

Abstract
Mammals express seven different catalytically active proteasome subunits. In order to determine the roles of the different proteolytically active subunits in antigen presentation and other cellular processes, highly specific inhibitors and activity-based probes that selectively target specific active sites are needed. In this work we present the development of fluorescent activity-based probes that selectively target the β1 and β5 sites of the constitutive proteasome.
Introduction
Proteasomes degrade the majority of proteins in mammalian cells and are established targets for the development of anticancer drugs. All eukaryotic proteasomes have three types of active sites. The site located on the β1 subunit is referred to as caspase-like, the site on subunit β2 as trypsin-like and the site on the β5 subunit as chymotryptisin-like, although the subunits are rather more promiscuous in their substrate preference than is suggested by this designation. Four additional proteolytically active sites are present on proteasomes in specific cell types in vertebrates. In cells of the immune system, the β1i, β2i and the β5i subunits replace the corresponding constitutive subunits in newly formed proteasome particles called immunoproteasomes.1 Cortical thymic epithelial cells express an additional proteolytic subunit, the β5t, which replaces β5i in immunoproteasomes to form the thymoproteasome.2
This diversity of active sites in eukaryotic proteasomes is in a stark contrast to the much simpler proteasomes in bacteria and archaea, which possess only one or two types of active sites. In prokaryotic proteasomes there are proteolytic active sites on all 14 β-subunits. The total number of active sites on eukaryotic proteasomes is 6 (two of each type). The reasons for such dramatic reduction in total number of active sites (14 to 6) in favor of more diverse specificity are not clear. Site-directed mutagenesis in yeast revealed that the different active sites play specific roles in protein degradation, and that their contribution to growth, viability and stress resistance of yeast strains are strikingly different.3 According to these studies, the β5 sites are the most important, whereas the β1 sites are apparently redundant. Thus, it is not clear why β1 sites are present in all eukaryotes from yeast to humans.
The proteasome inhibitor bortezomib (Velcade, PS-341)4 is being used for the treatment of multiple myeloma5 and mantle cell lymphoma,6 and at least three other inhibitors are tested clinically. The role of individual active sites as drug targets in cancer has not been fully elucidated. The β5 and β5i sites are the primary targets of all inhibitors used in the clinic, but the majority of them also target other sites. For example, bortezomib also inhibits β1 sites of the constitutive proteasome and β1i and β2i sites of the immunoproteasome.7,8 Whether or not the co-inhibition of β1/β1i and β2i sites contributes to anti-neoplastic activity of bortezomib is not clear. Using newly developed specific inhibitors of the β1 and β5 sites (Fig. 1A, NC-001 (1) and NC-005 (2), respectively), we have recently demonstrated that inhibition of the β1 sites sensitizes myeloma cells to β5 selective inhibitors.9 This suggests that the β1 sites, despite their apparent redundancy, have to be considered co-targets for anti-neoplastic drugs. Furthermore, proteasomes are involved in the generation of antigenic peptides loaded in MHC class I complexes, but the contribution of each separate active proteasome subunit to the epitope repertoire has not yet been defined, largely due to the lack of cell permeable site-specific inhibitors.
Fig. 1.

(A) Schematic representation of the eukaryotic 20S proteasome and previously described β1 and β5 specific proteasome inhibitors. The proteolytically active β subunits are marked in red. (B) Recognition elements I and II and warheads a–c resulting in 6 hybrid structures, Ia–c and IIa–c.
In order to determine the roles of the different proteolytically active subunits in antigen presentation and other cellular processes, highly specific inhibitors and activity-based probes that selectively target specific active sites are needed. In this study we use the inhibitors we previously reported as being selective for the β1 and β5 sites,9 Ac-Ala-Pro-Nle-LeuEK (1) and Napht-Tyr(OMe)-Phe-LeuEK (2), respectively, as the basis for the development of a panel of fluorescent site-specific activity-based probes.
Results and discussion
The majority of proteasome inhibitors are short peptides with an electrophilic group at the C-terminus.10 This electrophilic “warhead” reacts with the proteasome’s catalytic N-terminal threonines. Previously, we demonstrated that swapping the peptide portions and warheads of known proteasome inhibitors can alter active site specificities and potencies.11 We decided to test whether changing the warheads on compounds 1 and 2 improves their potency and specificity. The best characterized and most commonly used proteasome warheads are aldehydes, vinyl sulfones, boronates and epoxyketones. Of these, only vinyl sulfones and epoxyketones are suitable for use in activity-based probes, since only these form covalent irreversible bonds with the proteasome active sites that are not destroyed during heating under denaturing conditions, a mandatory step preceding analysis of probe-treated samples by SDS-PAGE. Therefore, we have synthesized derivatives of 1 and 2 armed with leucine phenol vinyl sulfone (LeuVS-PhOH),12 leucine methyl vinyl sulfone (LeuVS)13 and leucine epoxyketone (LeuEK)14 to arrive at a panel of 6 potential subunit specific proteasome inhibitors (Fig. 1B). All inhibitors used in the study were synthesized by coupling the recognition fragments to the leucine-derived warheads. In order to avoid epimerization at the P2 position, we used the azide coupling (Scheme 1). Hence, the synthesis commenced with the preparation of the hydrazides of the peptidic recognition elements. Fmoc-based solid phase peptide synthesis using HMPB-functionalized MBHA resin gave immobilized acetyl capped tripeptide 3. After mild acidic cleavage from resin, the crude peptide was treated with TMS–diazomethane to give methyl ester 5. Refluxing in methanol in the presence of an excess of hydrazine resulted in the β1 recognition element peptide hydrazide 6. The β5 recognition peptide hydrazide was prepared by condensation of Boc-protected tyrosine methyl ether 7 with phenylalanine methyl ester to give dipeptide 8. After acidic cleavage of the Boc protective group and capping with 2-(naphthalen-2-yl)-acetic acid, methyl ester 9 was reacted with hydrazine in refluxing methanol to give peptide hydrazide 10. The recognition element peptide hydrazides were treated with tert-butyl nitrite under acidic conditions to generate an acyl azide in situ. After addition of base, the warhead amines (synthesized according to literature procedures12–14) were reacted with the activated peptides to give the hybrid structures Ia–c and IIa–c.
Scheme 1.
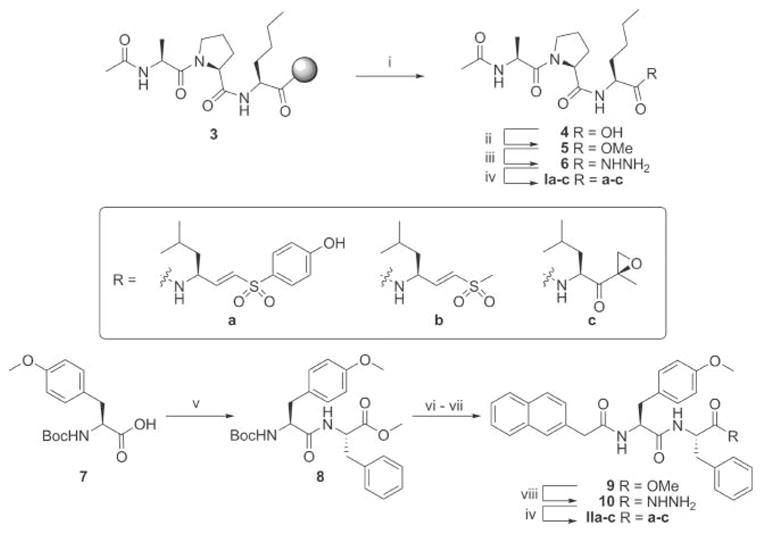
Synthesis of the hybrid structures. Reagents and conditions: (i) 1% TFA in DCM, 30 min, 3×. (ii) TMS–diazomethane (2 equiv.), 15 min., 85% from Fmoc-Nle-resin. (iii) Hydrazine monohydrate (60 equiv.), MeOH, reflux, 1.5 h, 92%. (iv) (a) HCl (2.8 equiv.), tBuONO (1.1 equiv.), DMF–EtOAc (1: 1, v/v), −25 °C, 4 h. (b) TFA·H-Ra–c (1.1 equiv.), DiPEA (3.8 equiv.), −25 °C to RT, 15 h, Ia 77%, Ib 34%, Ic 63%, IIa 77%, IIb 53%, IIc 66%. (v) HCl·H-Phe-OMe (1 equiv.), BOP (1 equiv.), DiPEA (2.2 equiv.), DCM, 15 h, quant. (vi) TFA/DCM 1: 1 (v/v), 15 min. (vii) 2-(Naphthalen-2-yl)-acetic acid (1 equiv.), BOP (1 equiv.), DiPEA (3.3 equiv.), DCM, 15 h, 68%. (viii) Hydrazine monohydrate (60 equiv.), MeOH, reflux, 2.5 h, 95%.
The proteasome inhibition profile of the panel of 6 modified oligopeptides was determined in competition experiments versus the fluorescent broad-spectrum activity-based proteasome probe MV151.15 Human Embryonic Kidney (HEK293T) cell lysates expressing the constitutive proteasome were exposed to increasing concentrations of the inhibitors for one hour. Residual proteasome activity was labeled with MV151, after which the proteins were denatured, separated on SDS-PAGE and visualized using a fluorescence scanner (Fig. 2A). Apparent IC50 values were determined by quantification of the fluorescent gel bands (Fig. 2A and Table 1). As reported, inhibitors Ia16 and Ic (1)9 selectively block the β1 subunit. The selectivity and potency of the methyl vinyl sulfone-equipped β1 recognition peptide Ib is in the same order of magnitude as the phenol vinyl sulfone Ia, but both are one order of magnitude less potent than the β1 selective epoxyketone Ic.
Fig. 2.
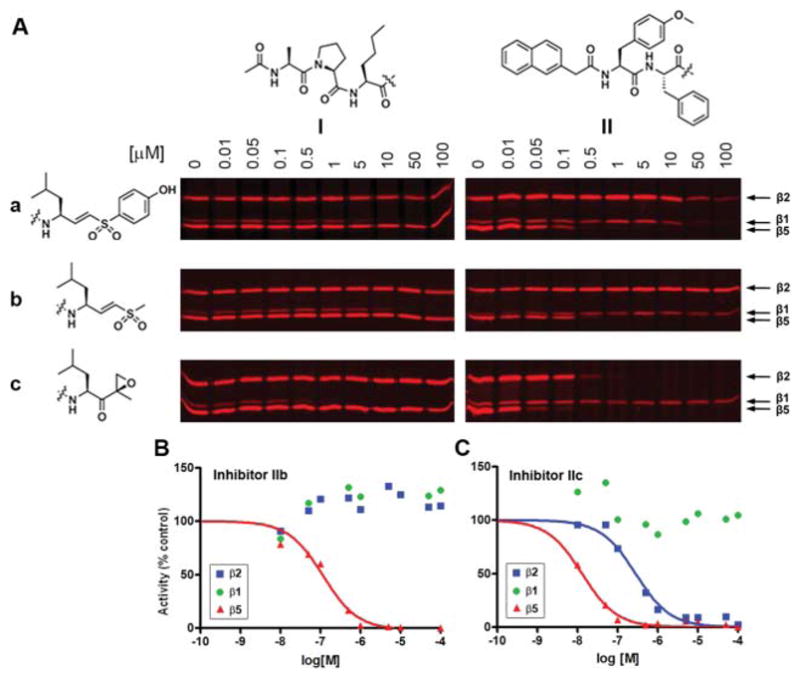
Proteasome profiling screen of the hybrids Ia–c and IIa–c using MV151. (A) HEK293T lysates (10 μg total protein) were incubated with the indicated concentrations of compounds Ia–c and IIa–c for 1 h at 37 °C. The residual proteasome activity was fluorescently labeled by incubation with 1 μM MV151 for 1 h at 37 °C. (B–C) The fluorescent band intensity for each subunit was quantified to give competition curves as shown in (B) for IIb and in (C) for IIc.
Table 1.
Apparent IC50 values (μM) for Ia–c and IIa–ca
| Subunit | β1 | β2 | β5 |
|---|---|---|---|
| Compound | |||
| Ia | 4.0 ± 0.67 | >100 | >100 |
| Ib | 7.8 ± 0.86 | >100 | >100 |
| Ic | 0.66 ± 0.12 | >100 | 373 ± 91 |
| IIa | 32 ± 16 | 78 ± 23 | 0.07 ± 0.01 |
| IIb | >100 | >100 | 0.11 ± 0.02 |
| IIc | >100 | 0.28 ± 0.04 | 0.013 ± 0.001 |
In the gels shown in Fig. 2, the fluorescent band intensity for each subunit was quantified in each lane to give competition curves, which were then used to calculate apparent IC50 values. IC50 values for irreversible inhibitors do not refer to inhibition constants (IC50 ≠ Ki). It is the observed 1/2 max inhibition concentration for the specific experimental setup.
In the panel of inhibitors containing the β5 recognition element, phenol vinyl sulfone IIa is a more selective inhibitor of the β5 subunit than the parent epoxyketone IIc (2), but still targets β1 and β2 at higher concentrations. However, vinyl sulfone IIb is a very potent and very selective inhibitor for the β5 subunit (Fig. 2B). Although about 10 times more potent for β5, the parent epoxyketone IIc (2) was much less selective, since it also blocks the β2 subunit, leaving β1 as the sole active proteasome subunit (Fig. 2C).
Three inhibitors were chosen as starting points for the development of fluorescent activity-based probes: Ic that is selective for β1, IIb as a selective and potent inhibitor for β5 and IIc inhibiting β2 and β5. It was reasoned that replacing the aromatic naphthyl acetic acid in inhibitors IIb and IIc (2) by azido-BODIPY17 would not affect the specificity of the resulting fluorescent probes. The synthesis of the β5 targeted probes commenced with the transformation of the Boc-protected dipeptide methyl ester 8 into the corresponding hydrazide 11 (Scheme 2). Azide coupling with LeuVS gave methyl vinyl sulfone 12. Acidic deprotection and subsequent capping of the free amine with azido-BODIPY-OSu (13) gave the fluorescent probe 14. Similarly, β2/β5 targeting probe 16 was constructed from Boc-protected tripeptide epoxyketone 15.
Scheme 2.

Synthesis of azido-BODIPY probes 14 and 16. Reagents and conditions: (i) Hydrazine monohydrate (60 equiv.), MeOH, reflux, 2 h, 88%. (ii) (a) HCl (2.8 equiv.), tBuONO (1.1 equiv.), DMF–EtOAc (1: 1, v/v), −25 °C, 4 h. (b) TFA·H-LeuVS (1.1 equiv.), DiPEA (3.8 equiv.), −25 °C to RT, 15 h, 75%. (iii) TFA/DCM 1: 1 (v/v), 30 min. (iv) 13 (1 equiv.), DiPEA (1 equiv.), DCM, 15 h, 14 54%, 16 27%. (v) (a) HCl (2.8 equiv.), tBuONO (1.1 equiv.), DMF–EtOAc (1: 1, v/v), −25 °C, 4 h. (b) TFA·H-LeuEK (1.1 equiv.), DiPEA (3.8 equiv.), −25 °C to RT, 15 h, 71%.
For the synthesis of a fluorescent β1 selective probe, we took advantage of the previously described green fluorescent acetylene functionalized BODIPY dye 22.18 The azide equipped intermediate 21 is easily accessible by replacing the acetyl in Ic with an azido glycine. Standard solid-phase peptide synthesis and subsequent capping with azido glycine afforded resin 17 (Scheme 3). Mild acidic cleavage, followed by reaction with TMS–diazomethane resulted in azido containing peptide methyl ester 19, which was converted to the corresponding hydrazide (20). An azide coupling with the LeuEK warhead resulted in the β1 selective two-step activity-based proteasome probe 21.9 Reaction with BODIPY dye 22 by a copper catalysed Huisgen [2 + 3] cycloaddition then afforded the fluorescently labeled probe 23 (Scheme 3).
Scheme 3.

Synthesis of the fluorescent β1 selective proteasome probe 23. Reagents and conditions: (i) 1% TFA in DCM, 30 min, 3×. (ii) TMS–diazomethane (2 equiv.), 15 min, 90% from Fmoc-Nle-resin. (iii) Hydrazine monohydrate (60 equiv.), MeOH, reflux, 1.5 h, quant. (iv) (a) HCl (2.8 equiv.), tBuONO (1.1 equiv.), DMF–EtOAc (1: 1, v/v), −25 °C, 4 h. (b) TFA·H-Leu-epoxyketone (1.1 equiv.), DiPEA (3.8 equiv.), −25 °C to RT, 15 h, 23%. (v) 22, CuSO4 (10 mol%), sodium ascorbate (15 mol%), Tol.–H2 O–tBuOH (1: 1: 1, v/v/v), 80 °C, 22 h, 65%.
Having synthesized the fluorescent derivatives of the subunit selective inhibitors, their proteasome labeling profile and cell-permeability were determined (Fig. 3). Fluorescently tagged epoxomicin analogues 2418 and 2519 (Fig. 3D), which label all proteolytically active proteasome subunits, were used as a reference. Probe 23, designed to specifically target the β1 active site, indeed shows a marked preference for β1 in HEK293T cell lysates (Fig. 3A, left panel). At higher concentrations it starts to label β5, presumably because of the increase in steric bulk and hydrophobicity due to BODIPY addition. To atest the cell permeability, living HEK293T cells were exposed to increasing concentrations of the probe (Fig. 3B, left panel). The fact that a similar labeling profile is observed shows that the introduction of the BODIPY dye has no dramatic effect on the cell permeability of the probe. Plotting of the fluorescent band intensity gave an apparent EC50 value of 62 ± 5 nM for β1 labeling (Fig. 3C, left panel). When using 1 μM probe, one can selectively saturate the labeling of roughly all β1 sites, leaving most of the β5 sites untouched.
Fig. 3.

Labeling profile of probe 14, 16 and 23. (A) HEK293T lysates (10 μg total protein) were incubated with the indicated concentrations of 23, 24 (left panel), 14 (middle panel) or 16 (right panel) for 1 h at 37 °C. (B) HEK293T cells (some 5 × 105 cells) were exposed to the indicated concentrations of 23 or 24 (left panel), 14 or 25 (middle panel) or 16 (right panel) for 2 h at 37 °C. (C) Quantification of fluorescent labeling of living cells treated with 23 (left panel), 14 (middle panel) and 16 (right panel). Corresponding gels are shown in Fig. 3B. (D) Structures of reference probes 24 and 25.
HEK293T cell lysates incubated with azido-BODIPY methyl vinyl sulfone probe 14 showed specific β5 labeling (Fig. 3A, middle panel), with only minimal labeling of what is presumably β1 at high concentrations. A similar labeling profile is observed when living HEK293T cells are exposed to the fluorescent probe (Fig. 3B, middle panel). Quantification of the fluorescent labeling revealed an apparent EC50 value of 163 ± 5 nM (Fig. 3C, middle panel). Probe 16, designed to target β5 and, at higher concentrations, the β2 site, indeed labeled both sites in lysates (Fig. 3A, right panel). In living cells β5 labeling appears to reach a maximum around 1 μM, after which the labeling intensity starts to decrease, whereas the labeling of the β2 subunit keeps increasing (Fig. 3B and C, right panel). The reason for this phenomenon remains to be investigated.
Poor resolution of β5 and β1 subunits is often a problem in the analysis of subunit-specific inhibition and labeling on SDS-PAGE. Using the probes labeled with different fluorophores developed in this work should eliminate this problem. In order to demonstrate that this is indeed the case, we have treated extracts simultaneously with the β1-specific probe 23 and either of β5-specific probes 14 and 16 (Fig. 4). Both subunits were detected and clearly resolved with 2-color imaging.
Fig. 4.
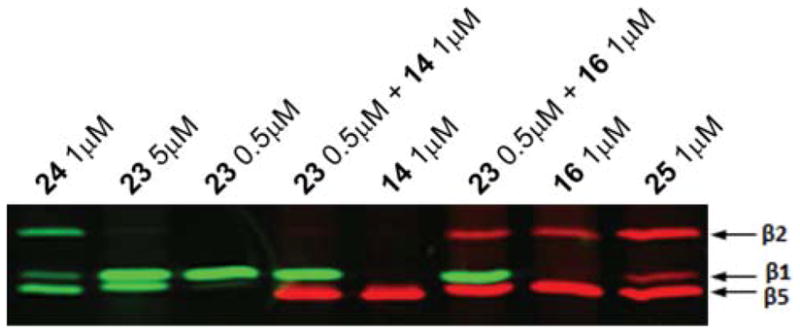
Labeling and, hence, chemical knock-down of specific proteasome active sites. HEK293T lysates (10 μg total protein) were incubated with the indicated concentrations of probes for 1 h at 37 °C. Merged image of fluorescence scanned at λex 488, λem 520 nm in green and λex 532, λem 560 nm in red.
Conclusions
Building on our expertise in developing site-specific proteasome inhibitors9,16 we describe here the development of fluorescent subunit-specific activity-based proteasome probes 14, 16 and 23. We have also made the intriguing observation that replacing the epoxyketone warhead in compound 2 for a vinyl sulfone increases the specificity of the β5-targeting inhibitors, with the methyl vinyl sulfone inhibitor IIb being the most β5 specific. An advantage of the fluorescent selective proteasome probes is the better resolution of the β1 and β5 subunits on the SDS-PAGE gels using 2 color imaging. The ability to chemically knock-down specific active sites and being able to validate and monitor this using the fluorescent readout will facilitate the elucidation of the roles of the individual proteasome active sites as drug targets in cancer and in other biological processes such as MHC class I antigen presentation.
Experimental
General
All reagents were commercial grade and were used as received unless indicated otherwise. Toluene (Tol.) (purum), ethyl acetate (EtOAc) (puriss.), diethyl ether (Et2O) and light petroleum ether (PetEt) (puriss.) were obtained from Riedel-de Haën. Dichloroethane (DCE), dichloromethane (DCM), dimethyl form-amide (DMF) and dioxane (Biosolve) were stored on 4 Å molecular sieves. Methanol (MeOH) and N-methylpyrrolidone (NMP) were obtained from Biosolve. Reactions were monitored by TLC-analysis using DC-alufolien (Merck, Kieselgel60, F254) with detection by UV-absorption (254 nm), spraying with 20% H2SO4 in ethanol followed by charring at ~150 °C, by spraying with a solution of (NH4)6Mo7O24·4H2O (25 g L−1) and (NH4)4Ce(SO4)4·2H2O (10 g L−1) in 10% sulfuric acid followed by charring at ~150 °C, or spraying with an aqueous solution of KMnO4 (20%) and K2CO3 (10%). Column chromatography was performed on Screening Di-vices Silica gel (0.040–0.063 nm). LC/MS analysis was performed on a LCQ Adventage Max (Thermo Finnigan) equipped with a Gemini C18 column (Phenomenex). The applied buffers were A: H2O, B: MeCN and C: 1.0% aq. TFA. HRMS were recorded on a LTQ Orbitrap (Thermo Finnigan). 1H- and 13C-APT-NMR spectra were recorded on a Jeol JNM-FX-200 (200/50 MHHz), Bruker DPX-300 (300/75 MHz), Bruker AV-400 (400/100 MHz) equipped with a pulsed field gradient accessory, a Bruker AV-500 (500/125 MHz) or a Bruker DMX-600 (600/150 MHz) with cryoprobe. Chemical shifts are given in ppm (δ) relative to tetramethylsilane as internal standard. Coupling constants are given in Hz. All presented 13C-APT spectra are proton decoupled. Optical rotations were measured on a Propol automatic polarimeter (sodium D line, λ = 589 nm). Boc-LeuVS-PhOH,12 Boc-LeuVS,13 Boc-LeuEK14 and were synthesized as described in literature.
Ac-Ala-Pro-Nle-OMe (5)
4-Methylbenzhydrylamine (MBHA)-functionalized polystyrene resin (7.14 g, 0.7 mmol g−1, 5 mmol) was washed with NMP (3×) followed by addition of a preactivated mixture of 4-(4-hydroxymethyl-3-methoxyphenoxy)-butyric acid (HMPB) linker (3.6 g, 15.0 mmol, 3 equiv.), BOP (6.6 g, 15 mmol, 3 equiv.) and DiPEA (5 mL, 30 mmol, 6 equiv.) in NMP. After 2 h of shaking, the resin was washed with NMP (3×) and DCM (3×), dried and used as such. Part of the resin (2 mmol) was transferred to a flask, coevaporated with DCE (2×), and condensed with Fmoc-Nle-OH (2.12 g, 6 mmol, 3 equiv.) under the influence of DIC (1.0 mL, 6.6 mmol, 3.3 equiv.) and DMAP (6.6 mg, 0.3 mmol, 5 mol%) in DCM for 2 h. The resin was filtered and washed with DCM (2×), followed by a second condensation cycle. The loading of the resin was determined to be 0.46 mmol g−1 (4.28 g, 1.97 mmol, 98%) by spectrophotometric analysis. The obtained resin was submitted to two cycles of Fmoc solid-phase synthesis with Fmoc-Pro-OH and Fmoc-Ala-OH, respectively, as follows: (a) deprotection with piperidine–NMP (1: 4, v/v, 20 min.), (b) wash with NMP (3×), (c) coupling of Fmoc amino acid (5 mmol, 2.5 equiv.) in the presence of BOP (2.2 g, 5 mmol, 2.5 equiv.) and DiPEA (0.99 ml, 6 mmol, 3 equiv.) in NMP and shaken for at least 2 h, (d) wash with NMP (3×) and DCM (3×), yielding resin-bound Fmoc-Ala-Pro-Nle. Couplings were monitored for completion by the Kaiser test. After Fmoc deprotection of 1.2 mmol, the resin-bound tripeptide was capped with acetic anhydride (0.57 ml, 6 mmol, 5 equiv.) and DiPEA (1.98 ml, 12 mmol, 10 equiv.) for 15 min. Mild acidic cleavage of resin 3 with 1% TFA in DCM (3 × 10 min.) resulted in Ac-Ala-Pro-Nle-OH 4 which was used without purification. The crude peptide 4 was dissolved in MeOH–Tol. (1: 1) and treated with TMS–diazomethane (1.2 ml 2 M in hexanes, 2.4 mmol, 2 equiv.) for 15 min before being coevaporated with Tol. (3×). Purification by flash column chromatography (DCM → 3% MeOH in DCM) yielded the title compound as a white solid (0.36 g, 1.0 mmol, 85%). 1H NMR (300 MHz, CDCl3): δ ppm 7.42 (d, J = 7.8 Hz, 1H), 7.14 (d, J = 7.5 Hz, 1H), 4.78 (m, 1H), 4.64 (m, 1H), 4.50 (m, 1H), 3.78 (m, 1H), 3.74 (s, 3H), 3.59 (m, 1H), 2.29 (m, 1H), 2.12 (m, 1H), 2.06–1.93 (m, 6H), 1.80 (m, 1H), 1.66 (m, 1H), 1.36 (d, J = 6.9 Hz, 3H), 1.28 (m, 4H), 0.87 (t, J = 6.8 Hz, 3H).
Ac-Ala-Pro-Nle-hydrazide (6)
Ac-Ala-Pro-Nle-OMe (5, 0.36 g, 1.0 mmol) was dissolved in MeOH. Hydrazine monohydrate (2.9 ml, 60 mmol, 60 equiv.) was added and the reaction mixture was refluxed for 1.5 h. Tol. was added and the resulting white solid was filtered to give the title compound (0.33 g, 0.92 mmol, 92%). 1H NMR (400 MHz, CDCl3): δ ppm 4.60 (q, J = 7.0 Hz, 1H), 4.46 (dd, J1 = 8.2, J2 = 4.6 Hz, 1H), 4.25 (dd, J1 = 8.4, J2 = 6.0 Hz, 1H), 3.85–3.77 (m, 1H), 3.69–3.60 (m, 1H), 2.26–2.13 (m, 1H), 2.11–2.01 (m, 1H), 2.00–1.91 (m, 5H), 1.82–1.71 (m, 1H), 1.71–1.60 (m, 1H), 1.42–1.24 (m, 5H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ ppm 174.12, 173.77, 173.45, 172.80, 61.49, 53.48, 48.50, 48.46, 32.88, 30.31, 28.97, 26.00, 23.36, 22.26, 16.88, 14.28.
Boc-Tyr(Me)-Phe-OMe (8)
HCl·H-Phe-OMe (2.16 g, 10 mmol) and Boc-Tyr(Me)-OH (7, 2.95 g, 10 mmol, 1 equiv.) were dissolved in DCM. BOP (4.42 g, 10 mmol, 1 equiv.) and DiPEA (3.64 ml, 16.5 mmol, 2.2 equiv.) were added and the reaction mixture was stirred for 15 h before being washed with 0.5 M HCl (aq.) and sat. aq. NaHCO3, separated and dried over MgSO4. Purification by flash column chromatography (20% EtOAc in PetEt → 40% EtOAc in PetEt) yielded the title compound as a white solid (4.6 g, 10 mmol, quant.). 1H NMR (200 MHz, CDCl3): δ ppm 7.32–7.17 (m, 4H), 7.15–7.05 (m, 3H), 7.04–6.94 (m, 2H), 6.81 (d, J = 8.7 Hz, 2H), 6.38 (d, J = 7.8 Hz, 1H), 4.78 (q, J = 5.9, 5.9, 5.9 Hz, 1H), 4.38–4.19 (m, 1H), 3.78 (s, 3H), 3.67 (s, 3H), 3.05 (dd, J = 5.9, 1.6 Hz, 2H), 2.96 (d, J = 6.7 Hz, 2H), 1.40 (s, 9H). 13C NMR (50 MHz, CDCl3): δ ppm 171.40, 158.15, 155.30, 135.62, 129.98, 128.86, 128.31, 128.10, 126.64, 113.54, 78.93, 55.41, 54.77, 53.13, 51.80, 37.46, 37.15, 27.78.
(Tyr(Me)-Phe-OMe)-2-(naphthalen-2-yl)-acetamide (9)
Boc-Tyr(Me)-Phe-OMe (8, 4.6 g, 10 mmol) was dissolved in TFA–DCM 1: 1 (v/v). The reaction mixture was stirred for 15 min before being co-evaporated with Tol. (3×). The crude TFA salt was dissolved in DCM and 2-(naphthalen-2-yl)-acetic acid (1.86 g, 10 mmol, 1 equiv.), BOP (4.42 g, 10 mmol, 1 equiv.) and DiPEA (5.46 ml, 33 mmol, 3.3 equiv.) were added. After being stirred for 15 h the reaction mixture was washed with 0.5 M HCl (aq.) and sat. aq. NaHCO3, separated and dried over MgSO4. Purification by flash column chromatography (PetEt → EtOAc, followed by a second column: DCM → 30% EtOAc in DCM), washing with H2O (3×) and drying over MgSO4 gave the pure title compound as a white solid (3.55 g, 6.8 mmol, 68%). 1H NMR (200 MHz, CDCl3): δ ppm 8.53 (d, J = 7.5 Hz, 1H), 8.27 (d, J = 8.7 Hz, 1H), 7.91–7.70 (m, 3H), 7.59 (s, 1H), 7.54–7.40 (m, 2H), 7.32–7.16 (m, 6H), 7.11 (d, J = 8.7 Hz, 2H), 6.71 (d, J = 8.7 Hz, 2H), 4.62–4.41 (m, 2H), 3.65 (s, 3H), 3.62 (d, J = 13.7 Hz, 1H), 3.58 (s, 3H), 3.49 (d, J = 14.0 Hz, 1H), 3.13–2.83 (m, 3H), 2.67 (dd, J1 = 13.7, J2 = 9.9 Hz, 1H).
(Tyr(Me)-Phe-hydrazinyl)-2-(naphthalen-2-yl)-acetamide (10)
To a solution of (Tyr(Me)-Phe-OMe)-2-(naphthalen-2-yl)-acetamide (9, 0.52 g, 1 mmol) in MeOH was added hydrazine monohydrate (2.91 ml, 60 mmol, 60 equiv.). The reaction mixture was refluxed for 2.5 h. The title compound precipitated as a white solid and was filtered off and washed with MeOH (0.5 g, 0.95 mmol, 95%). 1H NMR (600 MHz, CDCl3): δ ppm 9.22 (s, 1H), 8.25 (t, J = 7.9, 7.9 Hz, 2H), 7.86 (d, J = 7.8 Hz, 1H), 7.79 (d, J = 7.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.62 (s, 1H), 7.50–7.44 (m, 2H), 7.29–7.21 (m, 5H), 7.20–7.15 (m, 1H), 7.10 (d, J = 8.6 Hz, 2H), 6.71 (d, J = 8.6 Hz, 2H), 4.59–4.51 (m, 2H), 4.29 (s, 2H), 3.65 (s, 3H), 3.64 (d, J = 12.5 Hz, 1H), 3.53 (d, J = 14.0 Hz, 1H), 2.99 (dd, J1 = 13.7, J2 = 5.6 Hz, 1H), 2.94 (dd, J1 = 13.7, J2 = 3.9 Hz, 1H), 2.86 (dd, J1 = 13.7, J2 = 8.7 Hz, 1H), 2.70 (dd, J1 = 13.6, J2 = 10.1 Hz, 1H). 13C NMR (50 MHz, CDCl3): δ ppm 170.97, 170.00, 169.77, 157.66, 137.51, 133.91, 132.91, 131.71, 130.22, 129.50, 129.16, 128.07, 127.65, 127.41, 127.35, 127.21, 126.28, 125.93, 125.43, 113.30, 54.76, 53.98, 52.67, 42.34, 38.05, 36.70.
Synthesis of Ia–c and IIa–c; general procedure azide coupling
The peptide hydrazide was dissolved in DMF–EtOAc (1: 1, v/v) and cooled to −25 °C, before HCl (2.8 equiv., 4 M in 1,4-dioxane) and tBuONO (1.1 equiv.) were added. The reaction mixture was stirred for 4 h at −25 °C to form the corresponding acyl azide. Boc-protected Leucine derived warhead Boc-LeuVS-PhOH,12 Boc-LeuVS13 or Boc-LeuEK14 was dissolved in DCM–TFA (1: 1, v/v) and stirred for 30 min, before being coevaporated with Tol. (3×). The resulting warhead TFA-salt was dissolved in DMF and DiPEA (3.8 equiv.) was added, before the mixture was combined with the acyl azide mixture at −25 °C (NOTE: make sure the pH is 8–9. If not, add more DiPEA). The reaction mixture was allowed to warm up to room temperature overnight. EtOAc and water were added and the organic layer was separated, dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography.
Ac-Ala-Pro-Nle-Leu-4-hydroxyphenyl-vinylsulfone (Ia)
Following the general procedure for azide coupling, the title compound was obtained from Boc-LeuVS-PhOH (61 mg, 0.17 mmol, 1.1 equiv.) and Ac-Ala-Pro-Nle-hydrazide (6, 53.3 mg, 0.15 mmol). Purification by flash column chromatography (DCM → 6% MeOH in DCM) gave Ia as colorless oil (68.4 mg, 0.12 mmol, 77%). 1H NMR (500 MHz, DMSO, T = 353 K): δ ppm 7.68 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 6.79 (dd, J1 = 15.0, J2 = 5.1 Hz, 1H), 6.54 (dd, J1 = 15.1, J2 = 1.4 Hz, 1H), 4.69–4.59 (m, 1H), 4.56 (q, J = 7.0 Hz, 1H), 4.38 (dd, J1 = 8.2, J2 = 5.1 Hz, 1H), 4.17 (dd, J1 = 8.2, J2 = 6.0 Hz, 1H), 3.84–3.73 (m, 1H), 3.68–3.56 (m, 1H), 2.26–2.09 (m, 1H), 2.07–1.84 (m, 6H), 1.81–1.59 (m, 3H), 1.58–1.49 (m, 1H), 1.47–1.38 (m, 1H), 1.36–1.24 (m, 7H), 0.97–0.83 (m, 9H). HRMS: calcd. for [C29H44N4O7SH]+ 593.30035, found 593.30046.
Ac-Ala-Pro-NLe-Leu-methyl-vinylsulfone (Ib)
Following the general procedure for azide coupling, the title compound was obtained from Boc-LeuVS (50.7 mg, 0.17 mmol, 1.1 equiv.) and Ac-Ala-Pro-NLe-hydrazide (6, 53.3 mg, 0.15 mmol). Purification by flash column chromatography (DCM → 4% MeOH in DCM) gave Ib as white solid (26.1 mg, 51 μmol, 34%). 1H NMR (500 MHz, DMSO, T = 353 K): δ ppm 7.84 (s, 1H), 7.63–7.53 (m, 2H), 6.74–6.58 (m, 2H), 4.63–4.48 (m, 2H), 4.39–4.31 (m, 1H), 4.16 (dd, J1 = 13.5, J2 = 7.7 Hz, 1H), 3.69–3.61 (m, 1H), 3.60–3.52 (m, 1H), 2.95 (s, 3H), 2.10–2.00 (m, 1H), 1.98–1.86 (m, 3H), 1.84 (s, 3H), 1.77–1.67 (m, 1H), 1.67–1.55 (m, 2H), 1.54–1.46 (m, 1H), 1.45–1.35 (m, 1H), 1.33–1.24 (m, 4H), 1.22 (d, J = 6.8 Hz, 3H), 0.93–0.84 (m, 9H). HRMS: calcd. for [C24H42N4O6SH]+ 515.28978, found 515.28961.
Ac-Ala-Pro-NLe-Leu-epoxyketone (Ic)
Following the general procedure for azide coupling, the title compound was obtained from Boc-LeuEK (47.4 mg, 0.17 mmol, 1.1 equiv.) and Ac-Ala-Pro-NLe-hydrazide (6, 53.3 mg, 0.15 mmol). Purification by flash column chromatography (DCM → 4% MeOH in DCM) gave Ib as colorless oil (47.1 mg, 95 μmol, 63%). 1H NMR (500 MHz, DMSO, T = 353 K): δ ppm 7.80 (s, 1H), 7.73 (d, J = 7.3 Hz, 1H), 7.50 (d, J = 5.6 Hz, 1H), 4.60–4.48 (m, 1H), 4.47–4.41 (m, 1H), 4.41–4.36 (m, 1H), 4.22 (dd, J1 = 13.6, J2 = 7.9 Hz, 1H), 3.72–3.59 (m, 1H), 3.57–3.47 (m, 1H), 3.18 (d, J = 5.2 Hz, 1H), 2.96 (d, J = 5.2 Hz, 1H), 2.08–1.95 (m, 1H), 1.95–1.86 (m, 2H), 1.83 (s, 3H), 1.73–1.61 (m, 2H), 1.56–1.46 (m, 1H), 1.42 (s, 3H), 1.41–1.31 (m, 2H), 1.30–1.23 (m, 4H), 1.20 (d, J = 6.7 Hz, 3H), 0.91 (d, J = 6.64 Hz, 1H), 0.88–0.83 (m, 6H). HRMS: calcd. for [C25H42N4O6H]+ 495.31771, found 495.31755.
(Tyr(Me)-Phe-Leu-4-hydroxyphenyl-vinylsulfone)-2-(naphthalen-2-yl)-acetamide (IIa)
Following the general procedure for azide coupling, the title compound was obtained from Boc-LeuVS-PhOH (61 mg, 0.17 mmol, 1.1 equiv.) and (Tyr(Me)-Phe-hydrazinyl)-2-(naphthalen-2-yl)-acetamide (10, 78.7 mg, 0.15 mmol). Crystallization from EtOAc with PetEt gave IIa as a white solid (88.1 mg, 0.12 mmol, 77%). 1H NMR (400 MHz, DMSO): δ ppm 10.64 (s, 1H), 8.29 (d, J = 8.0 Hz, 1H), 8.25 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 7.0 Hz, 1H), 7.80–7.72 (m, 2H), 7.62 (d, J = 8.7 Hz, 2H), 7.58 (s, 1H), 7.50–7.42 (m, 2H), 7.25–7.20 (m, 1H), 7.18–7.13 (m, 5H), 7.07 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.7 Hz, 2H), 6.71–6.61 (m, 3H), 6.22 (dd, J1 = 15.1, J2 = 1.3 Hz, 1H), 4.58–4.42 (m, 3H), 3.64 (s, 3H), 3.59 (d, J = 13.9 Hz, 1H), 3.48 (d, J = 13.9 Hz, 1H), 2.98–2.75 (m, 3H), 2.64 (dd, J1 = 13.7, J2 = 10.0 Hz, 1H), 1.62–1.50 (m, 1H), 1.39–1.25 (m, 2H), 0.81 (dd, J1 = 12.6, J2 = 6.6 Hz, 6H). 13C NMR (100 MHz, DMSO): δ ppm 170.99, 170.17, 169.71, 161.95, 157.58, 145.37, 137.14, 133.85, 132.81, 131.61, 130.10, 129.96, 129.85, 129.61, 129.39, 128.96, 127.99, 127.55, 127.32, 127.31, 127.24, 127.10, 126.35, 125.85, 125.35, 115.86, 113.21, 54.72, 54.02, 53.82, 47.03, 42.17, 41.96, 37.51, 36.59, 23.99, 22.88, 21.31. HRMS: calcd. for [C44H47N3O7SH]+ 762.32075, found 762.32139.
(Tyr(Me)-Phe-Leu-methyl-vinylsulfone)-2-(naphthalen-2-yl)-acetamide (IIb)
Following the general procedure for azide coupling the title compound was obtained from Boc-LeuVS (32 mg, 0.11 mmol, 1.1 equiv.) and (Tyr(Me)-Phe-hydrazinyl)-2-(naphthalen-2-yl)-acetamide (10, 52 mg, 0.1 mmol). Column chromatography (DCM → 1.5% MeOH in DCM) gave the title compound as a white solid (36.2 mg, 53 μmol, 53%). 1H NMR (400 MHz, CDCl3): δ ppm 8.30 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.85 (d, J = 7.3 Hz, 1H), 7.81–7.73 (m, 2H), 7.60 (s, 1H), 7.52–7.42 (m, 2H), 7.31–7.15 (m, 6H), 7.07 (d, J = 8.1 Hz, 2H), 6.68 (d, J = 8.3 Hz, 2H), 6.60 (dd, J1 = 15.2, J2 = 4.9 Hz, 1H), 6.32 (d, J = 15.3 Hz, 1H), 4.61–4.43 (m, 3H), 3.65 (s, 3H), 3.61 (d, J = 13.9 Hz, 1H), 3.49 (d, J = 14.0 Hz, 1H), 3.01 (dd, J1 = 13.5, J2 = 6.6 Hz, 1H), 2.95 (s, 3H), 2.91–2.82 (m, 2H), 2.66 (dd, J1 = 14.6, J2 = 11.1 Hz, 1H), 1.68–1.55 (m, 1H), 1.47–1.29 (m, 2H), 0.85 (dd, J1 = 15.5, J2 = 6.5 Hz, 6H). HRMS: calcd. for [C39H45N3O6SH]+ 684.31018, found 684.31060.
(Tyr(Me)-Phe-Leu-epoxyketone)-2-(naphthalen-2-yl)-acetamide (IIc)
Following the general procedure for azide coupling the title compound was obtained from Boc-LeuEK (76 mg, 0.28 mmol, 1.1 equiv.) and (Tyr(Me)-Phe-hydrazinyl)-2-(naphthalen-2-yl)-acetamide (10, 0.13 g, 0.25 mmol). Column chromatography (DCM → 2% MeOH in DCM) gave the title compound as a white solid (0.11 g, 0.17 mmol, 66%). 1H NMR (400 MHz, CDCl3): δ ppm 7.86–7.75 (m, 1H), 7.73–7.67 (m, 2H), 7.52 (s, 1H), 7.49–7.44 (m, 2H), 7.20–7.09 (m, 3H), 7.01–6.91 (m, 3H), 6.68 (d, J = 8.6 Hz, 2H), 6.57–6.47 (m, 1H), 6.40 (d, J = 8.3 Hz, 2H), 4.79–4.66 (m, 2H), 4.58 (dt, J1 = 19.8, J2 = 3.0 Hz, 1H), 3.65–3.45 (m, 5H), 3.24 (d, J = 4.9 Hz, 1H), 3.01–2.85 (m, 1H), 2.84–2.77 (m, 4H), 1.59–1.51 (m, 1H), 1.49 (s, 3H), 1.47–1.37 (m, 1H), 1.33–1.19 (m, 1H), 0.88 (dd, J1 = 13.1, J2 = 6.4 Hz, 6H).13C NMR (100 MHz, CDCl3): δ ppm 207.99, 171.32, 170.57, 170.47, 158.50, 136.27, 133.56, 132.57, 131.70, 130.02, 129.36, 128.92, 128.87, 128.61, 128.56, 128.29, 128.25, 127.77, 127.72, 127.56, 127.12, 127.02, 126.98, 126.52, 126.19, 113.97, 79.36, 59.00, 55.11, 54.43, 54.01, 52.33, 50.11, 43.60, 39.94, 37.78, 36.31, 25.15, 23.30, 21.40, 16.71. HRMS: calcd. for [C40H45N3O6H]+ 664.33811, found 664.33838.
Boc-Tyr(Me)-Phe-hydrazide (11)
Boc-Tyr(Me)-Phe-methyl ester (8, 1.17 g, 2.56 mmol) was dissolved in MeOH and hydrazine monohydrate (7.47 ml, 154 mmol, 60 equiv.) was added. The reaction mixture was refluxed for 2 h, before being concentrated until white precipitate is formed. The resulting suspension was cooled and the product was filtered off (1.03 g, 2.25 mmol, 88%). 1H NMR (400 MHz, MeOD–CDCl3): δ ppm 7.26–7.08 (m, 5H), 7.03 (d, J = 8.6 Hz, 2H), 6.79 (d, J = 8.6 Hz, 2H), 4.55–4.50 (m, 1H), 4.18 (t, J = 6.6 Hz, 1H), 3.01 (dd, J1 = 13.7, J2 = 7.0 Hz, 1H), 2.96–2.85 (m, 2H), 2.73 (dd, J1 = 13.5, J2 = 8.1 Hz, 1H), 1.34 (s, 9H). NMR (100 MHz, MeOD–CDCl3): δ ppm 171.77, 170.37, 158.08, 135.87, 129.70, 128.62, 128.08, 127.96, 126.37, 113.40, 79.72, 55.67, 54.53, 52.52, 37.25, 36.74, 27.46.
Boc-Tyr(Me)-Phe-Leu-methyl-vinylsulfone (12)
Following the general procedure for azide coupling the title compound was obtained from Boc-LeuVS (0.24 g, 0.83 mmol, 1.1 equiv.) and Boc-Tyr(Me)-Phe-hydrazide (11, 0.39 g, 0.75 mmol). Purification by flash column chromatography (DCM → 1.5% MeOH in DCM) gave 12 (0.35 g, 0.56 mmol, 75%). 1H NMR (400 MHz, CDCl3): δ ppm 7.29–7.22 (m, 3H), 7.09 (d, J = 8.5 Hz, 2H), 7.03 (d, J = 6.1 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 6.82–6.65 (m, 2H), 6.47–6.37 (m, 2H), 4.84 (d, J = 3.0 Hz, 1H), 4.77–4.69 (m, 1H), 4.66 (dd, J1 = 13.0, J2 = 6.3 Hz, 1H), 4.16 (td, J1 = 7.8, J2 = 4.8 Hz, 1H), 3.80 (s, 3H), 3.32–3.23 (m, 1H), 3.03 (dd, J1 = 14.3, J2 = 5.0 Hz, 1H), 2.91 (s, 3H), 2.91–2.83 (m, 2H), 1.53–1.41 (m, 1H), 1.37–1.30 (m, 2H), 1.27 (s, 9H), 0.90 (d, J = 6.4 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H).
Azido-BODIPY-Tyr(Me)-Phe-Leu-methyl-vinylsulfone (14)
Boc-Tyr(Me)-Phe-LeuVS (12, 22 mg, 35 μmol) was dissolved in TFA–DCM 1: 1 (v/v). The reaction mixture was stirred for 30 min, before being co-evaporated with Tol. (3×). The crude TFA salt was dissolved in DCM and N3-BODIPY-OSu17 (13, 20 mg, 35 μmol, 1 equiv.) and DiPEA (6 μl, 35 μmol, 1 equiv.) were added. The reaction mixture was stirred for 5 h, before being concentrated. Purification by flash column chromatography (DCM → 1.5% MeOH in DCM) afforded the title compound as a purple solid (18.2 mg, 18.9 μmol, 54%). 1H NMR (400 MHz, CDCl3): δ ppm 7.87 (d, J = 8.8 Hz, 2H), 7.29–7.20 (m, 3H), 7.07 (s, 1H), 7.04 (dd, J1 = 7.4, J2 = 1.5 Hz, 2H), 7.00–6.93 (m, 5H), 6.79 (d, J = 8.5 Hz, 2H), 6.71 (dd, J1 = 15.1, J2 = 4.6 Hz, 1H), 6.54 (d, J = 4.1 Hz, 1H), 6.52–6.44 (m, 2H), 6.21 (dd, J1 = 15.1, J2 = 1.1 Hz, 1H), 6.10–6.04 (m, 1H), 4.72–4.57 (m, 2H), 4.45 (q, J = 6.4 Hz, 1H), 4.10 (t, J = 5.9 Hz, 2H), 3.70 (s, 3H), 3.53 (t, J = 6.6 Hz, 3H), 3.19 (dd, J1 = 13.8, J2 = 5.4 Hz, 1H), 2.95–2.89 (m, 3H), 2.89 (s, 3H), 2.65–2.50 (m, 2H), 2.48 (s, 3H), 2.19–2.13 (m, 3H), 2.13 (s, 3H), 2.11–2.03 (m, 2H), 1.52–1.38 (m, 1H), 1.38–1.26 (m, 2H), 0.87 (t, J = 6.7 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ ppm 172.52, 170.81, 169.95, 159.55, 158.86, 158.32, 156.04, 147.21, 139.32, 135.87, 135.13, 134.04, 134.02, 130.77, 130.73, 130.69, 130.08, 129.60, 129.25, 129.21, 128.85, 128.80, 128.39, 127.41, 127.31, 125.53, 123.01, 118.64, 118.60, 118.58, 114.29, 114.20, 64.45, 55.19, 54.25, 48.20, 47.91, 42.74, 42.42, 37.17, 36.37, 35.82, 28.74, 24.61, 22.80, 21.74, 19.59, 13.09, 9.57. HRMS: calcd. for [C50H59BF2N8O7SH]+ 965.43613, found 965.43837.
Boc-Tyr(Me)-Phe-Leu-epoxyketone (15)
Following the general procedure for azide coupling the title compound was obtained from Boc-LeuEK (0.22 g, 0.83 mmol, 1.1 equiv.) and Boc-Tyr(Me)-Phe-hydrazide (11, 0.39 g, 0.75 mmol). Purification by flash column chromatography (DCM → 1.5% MeOH in DCM) gave 15 (0.32 g, 0.54 mmol, 71%). 1H NMR (400 MHz, CDCl3): δ ppm 7.26–7.11 (m, 3H), 7.10–7.04 (m, 4H), 7.04–7.01 (m, 1H), 6.90 (s, 1H), 6.78 (d, J = 8.5 Hz, 2H), 5.31 (d, J = 7.7 Hz, 1H), 4.77 (q, J = 6.9 Hz, 1H), 4.56 (dt, J1 = 9.8, J2 = 3.2 Hz, 1H), 4.41 (s, 1H), 3.73 (s, 3H), 3.25 (d, J = 4.4 Hz, 1H), 3.02 (dd, J1 = 14.0, J2 = 6.8 Hz, 1H), 2.98–2.88 (m, 3H), 2.84 (d, J = 4.9 Hz, 1H), 1.58–1.51 (m, 1H), 1.49 (s, 3H), 1.48–1.38 (m, 1H), 1.36 (s, 9H), 1.27–1.18 (m, 1H), 0.88 (dd, J1 = 12.3, J2 = 6.4 Hz, 6H).
Azido-BODIPY-Tyr(Me)-Phe-Leu-epoxyketone (16)
Boc-Tyr(Me)-Phe-LeuEK (15, 21 mg, 35 μmol) was dissolved in TFA–DCM 1: 1 (v/v). The reaction mixture was stirred for 30 min, before being co-evaporated with Tol. (3×). The crude TFA salt was dissolved in DCM and N3-BODIPY-OSu17 (13, 20 mg, 35 μmol, 1 equiv.) and DiPEA (6 μl, 35 μmol, 1 equiv.) were added. The reaction mixture was stirred for 15 h, before being concentrated. Purification by flash column chromatography (DCM → 1% MeOH in DCM) afforded the title compound as a purple solid (8.8 mg, 9.3 μmol, 27%). 1H NMR (400 MHz, CDCl3): δ ppm 7.87 (d, J = 8.6 Hz, 2H), 7.27–7.19 (m, 3H), 7.11–7.05 (m, 3H), 7.00–6.92 (m, 5H), 6.76 (d, J = 8.2 Hz, 2H), 6.54 (d, J = 3.9 Hz, 1H), 6.32 (d, J = 7.6 Hz, 1H), 6.03 (d, J = 7.6 Hz, 1H), 5.88 (d, J = 7.0 Hz, 1H), 4.58–4.47 (m, 3H), 4.10 (t, J = 5.8 Hz, 2H), 3.73–3.70 (m, 3H), 3.53 (t, J = 6.6 Hz, 2H), 3.23 (d, J = 4.8 Hz, 1H), 3.08–2.89 (m, 4H), 2.88 (d, J = 4.8 Hz, 1H), 2.76–2.55 (m, 2H), 2.51 (s, 3H), 2.33–2.20 (m, 2H), 2.18 (s, 3H), 2.08 (td, J1 = 12.1, J2 = 6.1 Hz, 2H), 1.50 (s, 3H), 1.49–1.38 (m, 2H), 1.20–1.13 (m, 1H), 0.89 (dd, J1 = 13.7, J2 = 5.5 Hz, 6H). HRMS: calcd. for [C51H59BF2N8O7H]+ 945.46406, found 945.46639.
Az-Ala-Pro-Nle-OMe (19)
Resin-bound Fmoc-Ala-Pro-Nle (synthesis described above, 0.25 mmol) was deprotected with piperidine–NMP (1: 4, v/v, 20 min), washed with NMP (3×) and DCM (3×), and capped with azido acetic acid (63 mg, 0.63 mmol, 2.5 equiv.) under the influence of BOP (0.28 g, 0.63 mmol, 2.5 equiv.) and DiPEA (0.12 ml, 0.75 mmol, 3 equiv.) for 15 h. Mild acidic cleavage of resin 17 with 1% TFA in DCM (3 × 10 min.) and co-evaporation with Tol. (3×) resulted in the crude Az-Ala-Pro-NLe-OH 18 which was used without purification. The crude peptide was dissolved in MeOH–Tol. (1: 1) and treated with TMS–diazomethane (0.25 ml 2 M in hexanes, 0.5 mmol, 2 equiv.) for 15 min before being co-evaporated with Tol. (3×). Purification by flash column chromatography (DCM → 2.5% MeOH in DCM) yielded the title compound as a white solid (89.3 mg, 0.23 mmol, 90%). 1H NMR (400 MHz, CDCl3): δ ppm 7.25 (d, J = 7.4 Hz, 1H), 7.11 (d, J = 7.7 Hz, 1H), 4.77 (p, J = 7.0 Hz, 1H), 4.62 (dd, J1 = 8.1, J2 = 2.6 Hz, 1H), 4.50 (dt, J1 = 7.7, J2 = 5.5 Hz, 1H), 3.98 (d, J = 3.6 Hz, 2H), 3.74 (s, 3H), 3.70–3.55 (m, 2H), 2.39–2.29 (m, 1H), 2.23–2.08 (m, 1H), 2.08–1.99 (m, 1H), 1.98–1.89 (m, 1H), 1.87–1.76 (m, 1H), 1.71–1.59 (m, 1H), 1.40 (d, J = 6.9 Hz, 3H), 1.35–1.21 (m, 2H), 0.88 (t, J = 7.0 Hz, 3H).
Az-Ala-Pro-Nle-hydrazide (20)
Az-Ala-Pro-NLe-OMe (19, 89.3 mg, 0.23 mmol) was dissolved in MeOH. Hydrazine monohydrate (0.67 ml, 13.8 mmol, 60 equiv.) was added and the reaction mixture was refluxed for 2 h. Tol. was added and the resulting white solid was filtered to give the title compound (90 mg, 0.23 mmol, quant.).1H NMR (400 MHz, MeOD): δ ppm 4.65 (q, J = 7.0 Hz, 1H), 4.46 (dd, J1 = 8.2, J2 = 4.6 Hz, 1H), 4.23 (dd, J1 = 8.4, J2 = 6.0 Hz, 1H), 3.89 (s, 2H), 3.86–3.77 (m, 1H), 3.70–3.60 (m, 1H), 2.30–2.12 (m, 1H), 2.12–1.91 (m, 2H), 1.83–1.72 (m, 1H), 1.71–1.61 (m, 1H), 1.44–1.25 (m, 7H), 0.92 (t, J = 6.9 Hz, 3H).
Az-Ala-Pro-NLe-Leu-epoxyketone (21)
Following the general procedure for azide coupling the title compound was obtained from Boc-LeuEK (38.8 mg, 0.14 mmol, 1.1 equiv.) and Az-Ala-Pro-Nle-hydrazide (19, 53.2 mg, 0.13 mmol). Purification by flash column chromatography (DCM → 2% MeOH in DCM) gave 21 (15.9 mg, 30 μmol, 23%). 1H NMR (400 MHz, CDCl3): δ ppm 7.16 (d, J = 7.4 Hz, 1H), 7.08 (d, J = 7.7 Hz, 1H), 6.40 (d, J = 8.0 Hz, 1H), 4.77 (p, J = 6.9 Hz, 1H), 4.62–4.54 (m, 2H), 4.30 (dt, J1 = 7.8, J2 = 5.7 Hz, 1H), 3.98 (d, J = 5.0 Hz, 2H), 3.71–3.62 (m, 1H), 3.61–3.55 (m, 1H), 3.31 (d, J = 5.0 Hz, 1H), 2.89 (d, J = 5.0 Hz, 1H), 2.35–2.27 (m, 1H), 2.20–2.09 (m, 1H), 2.07–1.99 (m, 1H), 1.98–1.90 (m, 1H), 1.85–1.75 (m, 1H), 1.69–1.53 (m, 2H), 1.51 (s, 3H), 1.39 (d, J = 6.8 Hz, 3H), 1.36–1.20 (m, 6H), 0.94 (dd, J1 = 6.5, J2 = 2.4 Hz, 6H), 0.88 (t, J = 7.1 Hz, 3H). HRMS: calcd. for [C25H41N7O6H]+ 536.31911, found 536.31980.
Green fluorescent β1specific probe (23)
β1 selective probe 21 (7.7 mg, 0.014 mmol) was reacted with acetylene-functionalised BODIPY 2218 (4.7 mg, 0.014 mmol, 1.0 equiv.) catalysed by CuSO4 (0.05 ml 28 mM in H2O, 1.4 μmol, 10 mol%) and sodium ascorbate (0.05 ml 44 mM in H2O, 2.2 μmol, 15 mol%) in a mixture of Tol.–H2O–tBuOH (final ratio 1: 1: 1, v/v/v, 0.6 ml) at 80 °C for 22 h. The mixture was then allowed to cool to room temperature and concentrated in vacuo. Purification by column chromatography (DCM → 2% MeOH in DCM) gave the fluorescent probe 23 (8.6 mg, 9.3 μmol, 65%) as an orange solid. 1H NMR (400 MHz, DMSO): δ ppm 8.66 (d, J = 6.8 Hz, 1H), 8.09 (d, J = 7.2 Hz, 1H), 7.80–7.77 (m, 2H), 6.23 (s, 2H), 5.07 (s, 2H), 4.56–4.53 (s, 1H), 4.38–4.33 (m, 2H), 4.24–4.17 (m, 1H), 3.58–3.55 (m, 2H), 3.19–3.17 (m, 1H), 3.02–2.96 (m, 3H), 2.72 (t, J = 7.2, 7.2 Hz), 2.41 (s, 6H), 2.40 (s, 6H), 1.98–1.94 (m, 2H), 1.89–1.81 (m, 4H), 1.64–1.62 (m, 4H), 1.47–1.44 (m, 1H), 1.41 (s, 3H), 1.36–1.31 (m, 2H), 1.24–1.20 (m, 7H), 0.91 (d, J = 6.8 Hz, 6H), 0.88–0.81 (m, 3H). 13C NMR (100 MHz, DMSO): δ ppm 208.28, 171.87, 170.92, 170.39, 165.03, 153.06, 146.68, 140.90, 130.72, 123.50, 121.69, 59.18, 58.93, 51.99, 51.64, 51.22, 49.60, 46.70, 46.41, 38.33, 31.90, 30.81, 29.43, 28.78, 27.61, 27.20, 24.53, 24.40, 23.16, 21.92, 20.98, 17.07, 16.52, 15.83, 14.08, 13.92. HRMS: calcd. for [C44H64BF2N9O6H]+ 864.51134, found 864.51332.
Competition and labeling experiments in vitro
HEK293T cells were cultured on DMEM supplemented with 10% Fetal Calf Serum (FCS), 10 units ml−1 penicillin and 10 μg ml−1 streptomycin in a 7% CO2 humidified incubator at 37 °C. Cells were harvested, washed with PBS (2×) and permeated in digitonin lysis buffer (4× pellet volume, 50 mM Tris pH 7.5, 250 mM sucrose, 5 mM MgCl2, 5 mM DTT, 0.025% digitonin) for 5 min on ice and centrifuged at 16.100 rcf for 20 min at 4 °C. The supernatant containing the cytosolic fraction was collected and the protein content was determined by Bradford assay. 10 μg total protein per experiment was exposed to the inhibitors or fluorescent probes (10 × solution in DMSO) for 1 h at 37 °C prior to incubation with MV151 (1 μM) for 1 h at 37 °C in case of a competition experiment. Reaction mixtures were boiled with Laemmli’s buffer containing β-mercapto-ethanol for 3 min before being resolved on 12.5% SDS-PAGE. In-gel detection of fluorescently labeled proteins was performed in the wet gel slabs directly on the Typhoon Variable Mode Imager (Amersham Biosciences) using the Cy3/Tamra settings (λex 532, λem 560) for MV151 and the azido-BODIPY functionalized probes 14, 16 and 25 or λex 488 nm, λem 520 nm for probes 23 and 24.
Competition and labeling experiments in living cells
HEK293T cells were cultured on DMEM supplemented with 10% Fetal Calf Serum (FCS), 10 units ml−1 penicillin and 10 μg ml−1 streptomycin in a 7% CO2 humidified incubator at 37 °C. Some 5 × 105 HEK293T cells were seeded in 6 cm Petri dishes and allowed to grow O/N in 2 ml of medium. The cells were exposed to the indicated concentrations of the inhibitors or fluorescent probes (100 × solution in DMSO) for 2 h, before being washed with PBS (2×) and harvested. The cells were permeated in digitonin lysis buffer (4 × pellet volume, 50 mM Tris pH 7.5, 250 mM sucrose, 5 mM MgCl2, 5 mM DTT, 0.025% digitonin) for 5 min on ice and centrifuged at 16.100 rcf for 20 min at 4 °C. The supernatant containing the cytosolic fraction was collected and the protein content was determined by Bradford assay. In case of a competition experiment, the lysates were exposed to MV151 (1 μM) for 1 h at 37 °C. Some 10 μg protein/lane was boiled for 5 min in Laemli’s sample buffer containing beta-mercapto-ethanol and the proteins were resolved by 12.5% SDS-PAGE. Labeled proteasome subunits were visualised by in-gel fluorescence scanning on a Typhoon variable mode imager (Amersham Biosciences) using the Cy3/Tamra settings (λex 532, λem 560) for MV151 and the azido-BODIPY functionalized probes 14, 16 and 25 or λex 488 nm, λem 520 nm for probes 23 and 24.
Supplementary Material
Acknowledgments
This work was supported by The Netherlands Organization for Scientific Research (NWO), The Netherlands Genomics Centre Initiative (NGI) and the NCI (grant RO1CA124634). We thank Hans van den Elst and Nico Meeuwenoord for HPLC and LC-MS assistance.
Footnotes
Electronic supplementary information (ESI) available: 1H NMR spectra. See DOI: 10.1039/c001036g
References and notes
- 1.Griffin TA, Nandi D, Cruz M, Fehling HJ, Kaer LV, Monaco JJ, Colbert RA. J Exp Med. 1998;187:97–104. doi: 10.1084/jem.187.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murata S, Sasaki K, Kishimoto T, Niwa S, Hayashi H, Takahama Y, Tanaka K. Science. 2007;316:1349–1353. doi: 10.1126/science.1141915. [DOI] [PubMed] [Google Scholar]
- 3.(a) Arendt CS, Hochstrasser M. Proc Natl Acad Sci U S A. 1997;94:7156–7161. doi: 10.1073/pnas.94.14.7156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen P, Hochstrasser M. Cell. 1996;86:961–972. doi: 10.1016/s0092-8674(00)80171-3. [DOI] [PubMed] [Google Scholar]; (c) Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. J Biol Chem. 1997;272:25200–25209. doi: 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- 4.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL. Bioorg Med Chem Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kane RC, Bross PF, Farrell AT, Pazdur R. Oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]; (b) Bross PF, Kane R, Farrell AT, Abraham S, Benson K, Brower ME, Bradley S, Gobburu JV, Goheer A, Lee SL, Leighton J, Liang CY, Lostritto RT, McGuinn WD, Morse DE, Rahman A, Rosario LA, Verbois SL, Williams G, Wang YC, Pazdur R. Clin Cancer Res. 2004;10:3954–3964. doi: 10.1158/1078-0432.CCR-03-0781. [DOI] [PubMed] [Google Scholar]; (c) Adams J. Nat Rev Cancer. 2004;4:349–360. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]; (d) Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 6.Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, Pazdur R. Clin Cancer Res. 2007;13:5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 7.Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H, Galardy PJ. Nat Methods. 2005;2:357–362. doi: 10.1038/nmeth759. [DOI] [PubMed] [Google Scholar]
- 8.(a) Altun M, Galardy PJ, Shringarpure R, Hideshima T, LeBlanc R, Anderson KC, Ploegh HL, Kessler BM. Cancer Res. 2005;65:7896–7901. doi: 10.1158/0008-5472.CAN-05-0506. [DOI] [PubMed] [Google Scholar]; (b) Kisselev AF, Callard A, Goldberg AL. J Biol Chem. 2006;281:8582–8590. doi: 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]
- 9.Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M, Tokhunts R, Amir O, Goddard A, Pelphrey P, Wright DL, Overkleeft HS, Kisselev AF. Chem Biol. 2009;16:1278–1289. doi: 10.1016/j.chembiol.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For a recent review see: Verdoes M, Florea BI, Van der Marel GA, Overkleeft HS. Eur J Org Chem. 2009:3301–3313.
- 11.Verdoes M, Florea BI, Van Der Linden WA, Renou D, Van Den Nieuwendijk AM, Van Der Marel GA, Overkleeft HS. Org Biomol Chem. 2007;5:1416–1426. doi: 10.1039/b702268a. [DOI] [PubMed] [Google Scholar]
- 12.Bogyo M, Shin S, McMaster JS, Ploegh HL. Chem Biol. 1998;5:307–320. doi: 10.1016/s1074-5521(98)90169-7. [DOI] [PubMed] [Google Scholar]
- 13.Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh HL. Proc Natl Acad Sci U S A. 1997;94:6629–6634. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Bioorg Med Chem Lett. 1999;9:2283–2288. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- 15.Verdoes M, Florea BI, Menendez-Benito V, Maynard CJ, Witte MD, Van Der Linden WA, Van Den Nieuwendijk AMCH, Hofmann T, Berkers CR, van Leeuwen FW, Groothuis TA, Leeuwenburgh MA, Ovaa H, Neefjes JJ, Filippov DV, Van Der Marel GA, Dantuma NP, Overkleeft HS. Chem Biol. 2006;13:1217–1226. doi: 10.1016/j.chembiol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 16.van Swieten PF, Samuel E, Hernández RO, Van Den Nieuwendijk AM, Leeuwenburgh MA, Van Der Marel GA, Kessler BM, Overkleeft HS, Kisselev AF. Bioorg Med Chem Lett. 2007;17:3402–3405. doi: 10.1016/j.bmcl.2007.03.092. [DOI] [PubMed] [Google Scholar]
- 17.Verdoes M, Florea BI, Hillaert U, Willems LI, Van Der Linden WA, Sae-Heng M, Filippov DV, Kisselev AF, Van Der Marel GA, Overkleeft HS. ChemBioChem. 2008;9:1735–1738. doi: 10.1002/cbic.200800231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verdoes M, Hillaert U, Florea BI, Sae-Heng M, Risseeuw MD, Filippov DV, Van Der Marel GA, Overkleeft HS. Bioorg Med Chem Lett. 2007;17:6169–6171. doi: 10.1016/j.bmcl.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 19.Florea BI, Verdoes M, Li N, Van der Linden WA, Geurink PP, Van den Elst H, Hofmann T, de Ru A, Van Veelen P, Tanaka K, Sasaki K, Murata S, Den Dulk H, Brouwer J, Ossendorp F, Overkleeft HS. manuscript submitted for publication [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.