

Abstract
Adipocytokines may be the molecular link between obesity and vascular disease. However, the effects of these factors on coronary vascular function have not been discerned. Accordingly, the goal of this investigation was to delineate the mechanisms by which endogenous adipose-derived factors affect coronary vascular endothelial function. Both isolated canine coronary arteries and coronary blood flow in anesthetized dogs were studied with and without exposure to adipose tissue. Infusion of adipose-conditioned buffer directly into the coronary circulation did not change baseline hemodynamics; however, endothelial-dependent vasodilation to bradykinin was impaired both in vitro and in vivo. Coronary vasodilation to sodium nitroprusside was unaltered by adipose tissue. Oxygen radical formation did not cause the impairment because quantified dihydroethidium staining was decreased by adipose tissue and neither a superoxide dismutase mimetic nor catalase improved endothelial function. Inhibition of nitric oxide (NO) synthase with L-NAME diminished bradykinin-mediated relaxations and eliminated the subsequent vascular effects of adipose tissue. In vitro measurement of NO demonstrated that adipose tissue exposure quickly lowered baseline NO and abolished bradykinin-induced NO production. The results indicate that adipose tissue releases factor(s) that selectively impair endothelial-dependent dilation via inhibition of NO synthase-mediated NO production.
Keywords: coronary circulation, perivascular adipose tissue, vascular endothelium, adipocytokine, nitric oxide
INTRODUCTION
Adipose tissue is an active endocrine and paracrine organ that releases a variety of cytokines that influence many physiologic and pathophysiologic conditions (20). Recent studies have implicated perivascular adipose tissue in the pathogenesis of vascular dysfunction and disease (12; 13; 3; 17). Adipocyte production of pathogenic adipocytokines and/or chemokines have been shown to stimulate chemotaxis (12), inflammation (15), smooth muscle proliferation (1) and activate key mediators of atherogenesis (20). These potentially harmful adipocytokines have been speculated to promote coronary atherogenesis via local paracrine and vasocrine pathways (30). In addition, perivascular adipose tissue has also been shown to significantly attenuate contractile responses of rat aorta (6; 10; 21; 34), rat mesenteric (9; 36) and human internal thoracic arteries (11) to a variety of vasoconstrictor compounds. However, there are also abnormalities of vasodilation associated with adipocytokines. Our laboratory recently demonstrated that the adipocytokines leptin and resistin significantly impair canine coronary endothelial-dependent vasodilation both in vivo and in vitro (4; 16) in normal animals. As might be expected, we have also demonstrated that obesity and insulin resistance alter the control of coronary blood flow and significantly impair the balance between oxygen delivery and myocardial metabolism (32). This vascular dysfunction is related to sensitization of key coronary vasoconstrictor pathways (5; 17; 38), some of which could be influenced by factors released from adipose tissue.
The goal of the present investigation was to delineate the mechanisms by which endogenous adipose-derived factors affect coronary vascular endothelial production of nitric oxide (NO) at the level of both microvessels and conduit arteries. Potential mechanisms were examined by in vitro studies in isolated canine coronary arteries with or without perivascular adipose tissue as well as in vivo experiments in open-chest anesthetized dogs to evaluate microvascular resistance regulation by endothelial vasodilators before and during treatment with adipose-conditioned buffer. This approach was used to document the effects of adipose tissue on coronary vascular reactivity in large arteries in vitro where coronary disease predominantly occurs as well as coronary flow responses in vivo which reflect alterations in function of microvascular resistance vessels.
MATERIALS AND METHODS
This investigation was approved by the Institutional Animal Care and Use Committee in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85–23, Revised 1996). All dogs studied were lean mongrel dogs weighing between 20 and 30 kg.
Functional assessment of isolated epicardial coronary rings
Isolated coronary artery studies were performed as previously described (4; 16). Briefly, left circumflex coronary arteries from lean dogs were dissected from the heart with or without the naturally occurring perivascular adipose tissue surrounding the conduit artery. Representative coronary arteries with or without perivascular adipose tissue were stained with Sudan IV and are shown in Fig 1. The arteries were cut into 3 mm rings and mounted in organ baths for isometric tension studies. Perivascular adipose tissue was either rigorously removed from the arterial rings or allowed to remain intact (approximately 0.25 g adipose per ring). Optimal length was found by assessing contraction to 60 mM KCl. Passive tension was increased in gram increments until there was < 10% change in active KCl contractions.
Fig 1.
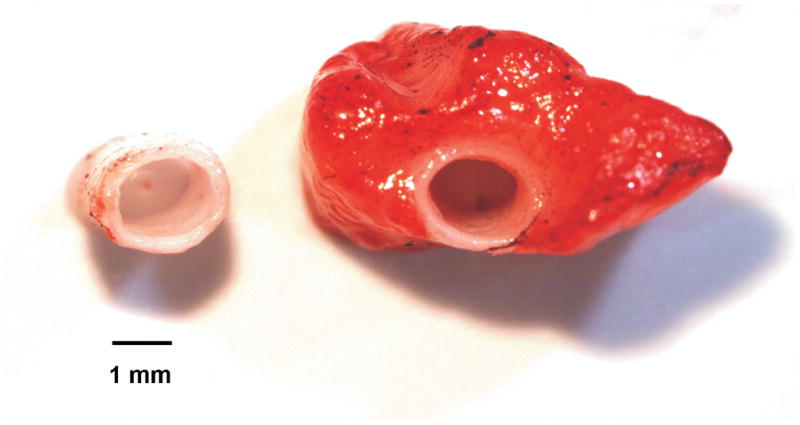
Representative isolated left circumflex coronary arteries with or without perivascular adipose tissue stained with Sudan IV (adipose tissue staining red).
Endothelial function was assessed by the addition of graded concentrations of bradykinin (0.1 nM/L–10 μM/L, n = 5) or sodium nitroprusside (1.0 nM/L–0.1mM/L, n = 3) to the tissue bath. In additional studies, bradykinin concentration responses were conducted in the presence of the NO synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME, 300 μM/L, n = 7), the superoxide dismutase mimetic tempol (10 μM/L, n = 3), and the H2O2 degrading enzyme catalase (1000 U/ml, n = 4). All results obtained during bradykinin and sodium nitroprusside dose response experiments are reported as the percent relaxation for each animal (Fig 3, 4, 5 and 6). 100 percent relaxation is defined as a return to the level of tension prior to U46619 contraction.
Fig 3.

Adipose tissue significantly attenuates coronary endothelial-dependent vasodilation to bradykinin in vivo (3A, n = 6) and in isolated coronary arteries (3B, n = 5). * = P < 0.01.
Fig 4.
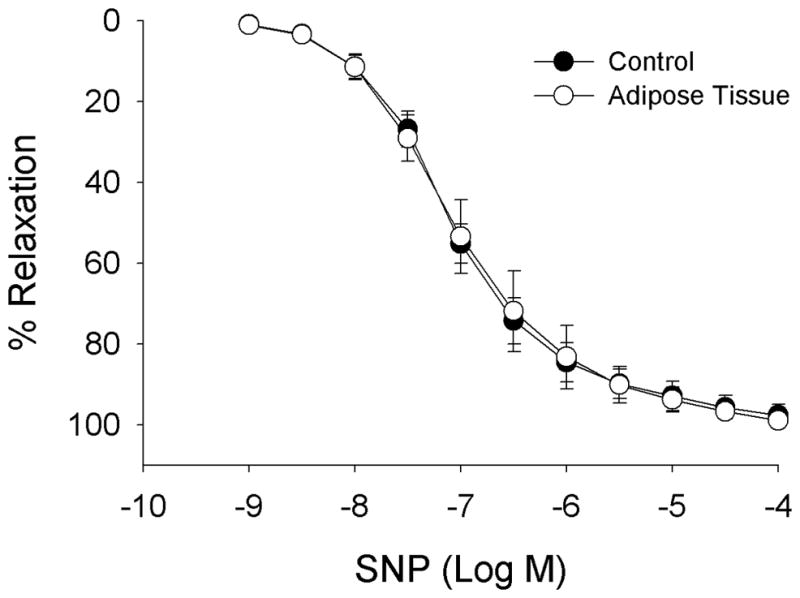
Adipose tissue has no significant effect on coronary endothelial-independent vasodilation to sodium nitroprusside (SNP) n = 3.
Fig 5.

DHE staining showed a significant decrease in fluorescence in coronary arteries with perivascular adipose tissue (Representative pictures A and B; inset shows average DHE fluorescence). The superoxide dismutase mimetic tempol did not improve reactivity of coronary arteries with perivascular adipose tissue (D); rather it significantly impaired reactivity of arteries without (C) and with (D) adipose tissue (n = 3). In addition, tempol also failed to improve reactivity in vivo (E, n = 5). * P < 0.001. Enzymatic degradation of peroxide (H2O2) with catalase also did not reverse the endothelial impairment *P < 0.01 (F, n = 4).
Fig 6.
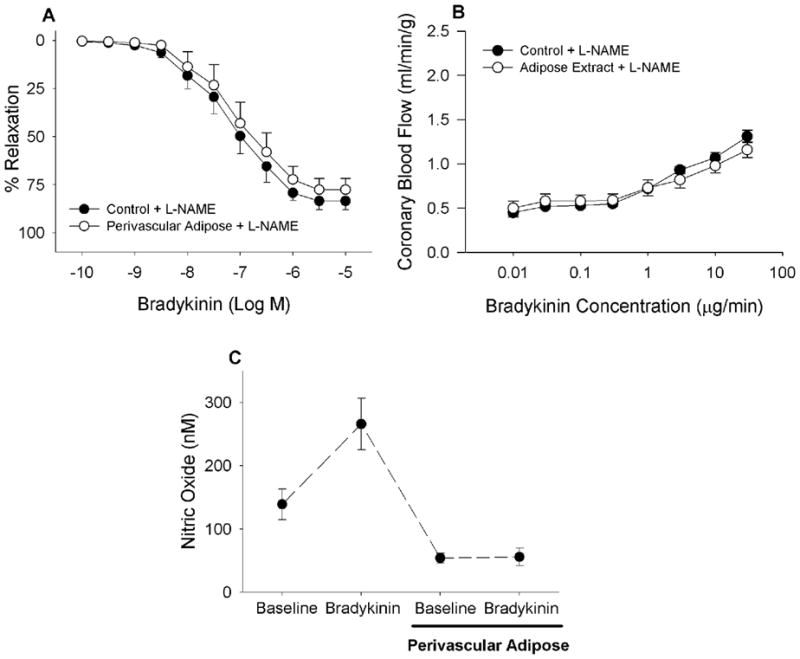
Inhibition of nitric oxide synthase with L-NAME reversed the effect of adipose tissue on bradykinin-mediated vasodilation in isolated coronary arteries (A, n = 7) and in open-chest anesthetized dogs (B, n = 5). Perivascular adipose tissue markedly impaired bradykinin (400 nM)-mediated increases in NO production (C, n = 3).
Coronary blood flow evaluation of microvascular performance
Dogs were initially sedated with morphine (3 mg/kg, sc) and anesthetized with α-chloralose (100 mg/kg, iv). The animals were then intubated and mechanically ventilated (Harvard respirator) with room air supplemented with oxygen. A catheter was placed in the right femoral vein for intravenous administration of supplemental anesthetic and sodium bicarbonate. The femoral artery was then cannulated to supply blood to an extracorporeal perfusion system which subsequently perfused the left anterior descending coronary artery (LAD) at a controlled pressure. A left lateral thoractomy was performed to expose the heart, and the LAD was isolated distal to its first major diagonal branch. After the administration of heparin (500 U/kg, iv) the LAD was cannulated with a stainless steel cannula (3 mm external diameter, 2.2 mm internal diameter). Coronary perfusion pressure was measured through a saline filled catheter advanced to the orifice of the LAD cannula. The pressure of the perfusion system was held constant at 100 mmHg by a servo-controlled roller pump. Similarly, an in line Transonic Systems flow transducer (Ithaca, NY) was used in the perfusion system to measure coronary blood flow. Data were continuously recorded on IOX data acquisition software from Emka Technologies (Falls Church, VA). The preparation was allowed a ~30 minute recovery time before data were recorded for analysis.
In order to test the effects of endogenous adipose-derived factors on coronary microvascular endothelial function in vivo, bradykinin was infused (0.3–3.0 μg/min) in the absence and presence of adipose conditioned buffer (n = 6). The adipose-conditioned buffer was prepared in phosphate buffered saline that was allowed to shake and mix with parietal pericardial adipose tissue (3 g/ml) for 30 min at 37°C in a shaking water bath. The conditioned buffer was then filtered (0.2 μm) and infused directly into the coronary circulation via the perfusion system (0.3 ml/min). In additional studies, bradykinin was simultaneously infused with either L-NAME (150 μg/min, n = 5) or tempol (10 mg/min, n = 5). Lastly, studies were also conducted to determine if adipose-conditioned buffer had any direct effect on coronary hemodynamics. During these studies, adipose-conditioned buffer was infused at various rates (0.3–3.0 ml/min, n = 3) without the administration of bradykinin.
Nitric oxide measurements
NO concentration was measured in isolated coronary arteries (n = 3) before and during exposure to 400 nM bradykinin with or without the addition of perivascular adipose tissue. This concentration of bradykinin was a supramaximal concentration for NO production by isolated arterial segments. NO was evaluated by a polarographic technique, using a carbon fiber, recessed-tip glass microelectrodes as previously described (37). The microelectrodes had a sharpened outer tip diameter of 7–10 μm and were polarized at +0.7 or +0.9 V relative to either a World Precision Instruments carbon fiber reference electrode (Sarasota, FL)or a simple silver-silver chloride electrode. The currents generated ranged from 0–20 pA. A calibration curve was established by measurement of the microelectrode current at NO concentrations of 0, 600, and 1,200 nM. These concentrations were based on the composition of the NO-N2 precision calibration gases in saline at 37.5°C. The working resolution of the microelectrodes is typically<10 nM, allowing for random noise and current drift. The microelectrodes are completely insensitive to oxygen when positively polarized.
Rings of vessel were placed in a perfused bath (5 ml/min, 5 ml bath volume) of media similar to that used for tension studies and equilibrated with 95% oxygen and 5% carbon dioxide. The tip of the NO microelectrode was placed in the lumen of the vessel and touched an endothelial surface. The baseline concentration of NO was measured followed by the response to bradykinin (400 nM). Thereafter, the adipose tissue removed from the arterial ring was placed upstream of the ring in the flowing media for 30 minutes before repeating the NO measurements at rest and during bradykinin exposure.
Dihydroethidium staining
Dihydroethidium (DHE) staining for superoxide (O2−) was carried out as described previously (4). Left circumflex coronary arteries with (n = 5) and without (n = 5) perivascular adipose tissue were incubated with 10 μM DHE at 37°C for 30 min with or without tempol (10 μM/L) administration. The arteries were embedded in OCT and flash frozen in liquid nitrogen. Tissue sections (10 μm) were then prepared using a cryostat on thaw-mounted slides. Ethidium fluorescence was assessed with 508 nm excitation and 615 nm emissions. Scion Image for Windows was used to perform histogram analysis of brightness in images of DHE-stained arteries.
Adipokine content in adipose tissue extract
A multiplexed biomarker immunoassay from Linco Laboratories was used to measure the concentration of key adipokines in the adipose-conditioned buffer (Table 2). The assay was conducted according to the manufacturer’s specifications. Briefly, the assay functions by using antibody coated beads, which are selective for specific human adipokines. Adipose-conditioned buffer samples used during the in vivo studies were collected, filtered (0.2 μm) and individually measured in triplicate on a 96 well plate. The antibody coated beads were incubated with each individual sample and allowed to bind overnight. Biotinylated detection antibodies were added to each well, and streptavidin-phycoerythrin was subsequently added in order to detect the fluorescence on each bead. A Luminex Instrument was used to both identify which adipokines were expressed and to quantify their relative concentrations.
Table 2.
Adipokine expression in adipose-conditioned buffer. A multiplexed biomarker immunoassay was conducted in order to measure adipokine concentrations present within the adipose-conditioned buffer.
| Adipokines | Concentration in Adipose-conditioned buffer (pg/ml) |
|---|---|
| TNF-α | 0.04 ± 0.01 |
| IL-1β | 0.24 ± 0.02 |
| PAI-1(active) | 0.42 ± 0.08 |
| MCP-1 | 0.45 ± 0.01 |
| HGF | 1.64 ± 0.20 |
| Leptin | 2.40 ± 0.20 |
| Resistin | 5338.00 ± 1032.00 |
Values are mean ± SE for all measured samples (n = 10). IL-1 β–interleukin 1β, PAI-1 -plasminogen activator inhibitor 1, MCP-1 - monocyte chemoattractant protein-1, HGF -hepatocyte growth factor.
Statistical analyses
Data are presented as mean ± standard error. For both in vitro and in vivo studies, a two-way repeated measures ANOVA was used to test the effects of the presence or absence of adipose tissue or conditioned buffer (Factor A) and various doses of sodium nitroprusside or bradykinin (Factor B) on coronary physical or chemical responses (Sigma Stat 3.0 Software). Identical statistics were performed for studies conducted in the presence of L-NAME, tempol and catalase. For the in vitro experiments, data were analyzed per animal. When statistical differences were found by ANOVA, a Student-Newman-Keuls multiple comparison test was performed. The criterion for statistical significance was P < 0.05 in all tests.
RESULTS
Adipose tissue and baseline hemodynamics
Data shown in Figure 2 demonstrate that increases in the intracoronary infusion rate of the adipose-conditioned buffer (0.3–3.0 ml/min) did not significantly affect baseline coronary blood flow (P = 0.68). Effects of adipose-conditioned buffer, L-NAME and tempol on baseline coronary blood flow, mean aortic pressure and heart rate are given in Table 1. Intracoronary infusion of adipose-conditioned buffer (0.3 mL/min) did not significantly affect coronary blood flow (P = 0.58), mean aortic pressure (P = 0.96) or heart rate (P = 0.89). Baseline coronary hemodynamics were also unaffected by adipose-conditioned buffer in the presence of L-NAME (P = 0.58) or the superoxide dismutase mimetic tempol (P = 0.19). The average concentrations of adipokines in adipose-conditioned buffer are presented in Table 2. Notably, the concentration of resistin was substantially greater than the other adipokines.
Fig 2.
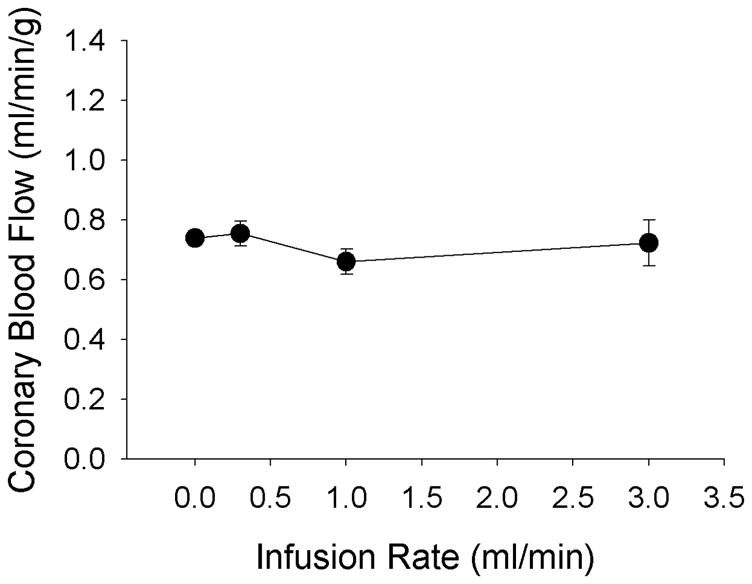
Direct infusion of endogenous adipose-derived factors from adipose-conditioned buffer into canine coronary circulation has no effect on baseline coronary blood flow (n = 3).
Table 1.
Effects of adipose-conditioned buffer, L-NAME and tempol on baseline hemodynamic data in anesthetized, open-chest dogs.
| Coronary Blood Flow (ml/min/g) | Aortic Pressure (mmHg) | Heart Rate (beats/min) | |
|---|---|---|---|
| Untreated | |||
| Control (n = 6) | 0.50 ± 0.01 | 103 ± 6 | 87 ± 13 |
| Adipose-conditioned buffer (n = 6) | 0.61 ± 0.05 | 96 ± 9 | 110 ± 24 |
| L-NAME | |||
| Control (n = 5) | 0.45 ± 0.05 | 105 ± 8 | 72 ± 9 |
| Adipose-conditioned buffer (n = 5) | 0.50 ± 0.08 | 103 ± 9 | 90 ± 11 |
| Tempol | |||
| Control (n = 5) | 0.69 ± 0.03 | 102 ± 9 | 77 ± 10 |
| Adipose-conditioned buffer (n = 5) | 0.94 ± 0.15 | 98 ± 12 | 94 ± 8 |
Values are mean ± SE. n = number of dogs.
Adipose tissue and coronary endothelial function
Adipose-conditioned buffer exposure significantly attenuated coronary endothelial-dependent vasodilation, as judged by blood flow responses, to the higher doses of bradykinin (1–3 μg/min; ~10 nM bradykinin) in open-chest anesthetized dogs (Fig 3A; P < 0.01). In isolated coronary arteries, arterial relaxation to bradykinin was also decreased by perivascular adipose tissue (Fig 3B; 1–100 nM; P < 0.01). In contrast, adipose tissue did not significantly affect endothelial-independent relaxation to sodium nitroprusside in isolated arteries (Fig 4).
Adipose tissue and coronary reactive oxygen species
DHE staining for coronary O2 showed a significant decrease (67 ± 7% of control) in tempol-sensitive fluorescence between arteries with perivascular adipose tissue relative to arteries fully cleaned of adipose tissue (Fig 5A and 5B). Therefore, vessel exposure to adipose tissue did not increase O2− formation. Additional studies demonstrated that the administration of tempol (10 μM) to scavenge oxygen radicals did not improve reactivity of arteries with perivascular adipose tissue to bradykinin (Fig 5D). Instead, tempol significantly impaired relaxation of arteries, with or without adipose tissue, to bradykinin (Fig 5C and 5D, P < 0.001). These findings were confirmed by additional in vivo studies in Figure 5E which found that tempol failed to reverse the adipose tissue-induced impairment of bradykinin-mediated coronary vasodilation in open-chest dogs (Fig 5E, P < 0.001). To test for H2O2 effects, catalase was applied prior to in vitro relaxation studies. Enzymatic degradation of H2O2 with catalase diminished the relaxation to bradykinin at 1 nM to 30 nM (Fig 5F) in comparison to the control relaxation shown in Figure 5C. However, arteries with perivascular adipose tissue continued to display further impairment (P < 0.01) to bradykinin.
Adipose tissue and nitric oxide production
Inhibition of NO synthase with L-NAME eliminated the relaxation differences between coronary artery rings with or without perivascular adipose tissue (Fig 6A, P = 0.32). Also note that compared to responses of untreated vessels in Figure 3B, L-NAME caused a right shift of the dose-relaxation curve indicating major suppression of endothelial dependent relaxation. These initial observations were further supported by in vivo studies demonstrating that pretreatment with L-NAME abolished the effect of adipose-conditioned buffer on bradykinin-mediated coronary vasodilation (Fig 6B; P = 0.24). Again, L-NAME caused a significant attenuation of the dose-blood flow response curve indicating major suppression of endothelial dependent relaxation in the in vivo coronary microvasculature (Fig. 3A vs. Fig 6B). Importantly, measures of coronary NO concentration with an NO sensitive microelectrode showed that adipose tissue both markedly lowered basal NO production by 60% and eliminated coronary endothelial NO production in response to bradykinin (Fig 6C, P < 0.05).
DISCUSSION
The present investigation was designed to examine the mechanisms by which endogenous adipose-derived factors might impair coronary endothelial function within both the coronary microcirculation and conduit arteries. The major new findings from this study are that factor(s) released from adipose tissue: 1) do not affect baseline coronary blood flow or systemic hemodynamics; 2) significantly impair endothelial-dependent vasodilation to bradykinin in both in vivo preparations and isolated coronary arteries; 3) do not alter coronary endothelial-independent vasodilation to sodium nitroprusside, i.e. vascular smooth muscle response to NO; 4) do not significantly increase coronary O2− production or H2O2-mediated vasodilation; and 5) markedly attenuate coronary artery endothelial production of NO in isolated vessels. Taken together, these data indicate that adipose tissue releases factor(s) that selectively impair endothelial-dependent dilation via inhibition of NO synthase-mediated NO production. This impairment is independent of alterations in coronary vascular smooth muscle response to NO, O2− mediated decreases in NO-bioavailability or H2O2-mediated coronary vasodilation.
Adipose tissue and baseline coronary hemodynamics
Before trying to interpret any effect of adipose tissue, it was necessary to test the direct effect of adipose-derived factors on baseline coronary blood flow, arterial pressure and heart rate (Fig. 2 and Table 1). Our results indicate that factors released by adipose tissue do not directly affect overall coronary microvascular resistance under normal resting conditions. Importantly, the present findings are consistent with earlier studies and the current L-NAME studies (Table 1) which demonstrated that blockade of NO synthase-mediated NO production has little effect on baseline coronary blood flow (7; 35).
Effects of adipose tissue on coronary endothelial function
Data from this study are the first to document that endogenous factors released from adipose tissue selectively inhibit coronary endothelial-dependent dilation to bradykinin judged both by in vivo measures of coronary blood flow (regulated predominantly by microvessels; Figs 3, 5, and 6), by relaxation of isolated coronary arteries (where atherosclerotic disease predominantly occurs; Fig 3) and by in vitro measurement of NO (Fig 6C). Our findings indicate that the adipose-induced impairment of coronary endothelial function is mediated by a selective impairment of NO synthase mediated NO production because the effect of adipose tissue on bradykinin-mediated dilation was reversed by L-NAME (Fig 6A–B) and direct measures of NO demonstrated a decline in both basal NO and bradykinin stimulated NO production (Fig 6C). We propose that the impaired relaxation is specific to NO generation as we did not detect any effect of adipose tissue on relaxations to sodium nitroprusside (Fig 4) or to bradykinin in the presence of L-NAME (Fig 6), suggesting that adipose tissue does not affect dilation to prostacyclin or endothelial-derived hyperpolarizing factors.
The findings documented in this investigation are potentially marred by a few concerns. First, there was a potential that the measured endothelial impairment was simply an artifact of decreasing responses to bradykinin after repeated doses (i.e. tachyphylaxis). However, a recent study from our laboratory demonstrated no tachyphylaxis to repeated treatments of bradykinin both in vivo in open-chest anesthetized dogs and in vitro in isolated coronary arteries (4). Hence any measured experimental differences in bradykinin-mediated dilation are not an artifact of repeated bradykinin dose-response curves. Second, the isometric tension and NO production measurements were done under circumstances where there was at most a trivial amount of hemoglobin, oxyhemoglobin or myoglobin. Therefore, these iron containing organ compounds were highly unlikely to suppress NO bioavailability (13; 14; 39). This conclusion is further supported by our in vivo coronary flow data.
Adipose tissue and coronary reactive oxygen species
Recently, many investigations have focused on the role of reactive oxygen species in regulating endothelial function (2; 29). Specifically, O2− has been shown to decrease NO bioavailability by reacting to produce peroxynitrite (22). Additional studies have also shown that H2O2 acts as an endothelium-derived hyperpolarizing factor and metabolic vasodilator of the coronary circulation (25; 28; 29; 31). Together, both O2− and H2O2 are potential direct and/or indirect mediators of the observed endothelial dysfunction caused by adipose tissue. The fact that DHE staining of arteries with perivascular adipose tissue showed a significant decrease in fluorescence (Fig 5A–B) argues that adipose tissue does not impair coronary endothelial-dependent vasodilation via increases in O2− production. This is supported by additional in vitro and in vivo studies with tempol that showed this O2− dismutase mimetic did not significantly improve the adipose tissue induced impairment of bradykinin-mediated coronary vasodilation (Fig 5C, 5D, 5E). In fact, administration of tempol significantly decreased bradykinin-mediated relaxation suggesting that O2− may function as a vasodilator, a hypothesis supported by other recent investigations (18; 19; 24; 33). Enzymatic degradation of H2O2 with catalase also failed to reverse adipose-induced endothelial impairment (Fig 5F). Taken together these important data argue against O2− or H2O2 as a mechanism of adipose-induced coronary endothelial dysfunction.
Identity of adipose tissue derived vasoactive factor(s)
We performed a “targeted” immunoassay to measure the expression of key adipocytokines in adipose-conditioned buffer (Table 2). This assay was in no way exhaustive of all potential adipose-derived mediators; however this approach allowed us to identify candidate adipokines that could contribute to the adipose-induced endothelial impairment. It is important to point out that the use of anti-human antibodies for this canine adipose tissue assay was an unavoidable limitation of this assay. However, it should be pointed out that there was no clear correlation between the % homology of human vs. canine adipokine protein sequences (ranged from 63–92%) and the measured adipokine concentrations (Table 2) which suggests that antibody specificity alone was not the reason for the low concentrations measured.
Using this targeted immunoassay we found that resistin levels were markedly high relative to the other measured adipokines (5.9 ± 1.4 ng/ml, Table 2). This resistin concentration results in an estimated average plasma concentration of 0.19 ± 0.09 ng/ml, which is significantly lower than concentrations reported in human plasma (23; 27) and the 10 ng/ml that our laboratory (4) as well as others (17; 33) previously demonstrated to impair coronary endothelial-dependent vasodilation. Additionally, the present study found that L-NAME reversed the effect of adipose tissue on bradykinin-mediated dilation (Fig 6A and 6B) but an early study from our laboratory showed that L-NAME failed to inhibit resistin-induced suppression of bradykinin dilation (4). These findings do not support resistin as the primary mechanism of adipose-induced coronary endothelial dysfunction.
Other adipokines could potentially contribute the observed coronary effects of adipose tissue. Recently, our laboratory found that leptin induces significant impairment of coronary endothelial-dependent vasodilation (16). However, the measured concentration of leptin in the adipose-conditioned buffer (2.40 ± 0.20 pg/ml) is substantially lower than the ~10 ng/ml previously found to be necessary for leptin-induced coronary endothelial dysfunction (16). Therefore, as was the case for resistin, the functional concentration for leptin was below the pathologic concentration that negatively influenced endothelial-dependent dilation. Alternatively, TNF-α and long chain fatty acids have both been shown to attenuate endothelial-dependent vasodilation, albeit via mechanisms that elevate oxidative stress/O2− production (3; 8; 26) which is inconsistent with our findings with adipose tissue (Fig 5). Therefore, at present it is difficult to attribute the adipose-induced impairment to any known adipocytokine. However, it is plausible that several different adipose-derived factors could act in an additive and/or synergistic manner to diminish coronary NO production. Clearly future studies are needed to identify the exact adipose-derived factor(s) and cellular/molecular mechanisms involved.
In conclusion, results from this study are the first to demonstrate that endogenous adipose-derived factors diminish production of coronary endothelial NO as judged by pharmacological challenges and direct measurements of endothelial NO. The overall findings implicate that adipocytokines have the ability to rapidly depress coronary microvascular and arterial NO generation. It is important to note that during in vivo circumstances, perivascular adipose tissue has little or no access to the endothelial cells of either arteries or arterioles. We are only demonstrating that adipose tissue contains factors that when released and circulated throughout the heart cause potentially dire consequences for endothelial mediated dilation. These results provide us with a new potential explanation of how alterations in adipokine expression in obesity may contribute to the development of vascular dysfunction and coronary atherosclerosis.
Acknowledgments
The authors wish to thank Eli Lilly Company for their assistance with the multiplexed biomarker immunoassay from Linco Laboratories. Additionally, the authors wish to thank Falon Greer for technical assistance with these studies. This work was supported by HL67804 (JDT) and HL20605 (HGB).
References
- 1.Barandier C, Montani JP, Yang Z. Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: effects of aging and obesity. Am J Physiol Heart Circ Physiol. 2005;289:H1807–H1813. doi: 10.1152/ajpheart.01259.2004. [DOI] [PubMed] [Google Scholar]
- 2.Barlow RS, El-Mowafy AM, White RE. H(2)O(2) opens BK(Ca) channels via the PLA(2)-arachidonic acid signaling cascade in coronary artery smooth muscle. Am J Physiol Heart Circ Physiol. 2000;279:H475–H483. doi: 10.1152/ajpheart.2000.279.2.H475. [DOI] [PubMed] [Google Scholar]
- 3.Christon R, Marette A, Badeau M, Bourgoin F, Melancon S, Bachelard H. Fatty acid-induced changes in vascular reactivity in healthy adult rats. Metabolism. 2005;54:1600–1609. doi: 10.1016/j.metabol.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Dick GM, Katz PS, Farias M, III, Morris M, James J, Knudson JD, Tune JD. Resistin impairs endothelium-dependent dilation to bradykinin, but not acetylcholine, in the coronary circulation. Am J Physiol Heart Circ Physiol. 2006;291:H2997–H3002. doi: 10.1152/ajpheart.01035.2005. [DOI] [PubMed] [Google Scholar]
- 5.Dincer UD, Araiza AG, Knudson JD, Molina PE, Tune JD. Sensitization of coronary alpha-adrenoceptor vasoconstriction in the prediabetic metabolic syndrome. Microcirculation. 2006;13:587–595. doi: 10.1080/10739680600885228. [DOI] [PubMed] [Google Scholar]
- 6.Dubrovska G, Verlohren S, Luft FC, Gollasch M. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1107–H1113. doi: 10.1152/ajpheart.00656.2003. [DOI] [PubMed] [Google Scholar]
- 7.Duncker DJ, Merkus D. Acute adaptations of the coronary circulation to exercise. Cell Biochem Biophys. 2005;43:17–35. doi: 10.1385/CBB:43:1:017. [DOI] [PubMed] [Google Scholar]
- 8.Esenabhalu VE, Schaeffer G, Graier WF. Free fatty acid overload attenuates Ca2+ signaling and NO production in endothelial cells. Antioxid Redox Signal. 2003;5:147–153. doi: 10.1089/152308603764816505. [DOI] [PubMed] [Google Scholar]
- 9.Galvez B, de CJ, Herold D, Dubrovska G, Arribas S, Gonzalez MC, Aranguez I, Luft FC, Ramos MP, Gollasch M, Fernandez Alfonso MS. Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 2006;26:1297–1302. doi: 10.1161/01.ATV.0000220381.40739.dd. [DOI] [PubMed] [Google Scholar]
- 10.Gao YJ, Lu C, Su LY, Sharma AM, Lee RM. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151:323–331. doi: 10.1038/sj.bjp.0707228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao YJ, Zeng ZH, Teoh K, Sharma AM, Abouzahr L, Cybulsky I, Lamy A, Semelhago L, Lee RM. Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J Thorac Cardiovasc Surg. 2005;130:1130–1136. doi: 10.1016/j.jtcvs.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 12.Henrichot E, Juge-Aubry CE, Pernin A, Pache JC, Velebit V, Dayer JM, Meda P, Chizzolini C, Meier CA. Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol. 2005;25:2594–2599. doi: 10.1161/01.ATV.0000188508.40052.35. [DOI] [PubMed] [Google Scholar]
- 13.Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res. 1987;61:866–879. doi: 10.1161/01.res.61.6.866. [DOI] [PubMed] [Google Scholar]
- 14.Joshi MS, Ferguson TB, Jr, Han TH, Hyduke DR, Liao JC, Rassaf T, Bryan N, Feelisch M, Lancaster JR., Jr Nitric oxide is consumed, rather than conserved, by reaction with oxyhemoglobin under physiological conditions. Proc Natl Acad Sci U S A. 2002;99:10341–10346. doi: 10.1073/pnas.152149699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juge-Aubry CE, Henrichot E, Meier CA. Adipose tissue: a regulator of inflammation. Best Pract Res Clin Endocrinol Metab. 2005;19:547–566. doi: 10.1016/j.beem.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 16.Knudson JD, Dincer UD, Zhang C, Swafford AN, Jr, Koshida R, Picchi A, Focardi M, Dick GM, Tune JD. Leptin Receptors are Expressed in Coronary Arteries and Hyperleptinemia Causes Significant Coronary Endothelial Dysfunction. Am J Physiol Heart Circ Physiol. 2005 doi: 10.1152/ajpheart.01159.2004. [DOI] [PubMed] [Google Scholar]
- 17.Knudson JD, Rogers PA, Dincer UD, Bratz IN, Araiza AG, Dick GM, Tune JD. Coronary vasomotor reactivity to endothelin-1 in the prediabetic metabolic syndrome. Microcirculation. 2006;13:209–218. doi: 10.1080/10739680600556894. [DOI] [PubMed] [Google Scholar]
- 18.Kontos HA. Oxygen radicals from arachidonate metabolism in abnormal vascular responses. Am Rev Respir Dis. 1987;136:474–477. doi: 10.1164/ajrccm/136.2.474. [DOI] [PubMed] [Google Scholar]
- 19.Kontos HA, Wei EP, Kukreja RC, Ellis EF, Hess ML. Differences in endothelium-dependent cerebral dilation by bradykinin and acetylcholine. Am J Physiol. 1990;258:H1261–H1266. doi: 10.1152/ajpheart.1990.258.5.H1261. [DOI] [PubMed] [Google Scholar]
- 20.Lau DC, Dhillon B, Yan H, Szmitko PE, Verma S. Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005;288:H2031–H2041. doi: 10.1152/ajpheart.01058.2004. [DOI] [PubMed] [Google Scholar]
- 21.Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002;16:1057–1063. doi: 10.1096/fj.02-0024com. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Jaramillo P, Casas JP, Bautista L, Serrano NC, Morillo CA. An integrated proposal to explain the epidemic of cardiovascular disease in a developing country. From socioeconomic factors to free radicals. Cardiology. 2001;96:1–6. doi: 10.1159/000047379. [DOI] [PubMed] [Google Scholar]
- 23.Monzillo LU, Hamdy O, Horton ES, Ledbury S, Mullooly C, Jarema C, Porter S, Ovalle K, Moussa A, Mantzoros CS. Effect of lifestyle modification on adipokine levels in obese subjects with insulin resistance. Obes Res. 2003;11:1048–1054. doi: 10.1038/oby.2003.144. [DOI] [PubMed] [Google Scholar]
- 24.Oltman CL, Kane NL, Miller FJ, Jr, Spector AA, Weintraub NL, Dellsperger KC. Reactive oxygen species mediate arachidonic acid-induced dilation in porcine coronary microvessels. Am J Physiol Heart Circ Physiol. 2003;285:H2309–H2315. doi: 10.1152/ajpheart.00456.2003. [DOI] [PubMed] [Google Scholar]
- 25.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol. 2007;292:H93–100. doi: 10.1152/ajpheart.00819.2006. [DOI] [PubMed] [Google Scholar]
- 26.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99:69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 27.Pilz S, Weihrauch G, Seelhorst U, Wellnitz B, Winkelmann BR, Boehm BO, Marz W. Implications of resistin plasma levels in subjects undergoing coronary angiography. Clin Endocrinol (Oxf) 2007;66:380–386. doi: 10.1111/j.1365-2265.2007.02743.x. [DOI] [PubMed] [Google Scholar]
- 28.Rogers PA, Chilian WM, Bratz IN, Bryan RM, Jr, Dick GM. H2O2 activates redox- and 4-aminopyridine-sensitive Kv channels in coronary vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2007;292:H1404–H1411. doi: 10.1152/ajpheart.00696.2006. [DOI] [PubMed] [Google Scholar]
- 29.Rogers PA, Dick GM, Knudson JD, Focardi M, Bratz IN, Swafford AN, Jr, Saitoh S, Tune JD, Chilian WM. H2O2-induced redox-sensitive coronary vasodilation is mediated by 4-aminopyridine-sensitive K+ channels. Am J Physiol Heart Circ Physiol. 2006;291:H2473–H2482. doi: 10.1152/ajpheart.00172.2006. [DOI] [PubMed] [Google Scholar]
- 30.Sacks HS, Fain JN. Human epicardial adipose tissue: a review. Am Heart J. 2007;153:907–917. doi: 10.1016/j.ahj.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 31.Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, Rogers PA, Knudson JD, Dick GM, Swafford A, Chilian WM. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol. 2006;26:2614–2621. doi: 10.1161/01.ATV.0000249408.55796.da. [DOI] [PubMed] [Google Scholar]
- 32.Setty S, Sun W, Tune JD. Coronary blood flow regulation in the prediabetic metabolic syndrome. Basic Res Cardiol. 2003;98:416–423. doi: 10.1007/s00395-003-0418-7. [DOI] [PubMed] [Google Scholar]
- 33.Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats. Evidence that reactive oxygen species activate K+ channels. Stroke. 1997;28:2290–2294. doi: 10.1161/01.str.28.11.2290. [DOI] [PubMed] [Google Scholar]
- 34.Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A. 1991;13:277–296. doi: 10.3109/10641969109042063. [DOI] [PubMed] [Google Scholar]
- 35.Tune JD, Gorman MW, Feigl EO. Matching coronary blood flow to myocardial oxygen consumption. J Appl Physiol. 2004;97:404–415. doi: 10.1152/japplphysiol.01345.2003. [DOI] [PubMed] [Google Scholar]
- 36.Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y, Gollasch M. Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension. 2004;44:271–276. doi: 10.1161/01.HYP.0000140058.28994.ec. [DOI] [PubMed] [Google Scholar]
- 37.Zani BG, Bohlen HG. Transport of extracellular l-arginine via cationic amino acid transporter is required during in vivo endothelial nitric oxide production. Am J Physiol Heart Circ Physiol. 2005;289:H1381–H1390. doi: 10.1152/ajpheart.01231.2004. [DOI] [PubMed] [Google Scholar]
- 38.Zhang C, Knudson JD, Setty S, Araiza A, Dincer UD, Kuo L, Tune JD. Coronary arteriolar vasoconstriction to angiotensin II is augmented in prediabetic metabolic syndrome via activation of AT1 receptors. Am J Physiol Heart Circ Physiol. 2005;288:H2154–H2162. doi: 10.1152/ajpheart.00987.2004. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Hogg N. Mixing artifacts from the bolus addition of nitric oxide to oxymyoglobin: implications for S-nitrosothiol formation. Free Radic Biol Med. 2002;32:1212–1219. doi: 10.1016/s0891-5849(02)00829-8. [DOI] [PubMed] [Google Scholar]