

Abstract
Long-range structural information derived from paramagnetic relaxation enhancement observed in the presence of a paramagnetic nitroxide radical is highly useful for structural characterization of globular, modular and intrinsically disordered proteins, as well as protein–protein and protein-DNA complexes. Here we characterized the conformation of a spin-label attached to the homodimeric protein CylR2 using a combination of X-ray crystallography, electron paramagnetic resonance (EPR) and NMR spectroscopy. Close agreement was found between the conformation of the spin label observed in the crystal structure with interspin distances measured by EPR and signal broadening in NMR spectra, suggesting that the conformation seen in the crystal structure is also preferred in solution. In contrast, conformations of the spin label observed in crystal structures of T4 lysozyme are not in agreement with the paramagnetic relaxation enhancement observed for spin-labeled CylR2 in solution. Our data demonstrate that accurate positioning of the paramagnetic center is essential for high-resolution structure determination.
Electronic supplementary material
The online version of this article (doi:10.1007/s10858-011-9471-y) contains supplementary material, which is available to authorized users.
Keywords: Spin label, Crystal structure, MTSL, Protein, NMR
Introduction
Long-range structural information may be derived from paramagnetic relaxation enhancement (PRE) of NMR signals observed in the presence of a paramagnetic nitroxide radical that has been specifically attached to a diamagnetic protein (Kosen 1989). PRE derived distances are highly useful for the structural characterization of globular (Donaldson et al. 2001; Feeney et al. 2001; Gaponenko et al. 2000) and intrinsically disordered proteins (Dyson and Wright 1998), as well as protein–protein (Iwahara and Clore 2006) and protein-DNA complexes (Iwahara et al. 2004).
Although broadening of NMR signals due to a covalently attached spin label can be measured to high accuracy, the encoded distance information is considered to be less precise because of the putative flexibility of the paramagnetic side chain. This has motivated efforts to rigidify the spin label (Iwahara et al. 2003; Keizers et al. 2008; Leonov et al. 2005; Wohnert et al. 2003). In addition, paramagnetic probe flexibility was accounted for via a multiple-conformer representation in simulated annealing calculations (Iwahara et al. 2004). In cases where prior structural knowledge is available, such as the monomeric structure in oligomeric assemblies or the structure of domains in multi-domain proteins, the position of the paramagnetic center might be obtained from intrasubunit PREs, followed by the determination of the intersubunit structure (Bermejo et al. 2009; Rumpel et al. 2008; Tang et al. 2006; Tang et al. 2007). This approach has the advantage that the sampling space of the spin label is reduced, i.e. the average position of the paramagnetic center is determined experimentally. Using this strategy a high-resolution structure of the homodimeric protein CylR2 (Rumpel et al. 2008) was determined previously using PREs from cysteine mutants that had been tagged with the spin label (1-oxy-2,2,5,5-tetramethyl-D-pyrroline-3-methyl)-methanethiosulfonate (MTSL, Toronto Research Chemicals Inc.).
Enterococcus faecalis is one of the major causes for hospital-acquired antibiotic-resistant infections. The 15.4 kDa homodimer CylR2 is part of a two-component system that regulates the production of the exotoxin cytolysin (Gilmore et al. 1990; Haas et al. 2002; Murray 1990). Apart from the NMR structure (Rumpel et al. 2008) we have solved the X-ray structure of CylR2 before (Rumpel et al. 2004). Here we have determined the crystal structure of MTSL-tagged CylR2 and combined it with NMR measurements and distance measurements from Double Electron Electron Resonance (DEER) (Jeschke and Polyhach 2007; Milov et al. 1984; Schiemann and Prisner 2007). This provided a unique opportunity for investigating the influence of the conformation and dynamics of the MTSL tag on paramagnetic relaxation enhancement observed in solution.
Materials and methods
Protein expression, purification and crystallization
Protein expression in E. coli and purification of CylR2 have been described elsewhere (Razeto et al. 2004). Also mutagenesis and MTSL labeling have been described in detail (Rumpel et al. 2008). 15N-labeled samples were prepared from E. coli cells grown in M9-based minimal medium containing 15NH4Cl. For our studies we further purified CylR2 55R1 (following the convention we call the amino acid resulting from the reaction of MTSL with cysteine R1) by reversed phase HPLC and verified by ESI mass spectrometry that all unbound MTSL had been removed. Crystallization of wildtype CylR2, CylR2 T55C and CylR2 55R1 was performed as described before (Razeto et al. 2004). Briefly, 50 μl protein solution (0.3 mM) in 50 mM HEPES pH 7.0, 2 M NaCl, was placed in the chamber of microdialysis buttons (Hampton Research) that were sealed with a semi-permeable membrane with 10 kDa MW cutoff. Dialysis was performed at 20°C by stepwise transferring the dialysis button every 24 h into the same buffer with decreasing NaCl concentrations (1, 0.5, 0.4, 0.3, 0.2 M NaCl). For CylR2 T55C the protein solution was supplemented with 5 mM Tris(2-carboxy-ethyl)phosphine hydrochloride (TCEP) and the dialysis buffers contained 2 mM TCEP. At 0.3 M NaCl crystals usually appeared. They formed long cuboids with typical dimensions of 1 mm × 0.1 mm × 0.1 mm.
Data collection and crystallographic analysis
For data collection all crystals were stepwise transferred to cryo-solution containing the reservoir solution supplemented with 10, 20 and 30% glycerol. After 1 min incubation in the cryo-solution with 30% glycerol the crystals were flash cooled in liquid nitrogen. Data were collected at the Swiss Light Source beamline PXII (Mar225 CCD detector), processed with XDS (Kabsch 1988) and scaled with SADABS (Bruker AXS). After scaling, the statistics were obtained and the space-group was determined with XPREP (Bruker AXS). Statistics for data collection and processing are summarized in Table S1.
Crystal structure determination and refinement
The structures were solved by molecular replacement using data up to 2.5 Å. The published structure of wildtype CylR2 (PDB code 1utx) was used as search model. The solutions for all crystals were obtained with the program Phaser (McCoy et al. 2005). After initial positional and B-factor refinement using the program REFMAC5 (Murshudov et al. 1997) manual model building was performed with Coot, including the mutation of threonine 55 to cysteine for CylR2 T55C and the placement of the MTSL model into the electron density for CylR2 55R1. The MTSL coordinates were obtained from PDB deposition 2w8h and restraints generated with SHELXPRO (Sheldrick 2008). The crystals of wildtype and CylR2 T55C diffracted to 1.2 Å and those of CylR2 55R1 diffracted to 1.5 Å. Therefore, for all structures the final refinement, including anisotropic B-factor refinement, was performed with SHELXL (Sheldrick 2008; Sheldrick and Schneider 1997). The statistics of the SHELXL refinement are shown in Table S2. Structural alignments were performed with the “align” command of PyMOL. All structural figures were generated with PyMOL (http://www.pymol.org).
EPR measurements
EPR measurements were carried out using a Bruker ELEXYS E580 X-band spectrometer with a dielectric ring resonator ER 4118X-MD5-W1 equipped with an Oxford flow cryostat. The sample contained 100 μM CylR2 55R1 in 50 mM HEPES pH 7.0, 600 mM NaCl, 10% (w/v) glycerol. The glycerol served as cryo-protectant. 100 μl of sample were placed into a 4 mm outer EPR tube and flash frozen in liquid nitrogen. Four-pulse DEER measurements were performed with the following pulse sequence π/2(υobs)-τ1-π-(υobs)-(τ1 + t)-π(υpump)-(τ2-t)-π(υobs)-τ2-echo. The resonator was strongly overcoupled to Q ~50 and the pump pulse (υpump) was set to 16 ns at the resonance mode of the resonator. The pump frequency was applied at the maximum of the nitroxide spectrum. π/2 and π pulses of detection sequence were set to 16 and 32 ns and applied at 65 MHz higher frequency. The amplitude of the observer pulses was adjusted to optimise refocused echo and a two-step phase cycle for the first observer π/2 pulse was used to eliminate any receiver offset. Proton modulation was suppressed by addition of eight spectra of variable delay τ1 time starting at 140 ns with an increment Δτ1 = 8 ns. The τ2 delay between the first Hahn echo and the fourth observer pulse was set to 1.8 μs. The measurement was performed at 20 K with a repetition time of 10 ms, a video amplifier bandwidth of 20 MHz and an amplifier video gain of 54 dB. All DEER traces were processed and analyzed with the DeerAnalysis2008 package (Jeschke et al. 2006).
NMR spectroscopy
The NMR sample contained ~0.3 mM of 15N-labeled CylR2 in 50 mM HEPES pH 7.0, 600 mM NaCl and 5% (v/v) D2O. Two-dimensional 1H/15N-HSQC spectra (Mori et al. 1995) were acquired at 298 K on a Bruker AVANCE 600 MHz spectrometer. The resonance assignment of CylR2 in the same conditions had been determined previously (Rumpel et al. 2004). Spectra were processed using NMRPipe/NMRDraw (Delaglio et al. 1995) and analyzed using Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco).
PRE intensity ratios were obtained from intensities of cross-peaks of backbone amide proton-nitrogen pairs in 15N-HSQC spectra of the paramagnetic (I para) and diamagnetic (I dia) state (i.e. after addition of ascorbic acid). Theoretical intensity ratios I para /I dia were calculated from the crystal structure of CylR2 55R1 according to (Battiste and Wagner 2000):
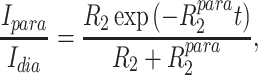 |
1 |
in which 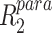 is the enhancement of the transverse relaxation rate by the unpaired electron, t is the total INEPT evolution time of the 15N-HSQC (~9.0 ms) and R
2 are the intrinsic transverse relaxation times of the amide protons. R
2 values of amide protons were approximated by experimental amide nitrogen R
2 values (Ishima and Torchia 2003), which were previously measured (Rumpel et al. 2008).
is the enhancement of the transverse relaxation rate by the unpaired electron, t is the total INEPT evolution time of the 15N-HSQC (~9.0 ms) and R
2 are the intrinsic transverse relaxation times of the amide protons. R
2 values of amide protons were approximated by experimental amide nitrogen R
2 values (Ishima and Torchia 2003), which were previously measured (Rumpel et al. 2008).
Assuming a rigid spin label (Iwahara et al. 2004), the enhancement of the transverse relaxation rate by the unpaired electron 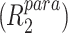 was calculated according to
was calculated according to
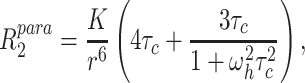 |
2 |
in which r is the distance between the unpaired electron and the amide proton, K is 1.23 × 10−32 cm6 s−2 and ω h is the Larmor frequency of the proton. τ c is the correlation time for the electron-nuclear interaction and was estimated from the T2 relaxation time of amide protons. The average T2 relaxation time of amide protons was measured using a one–one spin-echo experiment (Sklenar and Bax 1987), resulting in a τ c value of 8.5 ns, which is close to the value estimated by the program HydroNMR (Garcia de la Torre et al. 2000) from the crystal structure of CylR2 55R1 (τ c (HydroNMR) = 8.1–10.7 ns depending on simulation parameters).
Results
Crystal structures
Wild-type CylR2, untagged CylR2 T55C and CylR2 55R1 crystallized in space group P41 with very similar unit cell parameters (see Table S1). The structure of wild type CylR2 and the T55C mutant were solved at 1.21 and 1.23 Å resolution, respectively. The rmsd between the backbone of both structures is 0.13 Å, indicating that the mutation did not change the backbone structure. The structure of CylR2 55R1 was solved at 1.5 Å resolution and had an rms deviation of 0.173 and 0.157 Å from the structure of wild type and CylR2 T55C, respectively. Thus, the introduction of the spin label did not induce considerable structural changes beyond those caused already by the mutation at threonine 55. In both monomers the cysteine 55 side chain was modelled in two conformations into the electron density map, one being MTSL-bound the other one free. The electron density at the MTSL-bound conformer of cysteine 55 is well defined and allowed modelling of the MTSL tags in a single conformation in both monomers (Fig. 1). The occupancy of the MTSL tag was refined to 70% (chain A) and 84% (chain B).
Fig. 1.

Crystal structure of CylR2 55R1. a Side view; the 55R1 side chain is shown in stick representation for both monomers. b Top view; the distance between the nitroxide oxygens is indicated by a dashed line. c and d Electron density of the stick models of R1 in monomer a and b, respectively. σ-weighted map rendered at 1.0 σ
Wildtype CylR2, CylR2 T55C and CylR2 55R1 crystallized as one homodimer per asymmetric unit (Fig. 1). The structures of the two monomers show only small differences (0.58 Å rmsd for CylR2 55R1 Cα atoms). Notably, in all three structures the loop containing residues 38–41 in monomer A shows a double conformation, not observed in the published structure of wildtype CylR2 (Rumpel et al. 2004), while the same loop shows only a single conformation in monomer B. However, the two MTSL tags superpose very well (Fig. 2) in a single conformation stabilised by hydrophobic interactions with surrounding side chains, notably isoleucine 9. The distance between the nitroxide oxygens in the two monomers is 27.6 Å (Fig. 1b). The crystal structures of CylR2, CylR2 T55C and CylR2 55R1 have been submitted to the PDB with PDB IDs 2XI8, 2XJ3, and 2XIU, respectively.
Fig. 2.

Superposition of the R1 side chains from monomer a and b of CylR2 55R1. The superposition is based on the main chain atoms of residues 54–56
Intermonomer distance of the paramagnetic centers by EPR
To probe the distance between the paramagnetic nitroxide radicals in the CylR2 55R1 homodimer DEER measurements were conducted in frozen solution. Figure 3a shows experimental time-domain DEER data after subtraction of a homogeneous background function and distance profile after Tikhonov regularisation (Jeschke et al. 2006). An interspin distance of 26.0 Å with a standard deviation of 1.0 Å was observed (Fig. 3b). The distance is similar to the distance obtained between the nitroxide oxygens in the crystal structure of CylR2 55R1 (27.6 Å).
Fig. 3.

DEER-EPR data. a Background corrected DEER time-domain (black) and after Thikhonov regularization (red). b Distance distribution obtained after Thikhonov regularization
Comparison of NMR-detected paramagnetic relaxation enhancement with the crystal structure of CylR2
To obtain insight into the relevance of the crystal structure-derived conformation of the MTSL tag for measurements in solution at room temperature, we measured the PRE of amide protons in 15N-labeled CylR2 55R1. As both molecules of the CylR2 dimer are tagged with MTSL, intensity broadening results from spatial proximity within a molecule and between the two molecules of the dimer. However, due to the r−6 dependence of the PRE and the fact that the MTSL tag at T55C is not located directly at the dimer interface (Fig. 1a), intensity broadening in CylR2 55R1 is dominated by intramolecular distances (Fig. S1). In agreement with the position of the tag and the 3D structure of CylR2 (Fig. 1; (Rumpel et al. 2004) residues in direct vicinity to residue 55 and residue 8 are broadened beyond detection (Fig. 4a). Indeed, back-calculation of the PRE-induced broadening from the crystal structure of CylR2 55R1 resulted in an excellent fit with the experimental PRE profile (Fig. 4a). For most residues back-calculated and experimental PRE intensity ratios are identical within the experimental errors. A notable exception are residues 26–28 and 40, where experimental PREs would predict slightly shorter distances than observed in the crystal structure (Fig. 4a). A similarly good fit is obtained when comparing the distances calculated from the experimental PRE values and those observed in the crystal structure (Fig. 4b). The root-mean-square deviations between the two sets of distances are 2.8, 2.2 and 2.2 Å when considering only residues with crystal structure distances less than 17 Å (10 residues; excluding those that are broadened beyond detection), between 17 and 21 Å (30 residues) and between 17 and 25 Å (35 residues), respectively.
Fig. 4.

Comparison of experimental paramagnetic relaxation enhancement observed in 2D 1H-15N HSQC NMR spectra of CylR2 55R1 with values back-calculated from the crystal structure of CylR2 55R1. a Experimental (black) and back-calculated (grey) PRE intensity ratios, Iparam/Idia, as a function of residue number. Error bars were obtained from repeat measurements. Decreases in peak intensity ratios that occur far from the site of spin-labelling (>10 residues) are indicative of long-range contacts (<25 Å) between the spin-label and distant areas of sequence. b Comparison of intramolecular distances in the crystal structure of CylR2 55R1 with values calculated from the experimental PRE ratios. Residues, for which the signal was broadened beyond detection (upper distance value of 14 Å), are marked in grey. Black lines indicate ±5 Å
Next, we investigated the influence of the conformation of the MTSL tag on the PRE profile, which was back-calculated from the 3D structure. The dihedral angles within the MTSL tag were adjusted to values previously suggested and observed in crystal structures of T4 lysoszyme (T4L) tagged at sites 72 and 131 (Sezer et al. 2009). On the basis of the new MTSL conformations PRE values were predicted and compared to experimental values. χ2 values between calculated and experimental PRE intensity ratios varied between 2.10 and 7.69 (Table 1). The best matches were obtained for the C1 conformations of both site 72R1 (χ1 = −176°/χ2 = 49°/χ3 = 93°/χ4 = −82°/χ5 = 100°) and 131R1 (χ1 = −173°/χ2 = 173°/χ3 = −96°/χ4 = 72°/χ5 = −100°) of T4L with χ2 values of 2.10 and 2.26, respectively. These values, however, are still much higher than the χ2 value of 0.76 that is obtained when the MTSL conformation of the crystal structure of CylR2 55R1 is used (χ1 = −63°/χ2 = −61°/χ3 = −87°/χ4 = −77°/χ5 = 161° for subunit 1) (Table 1). The large variation in predicted PRE intensity ratios becomes clear when the positions of the spin centers for the different R1 conformations listed in Table 1 are mapped onto the 3D structure of CylR2 (Fig. 5a). The position of the paramagnetic oxygen for the different R1 conformations differs anywhere from 5.8 Å (C1 at site 72R1 of T4 lysozyme) to 11.6 Å (C2 at site 131R1 of T4L) from the one observed in the CylR2 55R1 crystal structure. Accordingly, large differences between the experimental PRE intensity profile and the one back-calculated on the basis of the 131R1 C2 conformation of T4L are observed (Fig. 5b).
Table 1.
Deviation between experimental NMR PRE intensity ratios and values back-calculated from the crystal structure of CylR2 55R1 for different conformations of the MTSL tag in T4L 72R1 (Sezer et al. 2009), T4L 131R1 (Sezer et al. 2009) and CylR2 55R1 (this work)
| Protein/conformers | Angle (degree) | χ2 | ||||
|---|---|---|---|---|---|---|
| χ1 | χ2 | χ3 | χ4 | χ5 | ||
| T4L 72R1 | ||||||
| C1 | −176 | 49 | 93 | − 82 | 100 | 2.26 |
| C2 | −168 | 48 | 85 | −159 | −70 | 3.54 |
| C3 | −169 | 48 | 78 | −144 | 20 | 3.44 |
| C4 | −63 | −66 | 94 | 172 | 57 | 3.91 |
| C5 | −55 | −44 | −91 | −178 | −93 | 3.51 |
| T4L 131R1a | ||||||
| C1 | −173 | 173 | −96 | 72 | −100 | 2.10 |
| C2 | −62 | −48 | 101 | −161 | 77 | 4.68 |
| C3 | −171 | 66 | −97 | 174 | −63 | 3.96 |
| C4 | −65 | −177 | −84 | −174 | −87 | 7.69 |
| C5 | −58 | −58 | 97 | 173 | −83 | 4.27 |
| CylR2 55R1 | ||||||
| Chain A/C1 | −63 | −61 | −87 | −77 | 161 | 0.76b |
| Chain B/C2 | −74 | −53 | −97 | −72 | 156 | |
aConformations observed in the crystal structure of T4 lysozyme 131R1 are underlined (Sezer et al. 2009)
bAverage χ2 value obtained for the two subunits of the CylR2 dimer
Fig. 5.

Influence of the conformation of the MTSL tag on paramagnetic relaxation enhancement in CylR2 55R1. a Theoretical positions of the paramagnetic oxygen in the 3D structure of CylR2 for conformations of the MTSL tag from the T4L structure. Left hand side: oxygen positions C1 (Magenta), C2 (orange), C3 (yellow), C4 (green) and C5 (blue) for the 72R1 conformations listed in Table 1. Right hand side: positions corresponding to 131R1. The oxygen location observed in the crystal structure of CylR2 55R1 is shown in red. Two views related by a 90° rotation are presented. b Comparison of the experimental PRE intensity profile (black) with the pattern calculated for the 131R1 C2 conformation (grey). Error bars were obtained from repeat measurements. Note the large deviation between the two profiles in comparison to Fig. 4
Discussion
Distance information derived from PREs of NMR signals observed in the presence of a paramagnetic nitroxide radical, which has been specifically attached to a diamagnetic protein, has three advantages over Nuclear Overhauser Enhancement: (i) It is long-range (up to 25 Å), (ii) it can be used in the case of fully deuterated proteins or for proteins for which no side chain assignment can be obtained and, (iii) the number of accessible distances might be increased by attaching spin labels to different sites in the protein.
For accurate PRE-based structure determination the variability and flexibility in the conformation of the spin label has to be taken into account (Iwahara et al. 2004). The crystal structure of CylR2 55R1 shows that the MTSL tags in both monomers of CylR2 are highly-ordered in the protein matrix (Fig. 1c, d). Such a well ordered MTSL tag has also been found in residue 115R1 of MTSL-tagged T4L (Guo et al. 2007). This residue is located at the protein surface, similar to the R1 residues in CylR2. The ordered, immobile state of T4L 115R1 has been attributed to hydrophobic interactions of the nitroxide ring. A close inspection of the CylR2 55R1 structure suggests that hydrophobic interactions, especially with isoleucine 9, are the reason for the ordered state of the R1 side chains in this protein. In line with this finding is the narrow distance distribution between both paramagnetic nitroxide radicals found by DEER measurements (Fig. 3b). A narrow distance distribution is expected when the paramagnetic centers are highly ordered as it was demonstrated for protein matrices, DNA and bi-radicals (Bennati et al. 2005; Endeward et al. 2009; Margraf et al. 2007; Marko et al. 2009; Schiemann et al. 2009). Thus, the preferred orientation of the tag in frozen solution is very similar to the orientation of the tag found in both monomers in the crystal structure of the CylR2 homodimer.
In case of the NMR data, a very good fit between experimental PRE intensity ratios and values back-calculated from the crystal structure was obtained (Fig. 4b), suggesting that the conformation seen in the crystal structure is also preferred in solution, in line with the EPR data. The root-mean-square deviation between PRE-derived distances and those observed in the crystal structure was ~2.5 Å. The remaining small deviations (outside of the experimental errors in the PRE intensity ratios) can have a variety of sources. (i) The average conformation of the spin label in solution deviates slightly from the one observed in the crystal structure. (ii) Some nuclei may effectively sense one region of the probe’s sampling space, while other nuclei may be affected by another region of such space, thus hindering the description of the effect in terms of a single probe position. (iii) Amide proton T 2 relaxation times were approximated by experimental amide nitrogen T 2 relaxation times (Ishima and Torchia 2005). (iv) Unspecific binding of MTSL to the protein.
The root-mean-square deviation between PRE-derived distances and those observed in the crystal structure is by a factor of two lower than the error boundaries (±5 Å) commonly used for PREs-derived distance restraints in structure calculations (Battiste and Wagner 2000). Thus, our study demonstrates that tighter error boundaries can be used, when the average position of the paramagnetic center can be determined from independent data, such as intrasubunit PREs. This will improve the quality of 3D structures determined with the help of PREs, as previously shown for the CylR2 homodimer (Rumpel et al. 2008) and a glutamine-binding protein (Bermejo et al. 2009). The situation is very similar to the case of pseudo-contact shifts induced by a lanthanide that is attached to the protein via a lanthanide tag. Use of the pseudo-contact shifts for structural and dynamic analysis requires prior determination of the metal position (for a review see Otting 2010). In both cases, it is generally best to refine directly against the experimentally observed PREs and not against PRE-derived distances (Iwahara et al. 2004).
Is it possible to restrict the position of the paramagnetic center without intrasubnit PREs? One strategy in this direction could be to implement dihedral angle potentials that favour conformations of the spin label that were previously observed in crystal structures of MTSL-tagged proteins, e.g. those shown for T4 lysozyme in Table 1. Figure 5a, however, demonstrates that this is dangerous because conformations of the spin label and thus the position of the paramagnetic center can change significantly when going from one protein to another protein or even when going from one site to another within the same protein. Importantly, incorrect positioning of the paramagnetic center might lead to severe distortions in structure (Fig. 5b).
In summary, the close agreement between the conformation of the spin label observed in the crystal structure of CylR2 with interspin distances measured by EPR and signal broadening in NMR spectra suggests that the conformation of the spin label seen in the crystal structure is also preferred in solution. This allowed for a cross-validation between the paramagnetic relaxation enhancement in solution and the X-ray crystal structure of this protein. Most importantly, our data demonstrate that accurate positioning of the paramagnetic center is essential for PRE-based high-resolution structure determination.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Christian Griesinger and George Sheldrick for generous support; Marina Bennati for the permission to record EPR data in her laboratory; Anuschka Pauluhn, Takahashi Tomizaki and Vincent Olieric for beamline assistance. This work was supported by the Max Planck Society. TG was supported by the “Fonds der Chemischen Industrie”; MZ by the DFG through a Heisenberg scholarship (ZW 71/2-2 and 3-2).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- MTSL
(1-oxy-2,2,5,5-tetramethyl-D-pyrroline-3-methyl)-methanethiosulfonate
- HSQC
Heteronuclear single quantum coherence
- EPR
Electron paramagnetic resonance
- DEER
Double electron electron resonance
- PRE
Paramagnetic relaxation enhancement
Footnotes
Tim Gruene and Min-Kyu Cho have contributed equally to this work.
Contributor Information
Markus Zweckstetter, Email: mzwecks@gwdg.de.
Stefan Becker, Email: sabe@nmr.mpibpc.mpg.de.
References
- Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Bennati M, Robblee JH, Mugnaini V, Stubbe J, Freed JH, Borbat P. EPR distance measurements support a model for long-range radical initiation in E. coli ribonucleotide reductase. J Am Chem Soc. 2005;127:15014–15015. doi: 10.1021/ja054991y. [DOI] [PubMed] [Google Scholar]
- Bermejo GA, Strub MP, Ho C, Tjandra N. Determination of the solution-bound conformation of an amino acid binding protein by NMR paramagnetic relaxation enhancement: use of a single flexible paramagnetic probe with improved estimation of its sampling space. J Am Chem Soc. 2009;131:9532–9537. doi: 10.1021/ja902436g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Donaldson LW, Skrynnikov NR, Choy WY, Muhandiram DR, Sarkar B, Forman-Kay JD, Kay LE. Structural characterization of proteins with an attached ATCUN motif by paramagnetic relaxation enhancement NMR spectroscopy. J Am Chem Soc. 2001;123:9843–9847. doi: 10.1021/ja011241p. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Equilibrium NMR studies of unfolded and partially folded proteins. Nat Struct Biol. 1998;5 Suppl:499–503. doi: 10.1038/739. [DOI] [PubMed] [Google Scholar]
- Endeward B, Butterwick JA, MacKinnon R, Prisner TF. Pulsed Electron-Electron Double-Resonance Determination of Spin-Label Distances and Orientations on the Tetrameric Potassium Ion Channel KcsA. J Am Chem Soc. 2009;131:15246–15250. doi: 10.1021/ja904808n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney J, Birdsall B, Bradbury AF, Biekofsky RR, Bayley PM. Calmodulin tagging provides a general method of using lanthanide induced magnetic field orientation to observe residual dipolar couplings in proteins in solution. J Biomol NMR. 2001;21:41–48. doi: 10.1023/A:1011924017938. [DOI] [PubMed] [Google Scholar]
- Gaponenko V, Howarth JW, Columbus L, Gasmi-Seabrook G, Yuan J, Hubbell WL, Rosevear PR. Protein global fold determination using site-directed spin and isotope labeling. Protein Sci. 2000;9:302–309. doi: 10.1110/ps.9.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia de la Torre J, Huertas ML, Carrasco B. HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J Magn Reson. 2000;147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- Gilmore MS, Segarra RA, Booth MC. An HlyB-type function is required for expression of the Enterococcus faecalis hemolysin/bacteriocin. Infect Immun. 1990;58:3914–3923. doi: 10.1128/iai.58.12.3914-3923.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Cascio D, Hideg K, Kalai T, Hubbell WL. Structural determinants of nitroxide motion in spin-labeled proteins: tertiary contact and solvent-inaccessible sites in helix G of T4 lysozyme. Protein Sci. 2007;16:1069–1086. doi: 10.1110/ps.062739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas W, Shepard BD, Gilmore MS. Two-component regulator of Enterococcus faecalis cytolysin responds to quorum-sensing autoinduction. Nature. 2002;415:84–87. doi: 10.1038/415084a. [DOI] [PubMed] [Google Scholar]
- Ishima R, Torchia DA. Extending the range of amide proton relaxation dispersion experiments in proteins using a constant-time relaxation-compensated CPMG approach. J Biomol NMR. 2003;25:243–248. doi: 10.1023/A:1022851228405. [DOI] [PubMed] [Google Scholar]
- Ishima R, Torchia DA. Error estimation and global fitting in transverse-relaxation dispersion experiments to determine chemical-exchange parameters. J Biomol NMR. 2005;32:41–54. doi: 10.1007/s10858-005-3593-z. [DOI] [PubMed] [Google Scholar]
- Iwahara J, Clore GM. Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature. 2006;440:1227–1230. doi: 10.1038/nature04673. [DOI] [PubMed] [Google Scholar]
- Iwahara J, Anderson DE, Murphy EC, Clore GM. EDTA-derivatized deoxythymidine as a tool for rapid determination of protein binding polarity to DNA by intermolecular paramagnetic relaxation enhancement. J Am Chem Soc. 2003;125:6634–6635. doi: 10.1021/ja034488q. [DOI] [PubMed] [Google Scholar]
- Iwahara J, Schwieters CD, Clore GM. Ensemble approach for NMR structure refinement against (1) H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J Am Chem Soc. 2004;126:5879–5896. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys Chem Chem Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. DeerAnalysis2006—a comprehensive software package for analyzing pulsed ELDOR data. Appl Magn Reson. 2006;30:473–498. doi: 10.1007/BF03166213. [DOI] [Google Scholar]
- Kabsch W. Automatic-indexing of rotation diffraction patterns. J Appl Crystallogr. 1988;21:67–71. doi: 10.1107/S0021889887009737. [DOI] [Google Scholar]
- Keizers PH, Saragliadis A, Hiruma Y, Overhand M, Ubbink M. Design, synthesis, and evaluation of a lanthanide chelating protein probe: CLaNP-5 yields predictable paramagnetic effects independent of environment. J Am Chem Soc. 2008;130:14802–14812. doi: 10.1021/ja8054832. [DOI] [PubMed] [Google Scholar]
- Kosen PA. Spin labeling of proteins. Meth Enzymol. 1989;177:86–121. doi: 10.1016/0076-6879(89)77007-5. [DOI] [PubMed] [Google Scholar]
- Leonov A, Voigt B, Rodriguez-Castaneda F, Sakhaii P, Griesinger C. Convenient synthesis of multifunctional EDTA-based chiral metal chelates substituted with an S-mesylcysteine. Chemistry. 2005;11:3342–3348. doi: 10.1002/chem.200400907. [DOI] [PubMed] [Google Scholar]
- Margraf D, Bode BE, Marko A, Schiemann O, Prisner TF. Conformational flexibility of nitroxide biradicals determined by X-band PELDOR experiments. Mol Phys. 2007;105:2153–2160. doi: 10.1080/00268970701724982. [DOI] [Google Scholar]
- Marko A, Margraf D, Yu H, Mu Y, Stock G, Prisner T. Molecular orientation studies by pulsed electron-electron double resonance experiments. J Chem Phys. 2009;130:064102. doi: 10.1063/1.3073040. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Milov AD, Ponomarev AB, Tsvetkov YD. Electron electron double-resonance in electron-spin echo—model biradical systems and the sensitized photolysis of decalin. Chem Phys Lett. 1984;110:67–72. doi: 10.1016/0009-2614(84)80148-7. [DOI] [Google Scholar]
- Mori S, Abeygunawardana C, Johnson MO, van Zijl PC. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J Magn Reson B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- Murray BE. The life and times of the Enterococcus. Clin Microbiol Rev. 1990;3:46–65. doi: 10.1128/cmr.3.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum- likelihood method. Acta Crystallogr Sect D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otting G. Protein NMR using paramagnetic ions. Annu Rev Biophys. 2010;39:387–405. doi: 10.1146/annurev.biophys.093008.131321. [DOI] [PubMed] [Google Scholar]
- Razeto A, Giller K, Haas W, Gilmore MS, Zweckstetter M, Becker S. Expression, purification, crystallization and preliminary crystallographic studies of the Enterococcus faecalis cytolysin repressor CylR2. Acta Crystallogr D Biol Crystallogr. 2004;60:746–748. doi: 10.1107/S0907444904002410. [DOI] [PubMed] [Google Scholar]
- Rumpel S, Razeto A, Pillar CM, Vijayan V, Taylor A, Giller K, Gilmore MS, Becker S, Zweckstetter M. Structure and DNA-binding properties of the cytolysin regulator CylR2 from Enterococcus faecalis. EMBO J. 2004;23:3632–3642. doi: 10.1038/sj.emboj.7600367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, Becker S, Zweckstetter M. High-resolution structure determination of the CylR2 homodimer using paramagnetic relaxation enhancement and structure-based prediction of molecular alignment. J Biomol NMR. 2008;40:1–13. doi: 10.1007/s10858-007-9204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann O, Prisner TF. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q Rev Biophys. 2007;40:1–53. doi: 10.1017/S003358350700460X. [DOI] [PubMed] [Google Scholar]
- Schiemann O, Cekan P, Margraf D, Prisner TF, Sigurdsson ST. Relative orientation of rigid nitroxides by PELDOR: beyond distance measurements in nucleic acids. Angew Chem Int Ed Engl. 2009;48:3292–3295. doi: 10.1002/anie.200805152. [DOI] [PubMed] [Google Scholar]
- Sezer D, Freed JH, Roux B. Multifrequency electron spin resonance spectra of a spin-labeled protein calculated from molecular dynamics simulations. J Am Chem Soc. 2009;131:2597–2605. doi: 10.1021/ja8073819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM, Schneider TR. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. doi: 10.1016/S0076-6879(97)77018-6. [DOI] [PubMed] [Google Scholar]
- Sklenar V, Bax A. Spin-echo water suppression for the generation of pure-phase two-dimensional NMR spectra. J Magn Reson. 1987;74:469–479. [Google Scholar]
- Tang C, Iwahara J, Clore GM. Visualization of transient encounter complexes in protein-protein association. Nature. 2006;444:383–386. doi: 10.1038/nature05201. [DOI] [PubMed] [Google Scholar]
- Tang C, Schwieters CD, Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449:1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- Wohnert J, Franz KJ, Nitz M, Imperiali B, Schwalbe H. Protein alignment by a coexpressed lanthanide-binding tag for the measurement of residual dipolar couplings. J Am Chem Soc. 2003;125:13338–13339. doi: 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.