; p < 0.05; Figure 1E). This was further quantified by determining plasma hydroxyproline by HPLC from baseline measurements to 3-weeks after injury. Hydroxyproline significantly increased at 1, 2, and 3 weeks compared to baseline values and significantly increased from 1 to 2 weeks after injury in plasma (p < 0.05; Figure 1F).
Figure 1.
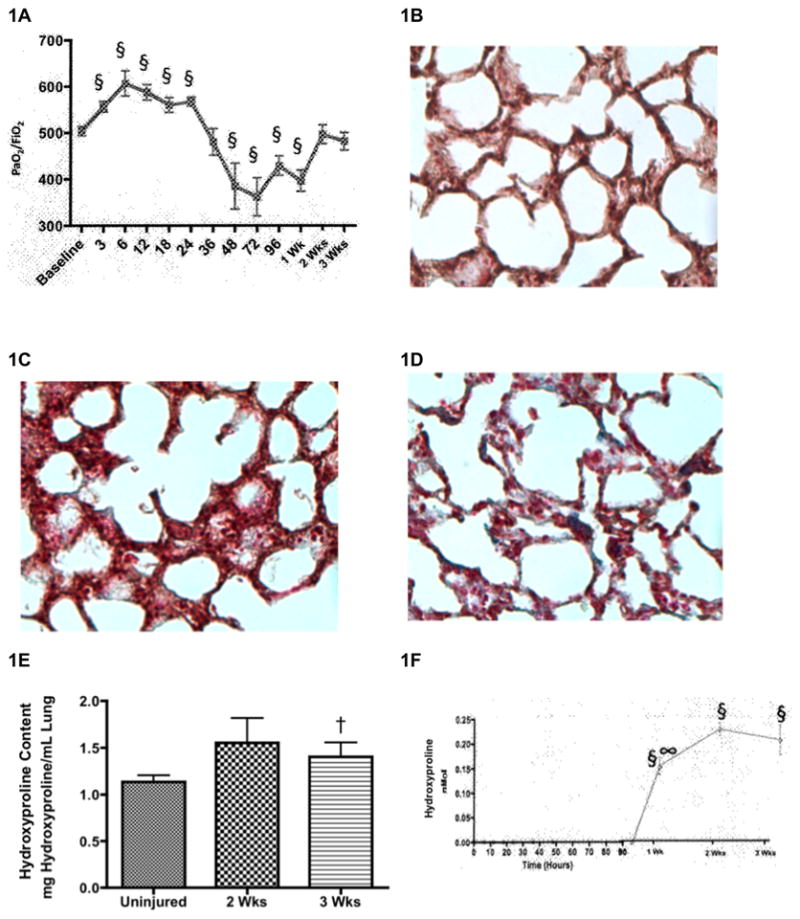
Burn and Inhalation Injury Causes Increased Collagen Deposition. After a 20% TBSA full thickness burn and a smoke inhalation injury, sheep were killed after two (n=6) and three (n=6) weeks. Results were compared to uninjured sham animals that were not injured but treated in the same manner. (A) Pulmonary function was measured in plasma using a ratio of the partial pressure of arterial oxygen (PaO2) to the fractional concentration of oxygen in inspired air (FiO2) over time until two or three weeks after the injury. The qualitative assessment of collagen deposition was illustrated by the Masson’s trichrome stain for collagen deposition on 4-μm paraffin sections. Blue color indicates collagen deposition. Representative sections from (B) uninjured animals (original magnification: x20), (C) injured animals after 2 wks, and (D) injured animals after 3 wks indicate increased collagen deposition in the lung after time. (E) The quantitative assessment of collagen used a hydroxyproline colorimetric assay in lung homogenate supernatant, showing significant collagen deposition in the lung. (F) Plasma was collected over time until two or three weeks before and after the injury to measure hydroxyproline. Data are shown as means ± SEM. §P < 0.05 versus baseline data, †P < 0.05 versus uninjured animals, ∞P < 0.05 versus injured animals sacrificed after 2 wks.
Increased Arginase Activity was Associated with Increased ODC and OAT Expression
Arginase activity was assayed in lung homogenates because arginase is a key enzyme influencing collagen synthesis in fibroblasts. Arginase activity significantly increased 3-weeks after injury (0.18 ± 0.009 μMol urea/μg protein) compared to uninjured animals (0.14 ± 0.01 μMol urea/μg protein; p < 0.05; Figure 2A). Arginase is the enzyme responsible for the conversion of arginine to ornithine. We initially analyzed the concentration of plasma ornithine from baseline values to 3-week values after burn and inhalation injury. Our data indicated that ornithine levels initially decrease until 48 hours, but after 48 hours ornithine concentrations increased to reach baseline values (Figure 2B). Therefore, the expression of ornithine’s converting enzymes were measured by western blot and showed significant increases after 2 and 3 weeks. ODC is significantly increased 3-fold after 2 and 3 weeks in injured animals compared to uninjured animals (6231 ± 1352 in uninjured animals versus 21520 ± 10554 in 3-week injured animals; p < 0.05; Figure 2C). OAT is significantly increased after 3 weeks in injured animals compared to uninjured animals (7747 ± 796 in uninjured animals versus 11880 ± 1401 in 3-week injured animals; p < 0.05; Figure 2D). The significant increases in ornithine’s converting enzymes provide a possible explanation for the level of ornithine returning to baseline values.
Figure 2.
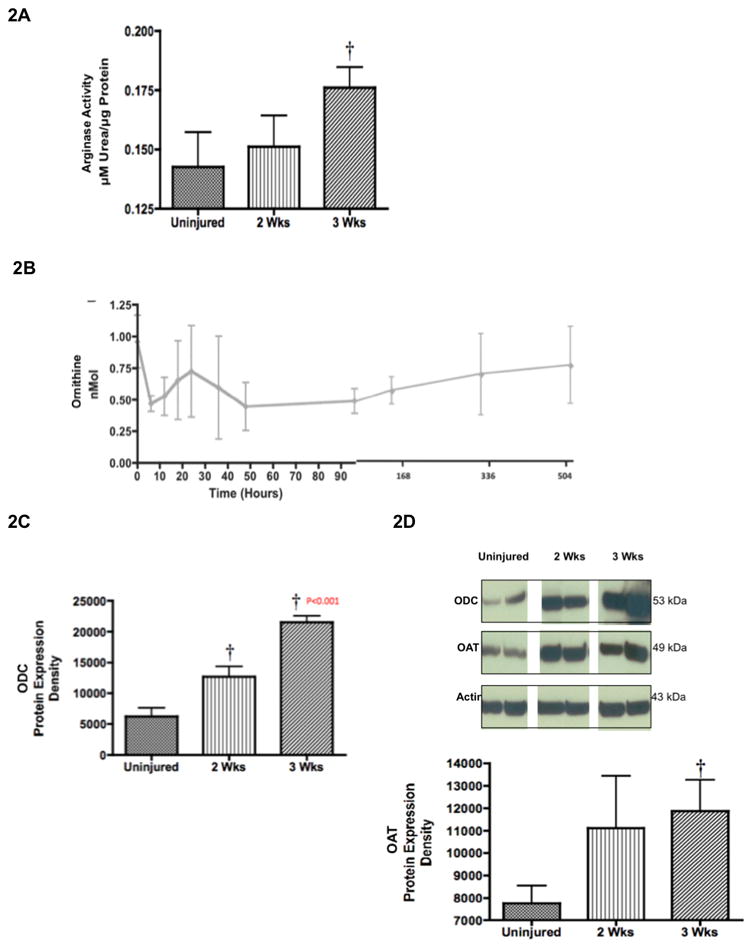
Increased Arginase Activity was Associated with Increased Collagen Deposition Precursors. Two (n=6) and three (n=6) weeks after 20% TBSA full thickness burn and a smoke inhalation injury, sheep were killed and (A) lung homogenate was used to measure arginase activity. Results were compared to uninjured sham animals that were not injured but treated in the same manner. (B) Plasma was collected over time until two or three weeks before and after the injury to measure ornithine. (C) Lung homogenate western blots were used to measure ODC expression, and (D) OAT expression. Equality of protein loading was confirmed by the expression of B-actin, and each band was quantified by densitometric analysis. Data are shown as means ± SEM. † P < 0.05 versus uninjured animals.
Decreased NO Synthesis was Associated with Increased ADMA Concentrations and Decreased DDAH-2 Expression
Given the intimate balance of the arginase and NOS pathways, we also assessed the effects of burn and inhalation injury on NOS activity. The stable end-products of NO, which include nitrates and nitrites (NOx), significantly increased 6 hours after the burn and inhalation injury in plasma compared to baseline values (0.079 uM ± 0.002 at baseline versus 0.061 ± 0.003 at 6 hours; p < 0.05; Figure 3A). Conversely, nitrates and nitrites subsequently decreased to baseline values over the remaining time. These findings illustrate that after 2 weeks, NOS activity initially peaks while lung arginase activity essentially remains at baseline, while at 3 weeks NOS activity decreases at the time that lung arginase activity peaks. Figure 3B shows the plasma concentrations of arginine over time. Initially arginine concentrations decrease after 6 hours, but increase over time to baseline levels.
Figure 3.
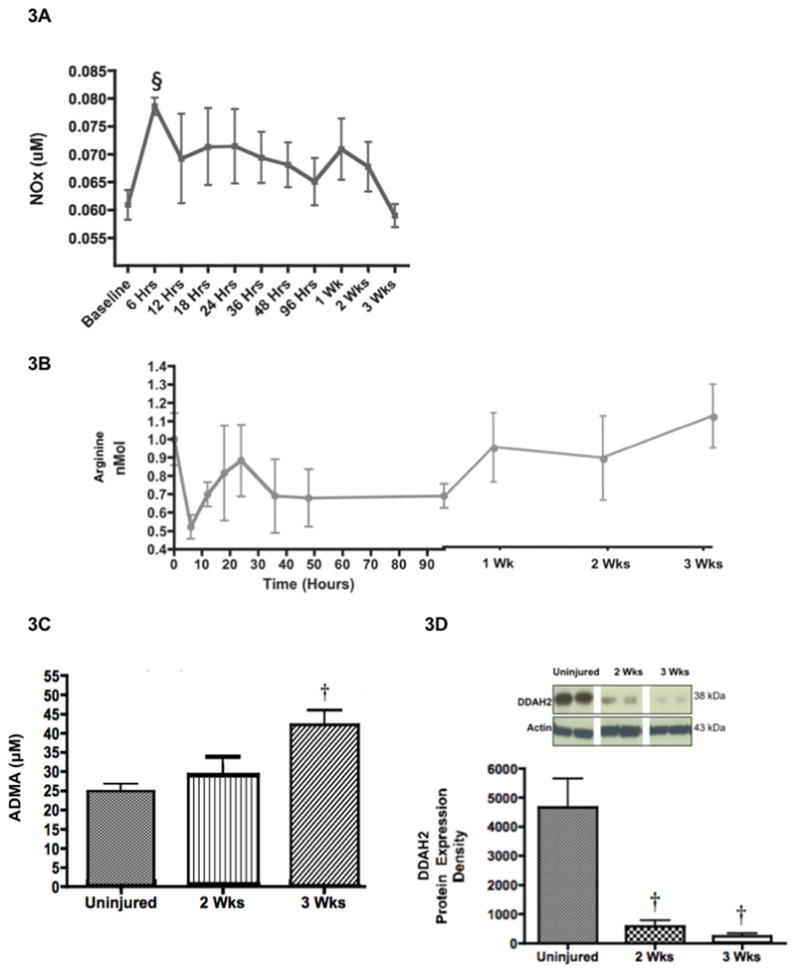
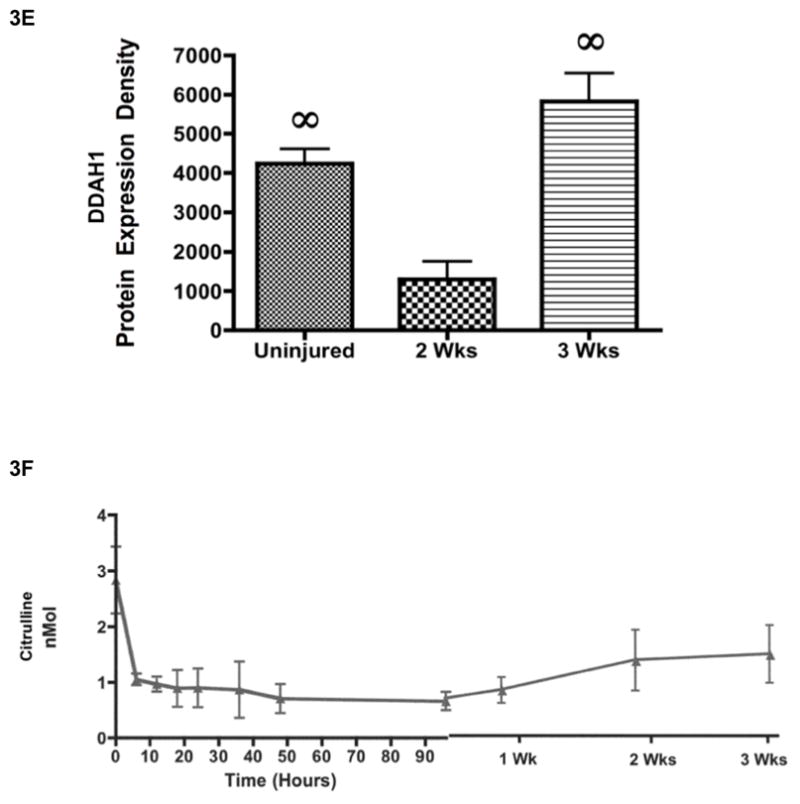
Decreased NO Synthesis was Associated with Increased ADMA Concentrations and Decreased DDAH-2 Expression. Two (n=6) and three (n=6) weeks after 20% TBSA full thickness burn and a smoke inhalation injury, sheep were killed and plasma was collected over time until two or three weeks before and after the injury to measure (A) the products of nitric oxide, nitrates and nitrites, and (B) arginine concentrations. (C) Lung homogenate was used to measure ADMA concentrations using LC-MS, and to measure (D) lung DDAH-2 expression and (E) lung DDAH-1 expression. The increase in ADMA is associated with the decrease in DDAH-2 and the decrease in NOx. (F) Plasma was collected over time until two or three weeks before and after the injury to measure citrulline concentrations. Results were compared to uninjured sham animals that were not injured but treated in the same manner. Equality of protein loading was confirmed by the expression of B-actin, and each band was quantified by densitometric analysis. Data are shown as means ± SEM. †P < 0.05 versus uninjured animals, §P < 0.05 versus baseline data, ∞P < 0.05 versus injured animals sacrificed after 2 wks.
ADMA is a known endogenous inhibitor of NOS in endothelial cells (9), and it may play a role in the lung by inhibiting NOS activity in epithelial cells (15). ADMA lung homogenate concentrations significantly increased over time in injured animals compared to uninjured animals in lung homogenate (p < 0.05; Figure 3C). The fall in NO coincides with increased ADMA. The elevation in ADMA could be a result in the reduction of its major hydrolase, DDAH-2 (22). Therefore, we determined DDAH-2 expression in an ovine lung tissue homogenate by Western blot. Expression of DDAH-2 in injured sheep was significantly lower, almost 20-fold, compared to uninjured sheep (4661 ± 995 in uninjured group versus 242 ± 97 in 3-week injured group; p < 0.05; Figure 3D).
The lung is a major source of ADMA (23). ADMA is also metabolized by another isoform of DDAH in the lung, DDAH-1. An unexpected finding is the significant decrease in lung DDAH-1 expression 2 weeks after the injury, followed by the significant increase in DDAH-1 three weeks after the injury (p < 0.05; Figure 3E). Because DDAH-2 expression in the lung (Figure 3D) was the lowest after 3 weeks while lung ADMA concentration was highest at 3 weeks, it suggests that DDAH-2 may be more important for ADMA regulation in our burn and smoke inhalation injury model. A second possibility is that DDAH-1 is significantly increasing after 3 weeks to compensate for elevated ADMA concentration.
Figure 3F illustrates plasma citrulline concentrations before and after the burn and smoke inhalation injury. An unexpected finding is the decrease in citrulline in plasma after the injury. The data does not coincide with the significant increases in plasma nitrate and nitrite concentration (Figure 3A), which was expected because arginine forms NO and citrulline by the action of NOS. This could simply reflect that total protein concentrations decrease after burn and smoke inhalation injury. Also, proteins such as fibrin are susceptible to citrullination during tissue inflammation, which can contribute to the increase towards baseline values 2 and 3 weeks after the injury.
Increased Lipid Peroxidation Product and Decreased Antioxidant Content was Associated with Decreased DDAH-2 Expression
DDAH-2, the enzyme that metabolizes ADMA, is oxidant sensitive (24). Therefore, we explored the possibility that the decrease in DDAH-2 was associated with a decrease in antioxidants and an increase in oxidants. Vitamin E is a peroxyl radical scavenger, and it has previously been found to be depleted in sheep after burn and smoke inhalation (25). Therefore, gamma- and alpha-tocopherol concentrations were measured 2 and 3 weeks after the injury. Relative to the gamma-tocopherol concentrations in the lungs of uninjured animals (0.62 nMol/g ± 0.11), concentrations decreased by more than half in the injured sheep after 2 weeks (0.25 ± 0.04; p < 0.05; Figure 4A). Lung alpha-tocopherol concentrations were depleted at 2 weeks and remained depleted at 3 weeks (6.40 ± 0.62 nMol/g) compared to uninjured animals (12.57 μMol/g ± 2.3; p < 0.05; Figure 4B). To determine whether decreased concentrations of gamma- and alpha-tocopherol was associated with a by-product of lipid peroxidation, malondialdehyde (MDA) was measured by HPLC (26). MDA concentrations were significantly increased in injured animals after 2 (197 ± 24 nMol/g) and 3 weeks compared to uninjured animals (114 nMol/g ± 9; p < 0.05; Figure 4C).
Figure 4.
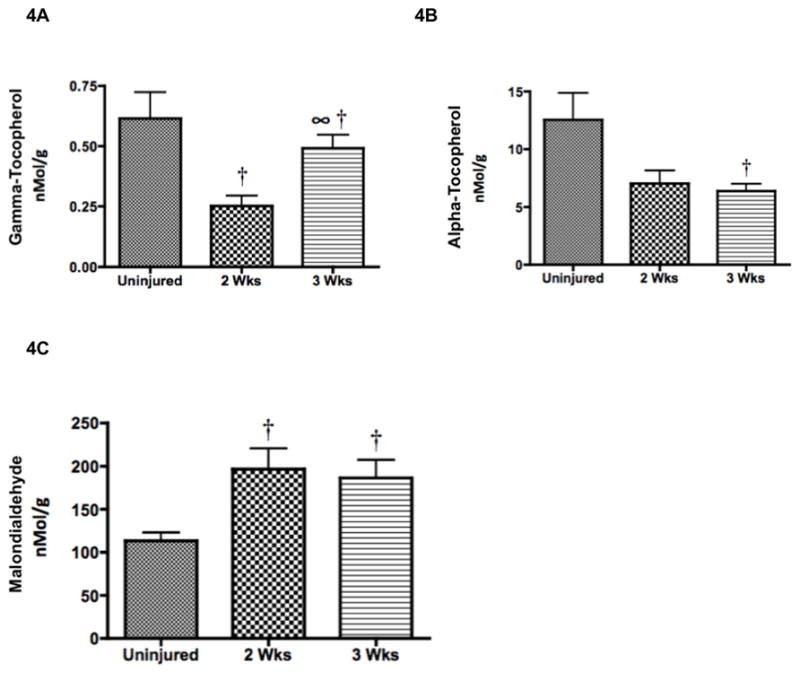
Increased Lipid Peroxidation Product and Decreased Antioxidant Content was Associated with Decreased DDAH-2 Expression. After a 20% TBSA full thickness burn and a smoke inhalation injury, sheep were killed after two (n=6) and three (n=6) weeks and lung homogenate was used to measure oxidative stress. Results were compared to uninjured sham animals that were not injured but treated in the same manner. Oxidative stress was evaluated by measuring (A) ROS and RNS scavenger gamma-tocopherol, (B) ROS scavenger alpha-tocopherol, and (C) lipid peroxidation product malondialdehyde. Oxidative stress was increased post-injury based on the decreases in tocopherol and the elevation in MDA. Increased oxidative stress is associated with decreased DDAH-2 (Figure 3D). Data are shown as means ± SEM. †P < 0.05 versus uninjured animals, ∞P < 0.05 versus injured animals sacrificed after 2 wks.
DISCUSSION
It is well established that NO, through the activity of the NOS enzymes, contributes to the development of ALI in sheep after burn and smoke inhalation injury (27). In the first 48 hours following burn and inhalation injury, there are significant increases in pulmonary fluid flux and edema that are associated with increases in oxidative stress (28). Although this inflammatory picture exits for the early changes observed in ALI, burn survivors are reported to have restrictive lung diseases in an 8-year follow-up study (2). Consequently, we decided to determine the etiology of excess collagen deposition in a long-term, 2 and 3 week ovine model of burn and smoke inhalation injury. NO is involved in various pulmonary processes, including vascular tone and anti-inflammatory properties (29). The recent report that the lung is a major source of the NOS inhibitor, ADMA, and that elevated circulating levels of ADMA can contribute to abnormal airway physiology suggests that ADMA may play a role in arginine metabolism in the pathophysiology of ALI (18, 23). In the present study, we explored the effects of burn and smoke inhalation injury on ADMA levels, DDAH-2 expression, and NOS activity. Results from this study demonstrate that increased collagen deposition is associated with increased oxidant stress, increased ADMA, and increased arginase activity.
The iNOS isoform produces large quantities of NO in the presence of pro-inflammatory cytokines, which contribute to ARDS. Our results show a significant increase and peak of nitric oxide products at 6 hrs, followed by their steady decline (Figure 2B). Our previous studies have shown that the use of the potent and highly selective iNOS inhibitor, BBS-2, after 48 hrs significantly attenuated the signs of ARDS after burn and inhalation injury including increased pulmonary edema, abnormal lung compliance, and extensive airway obstruction (30). The present findings are consistent with our previous studies because a NOS inhibitor initially attenuated the injury in the short term when NO products are high. However, after NO products decrease, collagen deposition becomes problematic because of decreased NO and increased arginase. We found that as the NOS inhibitor ADMA increases, lung NO products decrease and arginase activity and collagen deposition are significantly increased (Figures 3C, 3A, 2A, 1E). Another possible explanation of the indirect relationship between NOS and arginase is that ADMA blocks the formation of N (omega)-hydroxy-nor-L-arginine (NOHA), which is a potent inhibitor of arginase and an intermediate in the formation of NO. If NO is not formed, it is probable that NOHA will not form as well.
Acute respiratory distress syndrome (ARDS) is characterized by a PaO2/FiO2 ratio below 200. The PaO2/FiO2 ratio in our study significantly decreases from 6 hours up to 1 week after the burn and smoke inhalation injury compared to baseline values. However, the ratio begins to increase 2 weeks post-injury. A possible explanation is that the significant increase in NO causes a loss in hypoxic pulmonary vasoconstriction (HPV) in the ovine model (31). From 6 hours to 1 week, NO remains increased, which leads to the loss of HPV. After NO decreases to baseline values after 2 and 3 weeks, HPV is restored. In addition, Westphal, et.al. administered the neuronal NOS inhibitor, 7-nitroindazole, and significantly attenuated the loss of HPV. Because NO is heavily involved in the pathogenesis of burn and smoke inhalation injury, the decrease in NO and subsequent restoration of HPV allows the PaO2/FiO2 ratio to increase. Also, the sheep are currently limited on their movement. Future studies will more accurately assess pulmonary function while the animals are engaged in physical activity.
Based on our previous studies and our current findings, we propose a mechanism (Figure 5) whereby DDAH-2 is inactivated by oxidation, resulting in increased ADMA accumulation and a decrease in NOS activity. As a consequence, L-arginine has an increased availability for metabolism by arginase and arginase activity is increased, resulting in elevated ODC and OAT expression, hydroxyproline generation, and collagen deposition. This mechanism stresses that the combined effects of increased arginase activity and increased oxidative stress result in altered pulmonary structure.
Figure 5.
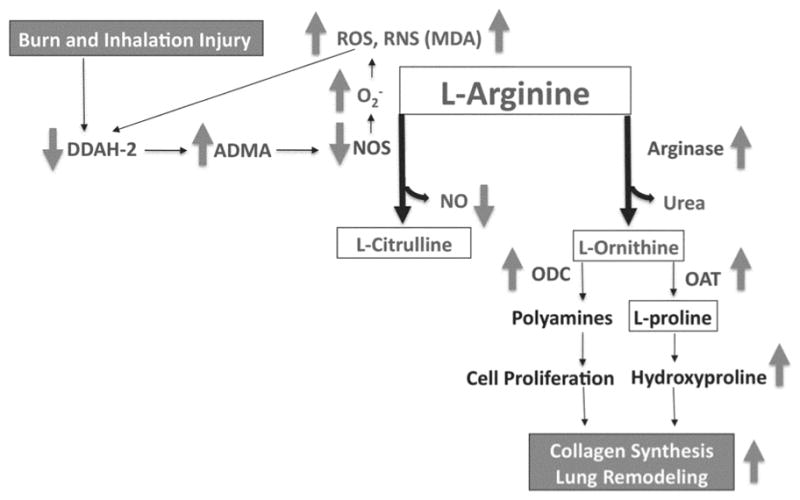
Model for the Role of Arginase Activity in Burn and Inhalation Injury. In this model, the burn and smoke inhalation injury and elevated oxidative stress decreases lung DDAH-2 expression, which (1) increases lung ADMA concentration, and (2) decreases the formation of NO. The increase in the NOS uncoupling increases oxidative stress, which (3) increases arginase activity, resulting in the increases in expression of lung ODC, OAT, and hydroxyproline. Increased hydroxyproline alters pulmonary structure because of increased collagen deposition.
Inhalation injuries are associated with increased levels of ROS and RNS (32). ROS and RNS modify cysteine and tyrosine residues and contribute to cell death, tissue injury, and organ dysfunction (33). Their detrimental sequelae can be reversed by gamma-tocopherol nebulization in the lung because it is a scavenger of ROS and RNS (17, 28, 34). Future studies will utilize gamma-tocopherol nebulization after burn and smoke inhalation injury as a tool to manage pulmonary changes and excess collagen deposition in the lung because of its actions as an ROS and RNS scavenger (35).
In summary, we present evidence that arginase activity is significantly increased in an ovine model of burn and inhalation injury, which is associated with increased collagen deposition in the lung. Although it is well known that the arginase and NOS enzymes compete for arginine, this is the first report of the arginase pathway being involved in burn and inhalation injury in the lung. These findings also provide the first evidence that elevated oxidative stress may contribute to lung dysfunction and structural changes and suggest that they may play a role in the excess deposition of collagen.
Acknowledgments
This work was supported by Grants GM66312 and GM60668 from the National Institute of General Medical Science, and Grants 8541 and 8450 from the Shrine Hospitals for Children (SHC). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the SHC.
The authors would like to thank the staff of the Investigational Intensive Care Unit at the University of Texas Medical Branch for their valuable assistance, especially E. Kraft, C. Moncebaiz, J. Jinkins, T. Walker, and C. Hallum. The authors also thank R. Connelly for consultation; K. Bansal, A. Esechie for assistance with molecular techniques; and Y. Larson, S. Assmussen and Y. Zhu for laboratory support.
Footnotes
DISCLOSURES: None
BIBLIOGRAPHY
- 1.Pruitt BA, JR, Goodwin CW, Mason AD., Jr . Epidemiology, demographic, and out come characteristics of burn injury. In: Herndon D, editor. Total Burn Care. 3. Philadelphia: Saunders/Elsevier Publishers; 2007. pp. 16–32. [Google Scholar]
- 2.Mlcak R, Desai MH, Robinson E, Nichols R, Herndon DN. Lung function following thermal injury in children—an 8-year follow up. Burns. 1998;24:213–216. doi: 10.1016/s0305-4179(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 3.Traber DL, Enkhabaatar P. Thermal lung injury and acute smoke inhalation. In: Fischaman DA, editor. Fischman’s Pulmonary Diseases and Disorders. 4. New York: McGraw-Hill Medical Publishing Company; 2008. pp. 1053–1064. [Google Scholar]
- 4.Selman M, Thannickal VJ, Pardo A, Zisman Da, Martinez FJ, Lynch JP., 3rd Idiopathic pulmonary fibrosis: Pathogenesis and therapeutic approaches. Drugs. 2004;64(4):405–430. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- 5.Moncada S, Palmer RM, Higgs EA. The discovery of nitric oxide as the endogenous nitrovasodilator. Hypertension. 1988;12:365–372. doi: 10.1161/01.hyp.12.4.365. [DOI] [PubMed] [Google Scholar]
- 6.Murakami K, Enkhbaatar P, Yu YM, Traber LD, Cox RA, Hawkins HK, Tompkins RG, Herndon D, Traber DL. L-arginine attenuates acute lung injury after smoke inhalation and burn injury in sheep. Shock. 2007;28(4):477–483. doi: 10.1097/shk.0b013e31804a59bd. [DOI] [PubMed] [Google Scholar]
- 7.Durante W, Liao L, Reyna SV, Peyton KJ, Schafer Al. Transforming growth factor-beta(1) stimulates I-arginine transport and metabolism in vascular smooth muscle cells: Role in polyamine and collagen synthesis. Circulation. 2001;103(8):1121–1127. doi: 10.1161/01.cir.103.8.1121. [DOI] [PubMed] [Google Scholar]
- 8.Bergeron C, Boulet LP, Page N, Laviolette M, Zimmermann N, Rothenberg ME, Hamid Q. Influence of cigarette smoke on the arginine pathway in asthmatic airways: Increase expression of arginase i. J Allergy Clin Immunol. 2007;119(2):391–397. doi: 10.1016/j.jaci.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 9.Vallance P, Leone A, Calver A, Collier J, Moncada S. Endogenous dimethylarginine as an inhibitor of nitric oxide synthesis. J Cardiovasc Pharmacol. 1992;20(Suppl 12):S60–62. doi: 10.1097/00005344-199204002-00018. [DOI] [PubMed] [Google Scholar]
- 10.Ueda S, Kato S, Matsuoka H, Kimoto M, Okuda S, Morimatsu M, Imaizumi T. Regulation of cytokine-induced nitric oxide synthesis by asymmetric dimethylarginine: Role of dimethylarginine dimethylaminohydrolase. Cir Res. 2003;92(2):226–233. doi: 10.1161/01.res.0000052990.68216.ef. [DOI] [PubMed] [Google Scholar]
- 11.Cantoni GL. Biological methylation: Selected aspects. Annu Rev Biochem. 1975;44:435– 451. doi: 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- 12.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339(8793):572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad T, Mabalirajan U, Ghosh B, Agrawal A. Altered asymmetrick dimethylarginine metabolism in allergically inflmed mouse lungs. Am J Respir Cell Mol Biol. 2010;42(1):3–8. doi: 10.1165/rcmb.2009-0137RC. [DOI] [PubMed] [Google Scholar]
- 14.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc Natl Acad Sci U S A. 1998;95(16):9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wells SM, Holian A. Asymmetric dimethylarginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am J Respir Cell Mol Biol. 2007;36(5):520–528. doi: 10.1165/rcmb.2006-0302SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitritie-mediated cellular injury. Proc Natl Acad Sci U S A. 1996;93(13):6770–6774. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soejima K, Traber LD, Schmalstieg FC, Hawkins H, Jodoin JM, Szabo C, Szabo E, Varig L, Salzman A, Traber DL. Role of nitric oxide in vascular permeability after combined burns and smoke inhalation injury. Am J Respir Crit Care Med. 2001;163:745–752. doi: 10.1164/ajrccm.163.3.9912052. [DOI] [PubMed] [Google Scholar]
- 18.Wells SM, Buford MC, Migliaccio CT, Holian A. Elevated asymmetric dimethylarginine alters lung function and induces collagen deposition in mice. Am J Respir Cell Mol Biol. 2008;40(2):179–188. doi: 10.1165/rcmb.2008-0148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gileltt AM, Wallace MJ, Gillespie MT, Hooper SB. Increased expansion of the lung stimulates calmodulin 2 expression in fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2002;2892(3):L440–447. doi: 10.1152/ajplung.00202.2001. [DOI] [PubMed] [Google Scholar]
- 20.Podda M, Weber C, Traber MG, Packer L. Simultaneous determination of tissue topcopherols, tocotrienols, ubiquinols, and ubiquinones. J Lipid Res. 1996;37(4):893–901. [PubMed] [Google Scholar]
- 21.Lykkesfeldt J. Determination of malondialdehyde as dithiobarbituric acid adduct in biological samples by hplc with fluorescence detection: Comparison with ultraviolet-visible spectrophotometry. Clin Chem. 2001;47(9):1725–1727. [PubMed] [Google Scholar]
- 22.Jiang DJ, Jia SJ, Yan J, Zhou Z, Yuan Q, Li YJ. Involvement of ddah/adma/nos pathway in nicotine-induced endothelial dysfunction. Biochem Biophys Res Commun. 2006;349(2):683–693. doi: 10.1016/j.bbrc.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 23.Bulau P, Zakrzewicz D, Kitowska K, Leiper J, Gunther A, Grimminger F, Eickelberg O. Analysis of methylarginine metabolism in the cardiovascular system identifies the lung as a major source of adma. Am J Physiol Lung Cell Mol Physiol. 2007;292(1):l18–24. doi: 10.1152/ajplung.00076.2006. [DOI] [PubMed] [Google Scholar]
- 24.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (ddah): Expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007;293(6):H3227–3245. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 25.Traber MG, Shimoda K, Murakami K, Leonard SW, Enkhbaatar P, Traber LD, Traber DL. Burn and smoke inhalation injury in sheep depletes vitamine e: Kinetic studies using deuterated tocopherols. Free Radic Biol Med. 2007;42(9):1421–1429. doi: 10.1016/j.freeradbiomed.2007.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marnett LJ. Lipid peroxidation-DNA damage by malondialdehyde. Mutat Res. 1999;424(102):83–95. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- 27.Cox RA, Jacob S, Oliveras G, Murakami K, Enkhbaatar P, Traber L, Schmalstieg FC, Herndon DN, Traber DL, Hawkins HK. Pulmonary exression of nitric oxide synthase isoforms in sheep with smoke inhalation and burn injury. Exp Lung Res. 2009;35(2):104–118. doi: 10.1080/01902140802446832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soejima K, McGuire R, Snyder N, Uchida T, Szabo C, Salzman A, Traber L, Traber DL. The effect of inducible nitric oxide synthase (inos) inhibition on smoke inhalation injury in sheep. Shock. 2000;13(4):561–266. doi: 10.1097/00024382-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Enkhbaatar P, Parkinson J, Traber D. Role inos-derived excessive nitric oxide in the pathogenesis of acute lung injury. In: Vincent JL, editor. Yearbook of Intensive Care and Emergency Medicine. Berlin, Heidelberg, New York: Springer Verlag; 2005. pp. 85–92. [Google Scholar]
- 30.Enkhbaatar P, Murakami K, Shimoda K, Mizutani A, Traber L, Phillips GB, Parkinson JF, Cox R, Hawkins H, Herndon D, Traber D. The inducible nitric oxide synthase inhibitor bbs-2 prevents acute lung injury in sheep after burn and smoke inhalation injury. Am J Respir Crit Care Med. 2003;167(7):1021–1026. doi: 10.1164/rccm.200209-1031PP. [DOI] [PubMed] [Google Scholar]
- 31.Westphal M, Cox RA, Traber LD, Morita N, Enkhbaatar P, Schmalstieg FC, Hawkins HK, Maybauer DM, Maybauer MO, Murakami K, Burke AS, Westphal-Varghese BB, Rudloff HE, Salsbury JR, Jodin JM, Lee S, Traber DL. Combined burn and smoke inhalation injury impairs ovine hypoxic pulmonary vasoconstriction. Crit Care Med. 2006;34(5):1428–1436. doi: 10.1097/01.CCM.0000215828.00289.B9. [DOI] [PubMed] [Google Scholar]
- 32.Rehberg S, Maybauer MO, Maybauer DM, Traber LD, Enkhbaatar P, Traber DL. The role of nitric oxide and reactive nitrogen specieis in experimental ards. Front Biosci (Schol Ed) 2010;2:18–29. doi: 10.2741/s43. [DOI] [PubMed] [Google Scholar]
- 33.Forman HJ, Fukuto JM, Torres M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiiol Cell Physiol. 2004;287(2):C246–256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 34.Hamahata A, Enkhbaatar P, Kraft ER, Lange M, Leonard SW, Traber MG, Cox RA, Schmalstieg FC, Hawkins HK, Whorton EB, Horvath EM, Szabo C, Traber LD, Herndon DN, Traber DL. Gamma-tocopherol nebulization by a lipid aerosolization device improves pulmonary function in sheep with burn and smoke inhalation injury. Free Radic Biol Med. 2008;45(4):425–433. doi: 10.1016/j.freeradbiomed.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leonard SW, Bruno RS, Paterson E, Schock BC, Atkinson J, Bray TM, Cross CE, Traber MG. 5-nitro-gamma-tocopherol increases in human plasma exposed to cigarette smoke in vitro and in vivo. Free Radic Biol Med. 2003;35(12):1560–1567. doi: 10.1016/j.freeradbiomed.2003.09.010. [DOI] [PubMed] [Google Scholar]